

Abstract
We explored the role of a chromatin-associated nuclear protein HMGB1 in apoptotic response to widely used anticancer drugs. A murine fibroblast model system generated from Hmgb1+/+ and Hmgb1−/− mice was used to assess the role of HMGB1 protein in cellular response to anticancer nucleoside analogs and precursors which act without destroying integrity of DNA. Chemosensitivity experiments with 5-fluorouracil (FU), cytosine arabinoside (araC), and mercaptopurine (MP) demonstrated that Hmgb1−/− MEFs were 3–10 times more resistant to these drugs compared with Hmgb1+/+ MEFs. Hmgb1-deficient cells showed compromised cell cycle arrest and reduced caspase activation after treatment with MP and araC. Phosphorylation of p53 at Ser12 (corresponding to Ser 9 in human p53) and Ser18 (corresponding to Ser 15 in human p53), as well as phosphorylation of H2AX after drug treatment was reduced in Hmgb1-deficient cells. Trans-activation experiments demonstrated diminished activation of pro-apoptotic promoters Bax, Puma, and Noxa in Hmgb1-deficient cells after treatment with MP or araC, consistent with reduced transcriptional activity of p53. For the first time, we demonstrated that Hmgb1 is an essential activator of cellular response to genotoxic stress caused by chemotherapeutic agents (thiopurines, cytarabine and 5-fluorouracil), which acts at early steps of antimetabolite-induced stress by stimulating phosphorylation of two DNA damage markers p53 and H2AX. This finding makes HMGB1 a potential target for modulating activity of chemotherapeutic antimetabolites. Identification of proteins sensitive to DNA lesions which occur without the loss of DNA integrity provides new insights into the determinants of drug sensitivity in cancer cells.
INTRODUCTION
HMGB1 is a versatile protein with intranuclear and extracellular functions: in the nucleus, it bends and plasticizes DNA; outside the cell, it acts as a cytokine mediator of inflammation. Despite its small size and a simple domain structure, HMGB1 facilitates numerous intranuclear processes including transcription, replication, V(D)J recombination, and transposition (Hock et al., 2007). This versatility is achieved through ability of HMGB1 to get involved into direct physical contacts with two distinct groups of macromolecules: HMGB1 reveals affinity with DNA cruciforms, bent, kinked, or chemically modified DNA; on the other hand, it interacts with a number of proteins including p53, steroid hormone receptors, general and specific transcription factors, NF-κB, DNA-PK etc (Bianchi and Agresti, 2005). These two distinct groups of binding substrates suggest that HMGB1 may provide a molecular link between distorted DNA, and proteins involved in DNA metabolism or genotoxic stress response. Therefore, HMGB1 is a potential modulator of anticancer therapy targeted against DNA.
Induction of apoptotic death in cancer cells via genotoxic stress by irradiation or chemotherapy remains the core of anticancer treatment. An important class of chemotherapeutic agents based on this principle, purine and pyrimidine antimetabolites, has been widely used for treatment of solid tumors and hematopoietic malignancies for several decades, though molecular triggers of apoptosis caused by these drugs remain elusive (Rich et al., 2004). For example, the overall response rate for FU as a single agent in advanced colorectal cancer is about 10–15%, though the combination of FU with newer chemotherapies including irinotecan and oxaliplatin has improved the response rate to 40–50%. With about 2 million people treated yearly with FU, new therapeutic strategies based on better understanding of mechanisms by which these agents induce cell death are urgently needed (Longley et al., 2003).
Several antimetabolite agents do not disrupt integrity of DNA and lead only to minute alterations in DNA geometry (Somerville et al., 2003;Sahasrabudhe et al., 1996). Instead, incorporation of these chemical moieties into DNA increases local flexibility of the double helix in the area surrounding the modification, and changes the DNA-protein interactions (Somerville et al., 2003;Krynetskaia et al., 2000;Seibert et al., 2005). Early evidence that chemotherapy-induced damage in DNA alters DNA-protein interactions in chromatin came from the work of Maybaum and Mandel, who described unilateral chromatid damage in cells treated with thiopurine (Maybaum and Mandel, 1983). DNA damage-induced changes in chromatin structure are hypothesized to serve as an initiating signal in ATM genotoxic response pathway (Bakkenist and Kastan, 2003).
Earlier, we isolated a nuclear complex with increased affinity to chemotherapy-damaged DNA (Krynetski et al., 2001;Krynetski et al., 2003). An essential component of this complex is high mobility group protein B1 (HMGB1). From our in vitro experiments, we concluded that DNA-bending protein HMGB1 plays a role of a sensor for nucleoside analogs deoxythioguanosine, deoxyfluorouridine, and cytosine arabinoside incorporated into DNA (Krynetski et al., 2001;Krynetski et al., 2003).
In contrast to other determinants of cellular sensitivity to antimetabolites, there is no known enzymatic activity for HMGB1. Here, we used a model system based on Hmgb1-knockout mouse embryonic fibroblast cells (MEFs) to elucidate the role of HMGB1 in cellular response to antimetabolite therapy. For the first time, we demonstrated that HMGB1 is an essential activator of cellular response to genotoxic stress caused by chemotherapeutic agents (thiopurines, cytarabine and 5-fluorouracil), which acts at early steps of antimetabolite-induced stress by stimulating phosphorylation of two DNA damage markers p53 and H2AX.
MATERIALS AND METHODS
Cell cultures, drug treatment, and plasmids
Mouse embryonic fibroblast cell lines (MEFs) deficient and proficient in Hmgb1 expression were generated as described previously and generously provided for this work by Dr. Bianchi (Calogero et al., 1999). Cells were maintained in DMEM medium (Fisher Scientific, Suwanee, GA) at 40–80% confluency. Treatment of MEFs was performed with drugs dissolved in DMEM medium without serum (Fisher Scientific, Suwanee, GA) as 20X stock solutions; drug concentrations (thioguanine, TG; mercaptopurine, MP; cytarabine, araC; 5-fluorouracil, FU; cladribine, CldA; fludarabine, FldA - Sigma, St. Louis, MO; clofarabine, ClfA - Genzyme, Cambridge, MA; gemcitabine, GZ - Eli Lilly, IN) were determined spectrophotometrically. Cell viability and cell count were determined by flow cytometry (Guava PCA, Guava, CA). Reporter plasmids p21-luc and pBax-luc were a generous gift from Dr. M. Oren, pNoxa-luc from Dr. T. Taniguchi, and pPuma-luc and pCMVp53 from Dr. B. Vogelstein.
Cell Growth and Chemosensitivity
Population doubling time of Hmgb1+/+ and Hmgb1−/− MEFs was determined in the middle of log phase of growth. Cells were trypsinized and counted using the ViaCount protocol implemented on Guava personal cell analyzer (Guava, CA). Chemosensitivity was evaluated using the MTT assay (CellTiter 96 cell proliferation kit, Promega, WI) after incubation of Hmgb1+/+ and Hmgb1−/− MEFs with a panel of anticancer drugs for 3–6 days as described earlier (Carmichael et al., 1987). Two thousand cells per well were plated into 96-well plates, and cultured for 3–6 days in the presence or absence of the following drugs: MP, TG, FU, araC, CldA, FldA, ClfA, and GZ. After incubation, MTT reagent was added to each well and endpoint data collected by a M2 microplate spectrophotometer (Molecular Device, CA) according to the manufacturer’s instructions. The IC50 values were calculated using GraphPad Prism (GraphPad Software Inc., CA) by fitting a sigmoid Emax model to the cell viability versus drug concentration data determined in triplicate from three independent experiments.
Incorporation of nucleoside analogs into DNA
2×106 Hmgb1+/+ or Hmgb1−/− cells were seeded at density 25,000 cell/cm2 and treated with [3H]-araC (1.6 Ci/mmol) at final concentration 1 μM or [14C]-MP (51 mCi/mmol) at final concentration 10 μM (Moravek Biochemicals, CA). After 14–28 hr incubation, cells were collected by trypsinization, and DNA extracted using DNAamp DNA Minikit (QIAGEN, CA) according to manufacturer’s instructions. 3H and 14C incorporation into DNA was determined in duplicate using a Beckman LS 6500 Scintillation counter (Beckman Coulter, CA).
Caspase activation
Caspase activity was assessed using fluorogenic substrates for caspases 2, 3, 8, 9 immobilized on BD Apoalert Caspase Assay plates (BD Biosciences, Palo Alto, CA). Hmgb1+/+ and Hmgb1−/− MEFs were grown in DMEM medium and treated with 10 μM MP or 0.5 μM araC for 14–28–42 hr. About 2×105 cells per sample were harvested by centrifugation and resuspended at final concentration 2×105 cells/50μl lysis buffer. Fluorogenic substrates VDVAD-AMC (Caspase 2), DEVD-AMC (Caspase 3), IETD-AMC (Caspase 8), and LEHD-AMC (Caspase 9) were used to assay activity of corresponding caspases in cell lysates. Actinomycin D (0.5 μg/ml) was used as a positive control. The plates were analyzed using a fluorescence microplate reader M2 (Molecular Dynamics, CA), and caspase activity was normalized per mg of total protein. Each experiment was performed in triplicate.
Cell cycle analysis
For cell-cycle analysis, cells were trypsinized, centrifuged and resuspended at a concentration of 1×106/ml in a propidium iodide (PI) staining solution (0.05 mg/ml PI, 0.1% sodium citrate, 0.1% Triton X-100). Each sample was treated with DNAse-free RNAse (5 ng/ml, Calbiochem, San Diego, CA) at room temperature for 30 minutes, filtered through 40-μm mesh, and analyzed by a FACScan flow cytometer (Becton Dickinson, San Jose, CA) collecting the fluorescence (wavelength range, 563 to 607 nm) from PI-bound DNA (approximately 15,000 cells). The percentages of cells within each phase of the cell cycle were computed by using ModFit software (Verity Software House, Topsham, ME).
Transfection of MEFs
About 25,000 cells per well were seeded in 24-well plates and transfected with plasmids containing Luc gene under control of p21, Bax, Puma, or Noxa promoters, and TP53 gene under control of CMV promoter using FuGene 6 transfection reagent (Roche, NJ). The next day, cells were treated with 10 μM MP or 0.5 μM araC and incubated for 28 hr. After incubation, cells were lysed and stored at (−20°C) until analysis. Luciferase activity was measured using a Dual-Luciferase Reporter Assay Kit (Promega, WI), as indicated in manufacturer’s instructions. Data were collected from a 96-well plate by Clarity luminescence microplate reader (Bio-Tek, VT). The expression of the promoter-driven firefly luciferase was normalized using the activity of constitutively expressed Renilla luciferase (Promega, WI).
For HMGB1 expression experiments, Hmgb1−/− MEFs were transfected with pCMV-SPORT6 plasmid with human HMGB1 cDNA insert (clone 1F5, Open Byosystems, Huntsville, AL). Control cells were transfected with a vector without HMGB1 cDNA. Transfection was performed with FuGene6 transfection reagent (Roche, NJ) as described above. Two days after transfection, the cells were treated with 10 μM MP, incubated for 42 hr and collected for Western blot analysis.
Western analysis was performed as described earlier (Krynetski et al., 2003). Briefly, 5×106 control cells and cells after drug treatment were rinsed with ice-cold PBS, lysed with TDLB buffer and scraped off the plates; lysates were sonicated 4×5 sec on ice and centrifuged in the microtubes for 10 min at 4°C. Concentration of protein in supernatant was determined by PlusOne2D Quant kit (Amersham Biosciences, NJ). 40 μg total protein was loaded onto 12% polyacrylamide gel and transferred to a PVDF or a nitrocellulose membrane (Invitrogen, Carlsbad, CA) in a Mini Trans-Blot electrotransfer cell (Bio-Rad, Hercules, CA). Membranes were developed with primary antibodies specific to p53 and p53 phosphorylated at Ser 12 and Ser 18 (Cell Signaling, Beverly, MA), γ-H2AX (H2AX phosphorylated at Ser 139) (Upstate, Temecula, CA), ATM phosphorylated at Ser 1981 (Abcam, MA), and ATR (Santa Cruz, CA). Anti-GAPDH antibody (Chemicon, Temecula, CA) and β-actin (Sigma, St. Louis, MO) were used for loading controls. For detection of ATM and ATR proteins, protein extract was separated using 7% Tris-acetate gels (Invitrogen, CA). Bands were visualized and quantified by PhosphorImager with the ImageQuaNT Software system (Molecular Dynamics, Sunnyvale, CA), using Blue Fluorescence/Chemifluorescence at 488 nm excitation, or Odissey Infrared Imaging system (LI-COR BioSciences, Lincoln, NE) using two-color fluorescence detection at 700 and 800 nm.
Gene expression experiments
Total cellular RNA was extracted with TriReagent (GIBCO BRL/Invitrogen, Carlsbad, CA) from untreated Hmgb1+/+ and Hmgb1−/− MEFs (about 5×106 cells per experiment, 7 replicates) and used for RT-PCR and DNA microarray experiments.
A. Analysis of Atm and Atr expression by Real-Time PCR
About 500 ng of total RNA was reverse transcribed using the TaqMan Reverse Transcription kit (Applied Biosystems, CA) according to manufacturer’s instructions. The level of Atm and Atr mRNA was evaluated using Relative Quantification protocol with murine β-actin as a normalization standard on ABI 7300 Real Time PCR instrument (Applied Biosystems, CA) according to manufacturer’s instructions. Data were collected from 3 independent experiments for each sample.
B. DNA microarray study
Total RNA was processed and hybridized to the GeneChip Murine Genome U74Av2 oligonucleotide microarray (Affymetrix, Santa Clara, CA) containing 12,489 probe sets per manufacturer’s instructions. For each experiment, consistency among replicate arrays was evaluated by measuring correlation coefficients among any given two chips’ (X,Y) intensity readings:
where
x̄ is mean intensity value of all genes in chip X
ȳ is mean intensity value of all genes in chip Y
xi is intensity reading of gene i in chip X
yi is intensity reading of gene i in chip Y
Matlab function “corrcoef” was used to measure correlation coefficient of two arrays’ intensities. A correlation coefficient value close to “1” between two arrays was considered indicative of good consistency of replicates, whereas value of “0” implied no correlation at all (Xie et al., 2007).
Selection of differentially expressed genes
Gene expression values were extracted using the Affymetrix Microarray Suite version 5.0. In the experiment (Hmgb1+/+ vs Hmgb1−/− MEFs), genes differentially expressed in untreated Hmgb1+/+ and Hmgb1−/− MEFs were defined as genes that had (i) > 2-fold difference between the cells and (ii) had p-value of the t-test <0.05 (Bolstad et al., 2003). Prior to statistical analysis, gene expression data were normalized by using quantile normalization method (Bolstad et al., 2003). Data quality in gene expression experiments was evaluated by calculating correlation coefficients among all arrays with Hmgb1+/+ and Hmgb1−/−MEFs (Xie et al., 2007). Gene expression values (log ratio) of selected genes were analyzed by hierarchical clustering analysis implemented in Spotfire statistical software (Spotfire, Somerville, MA).
Bioinformatics analyses and ontology classification of discriminative genes
Selected genes were imported into Ingenuity software to identify gene networks that are particularly enriched with the selected genes by searching against Ingenuity global molecular network (Ingenuity Systems, CA). Pathway annotations of genes were obtained from the Affymetrix annotation file. Fisher exact test (Ben-Shaul et al., 2005) was used to examine whether the selected differentially expressed genes were overrepresented within a given pathway.
Statistical analysis
Data are presented as the mean ± SE. The statistical analyses were carried out using Student’s t test with Statistica software program (StatSoft, OK). A P value <0.05 was considered statistically significant.
RESULTS
Hmgb1-deficient MEFs demonstrate delayed cytotoxicity after treatment with anticancer genotoxic drugs
Under conditions of experiments, the growth rates of Hmgb1-deficient and Hmgb1-proficient cells were similar: population doubling times in the middle of exponential phase for HMGB1+/+ and HMGB1−/− MEFs were 14.4 hr and 15.7 hr, respectively. Chemosensitivity to a panel of anticancer drugs (nucleoside analogs and their precursors) was assessed in HMGB1+/+ and HMGB1−/− MEFs (Table 1 and Fig. 1S). Hmgb1-deficient cells revealed 3–10 times higherviability by MTT assay after 3–6 days incubation with FU, araC, TG, and MP, although there was marginal or no difference in chemosensitivity of HMGB1+/+ and HMGB1−/− MEFs treated with cladribine, clofarabine, fludarabine and gemcitabine (Table 1). Actual proliferation plots are given in Supplemental Materials section (Fig. 1S). These results were consistent with our previous data on relative resistance of Hmgb1-deficient cells to thiopurines (Krynetski et al., 2003). MP, araC and FU were used in subsequent experiments to elucidate the role of HMGB1 in cell death after anticancer drug treatment.
Table 1.
Effect of chemotherapeutic agents on MEF cell viability (IC50). MEF (Hmgb1+/+ vs. Hmgb1−/−) were incubated with different drug concentrations for 5 days, and viability was measured by MTT assay.
| Drug | IC50, μMa | |
|---|---|---|
| Hmgb1+/+ MEF | Hmgb1−/− MEF | |
| Mercaptopurine | 0.8±0.12 | 8.9±0.65 |
| Thioguanine | 0.5±0.11 | 2.2±0.29 |
| Cytarabine | 0.4±0.02 | 1.7±0.08 |
| Fluorouracil | 1.3±0.35 | 6.2±0.17 |
| Fludarabine | 2.9±0.13 | 4.3±0.21 |
| Cladribine | 1.9±0.29 | 2.91±0.16 |
| Gemcitabine | 0.02±0.001 | 0.02±0.005 |
| Clofarabine | 0.6±0.15 | 0.7±0.07 |
Each point is mean of 3 replicates and error bars represent ±SD, p < 0.05.
Fig. 1.
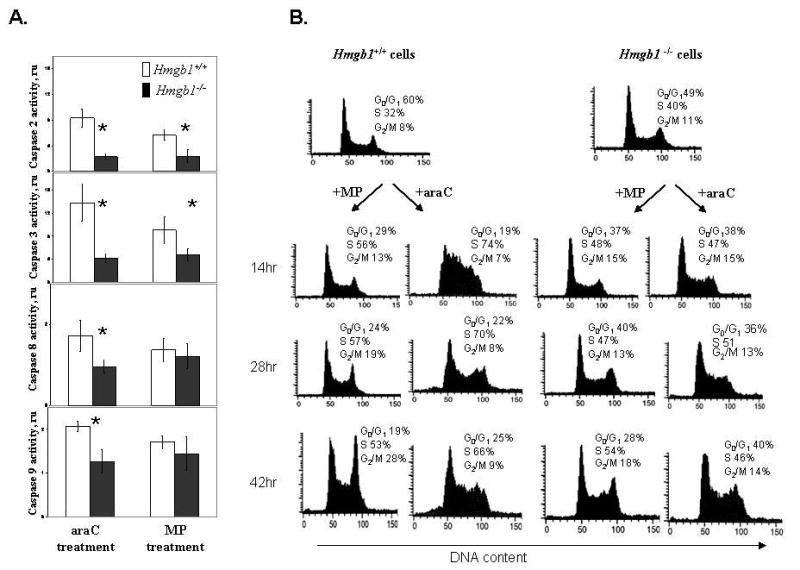
Hmgb1- deficient cells demonstrate decreased caspase activity and a compromised cell cycle arrest after genotoxic stress induced by antimetabolite drugs.
Panel A: Activation of caspases 2, 3, 8, and 9 in Hmgb1+/+ MEFs compared with Hmgb1−/− MEFs after 42 hr treatment with 10 μM MP or 0.5 μM araC for 42 hr. Activity of caspases was assayed using fluorogenic substrates as described in Materials and Methods. Relative activity of individual caspases in treated cells was expressed as a ratio to caspase activity in untreated cells after normalization per mg protein. Data are the mean ±SD generated from three independent experiments. ru, relative units. Asterisks denote significantly different levels of activity (p<0.05).
Panel B: Cell cycle arrest is induced by anticancer drugs in Hmgb1+/+ but not in Hmgb1−/−MEFs. Hmgb1+/+ (right) and Hmgb1−/− (left) MEFs were treated with 10 μM MP and 0.5 μM araC for 14 hr, 28 hr, and 42 hr, stained with propidium iodide, and DNA content was analyzed using flow cytometry after treatment with DNAse-free RNAse. The percentages of Hmgb1+/+ and Hmgb1−/− cells in G0/G1, S, and G2-M phases were determined by DNA histogram analysis.
Incorporation of radio-labeled nucleoside analogs araC and deoxythioguanosine (the product of metabolic activation of MP) into DNA occurred at similar levels in both cell lines (HMGB1+/+ and HMGB1−/− MEFs, p=0.78) irrespective of the status of Hmgb1 (Fig. 2S).
Fig. 2.
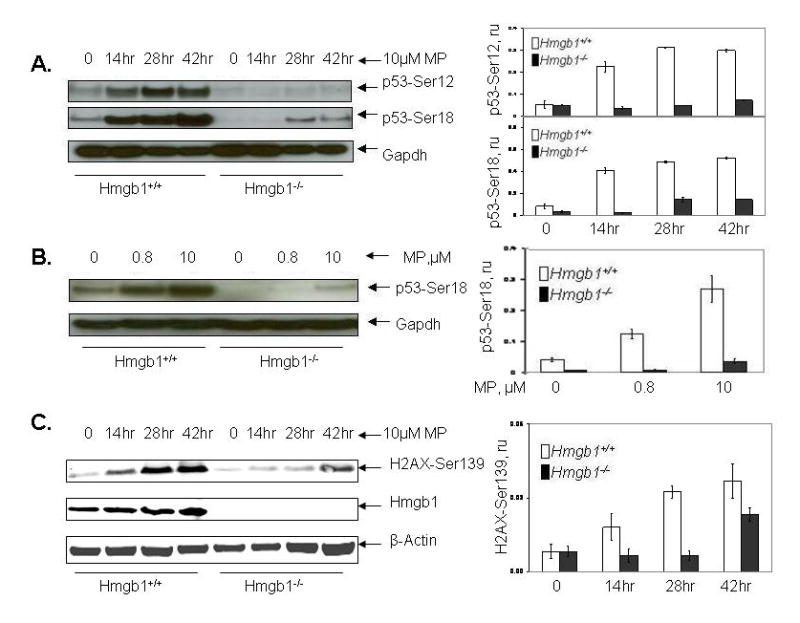
Phosphorylation of p53 and H2AX proteins in MEFs with different Hmgb1 status after MP treatment. Panel A: Western blotting analysis of p53 phosphorylation at Ser 12 and Ser 18 (left), and relative levels of p53 phosphorylation normalized vs GAPDH (right). Panel B: Induction of S18 phosphorylation in p53 increases with increased concentration of MP in both cell lines after 28 hr treatment. Panel C: Western blotting analysis of H2ax phosphorylation at Ser 139 (left), and relative levels of H2ax phosphorylation normalized vs β-actin (right). Cells were treated with 10 μM MP for 14-42 hr. Lysates from untreated cells were used as control. Analysis with anti-Hmgb1 antibody (Panel B, the central strip) confirmed the cell phenotype. 0, no drug treatment (control). Data are expressed as the mean ± S.D. of three independent experiments. Bars, SD. ru, relative units.
Caspase activation after drug treatment is reduced in Hmgb1-deficient cells
To gain further insight into cytotoxic effects of anticancer nucleosides in relation to Hmgb1 functions, we compared activity of apoptotic markers (caspases 2, 3, 8, and 9) in Hmgb1+/+ and Hmgb1−/− cells. Caspase activation was monitored using fluorogenic substrates after treatment with MP and araC for 14–42 hr. After treatment of HMGB1+/+ cells with 10 μM MP or 0.5 μM cytarabine for 42 hr, we detected 8-12-fold activation of caspases 2 and 3 in Hmgb1+/+ cells (Fig. 1A, open columns) vs 2–4-fold activation in Hmgb1−/− cells (Fig. 1A, black columns), thus confirming the induction of the apoptotic cascade after the genotoxic stress. Importantly, activation of caspases 2 and 3 was significantly lower in Hmgb1−/− cells compared with Hmgb1+/+ cells, suggesting a key role of Hmgb1 in induction of apoptosis in response to antimetabolite nucleoside analogs. In contrast, activation of caspases 8 and 9 after araC treatment for 42 hr in Hmgb1-proficient cells was less pronounced (1.5–2-fold) though still significantly higher than in Hmgb1-deficient cells. Difference in caspase 8 and caspase 9 activation after MP treatment did not reach significance (Fig. 1A).
Treatment with anticancer drugs MP and araC interrupts cell cycle progression in Hmgb1+/+ but not in Hmgb1−/− cells
We set up experiments to assess the effect of Hmgb1 status on cell cycle progression. Treatment of asynchronous HMGB1+/+ MEFs with 10 μM MP for 14–42 hr resulted in cell cycle arrest in G2/M phase (28% of treated cells vs 8% of untreated cells after 42 hr incubation) with a concomitant decrease of cells in G0/G1 phase (19% of treated cells vs 60% of untreated cells). On the other hand, in Hmgb1−/− cells this effect was less pronounced (18% of treated cells vs 11% in untreated cells in G2/M phase after 42 hr incubation) (Fig. 1B). Treatment of HMGB1+/+ MEFs with 0.5 μM araC resulted in cell cycle arrest in S phase after the first cell division. The percentage of HMGB1+/+ cells in S phase increased from 32% to 74% after 14 hr treatment, and remained at 70% after 28 hr incubation (Fig. 1B). In HMGB1−/− MEFs, the percentage of cells in S phase increased from 40% to 47% after 14 hr incubation with araC (Fig. 1B). These results support our hypothesis that Hmgb1 modulates the cellular response to antimetabolite nucleoside analogs.
Hmgb1 modulates phosphorylation of p53 and H2AX proteins after treatment with genotoxic chemotherapeutic drugs MP, FU, and araC
A. Accumulation of Ser-12- and Ser-18-phosphorylated p53
Transcriptional activation of p53 protein following DNA damage is accompanied by a series of post-translational modifications, among which phosphorylation of Ser 15 is thought to be the initial event in a series of subsequent modifications leading to stabilization and biochemical activation of p53 (Meek, 2004). We compared post-translational modification of p53 in Hmgb1+/+ and Hmgb1−/−cells in response to genotoxic stress caused by treatment with MP, araC, and FU. To monitor the status of p53, we used antibodies to phosphorylated forms of p53 (Soubeyrand et al., 2004). Ser-9 and Ser-15 in human p53 are evolutionary conserved residues, corresponding to Ser-12 and Ser-18 in murine p53; these residues can be phosphorylated by several protein kinases including ATM, ATR, and DNA-PK (Chao et al., 2003). After treatment of murine cells with 10 μM MP, 0.5 μM araC, and 10 μM FU, we observed accumulation of strong signals corresponding to p53 phosphorylated at Ser-12 and Ser-18 in Hmgb1-proficient cells. In contrast to Hmgb1-proficient cells, phosphorylation of p53 in Hmgb1-deficient cells was 6–8-fold decreased, and accumulation of phosphoserine 12 and 18 occurred at lower rate (Figs. 2A and 3A). A general inhibitor of PIKK-like kinases wortmannin decreased phosphorylation of Ser 18, suggesting that one of the DNA damage-activated kinases ATM, ATR, or DNA-PK catalyze phosphorylation of p53 following drug treatment (data not shown). Treatment of cells at two concentrations of drug (0.8 μM and 10 μM MP) revealed that the level of phosphorylated product paralleled drug concentration (Fig. 2B).
Fig. 3.
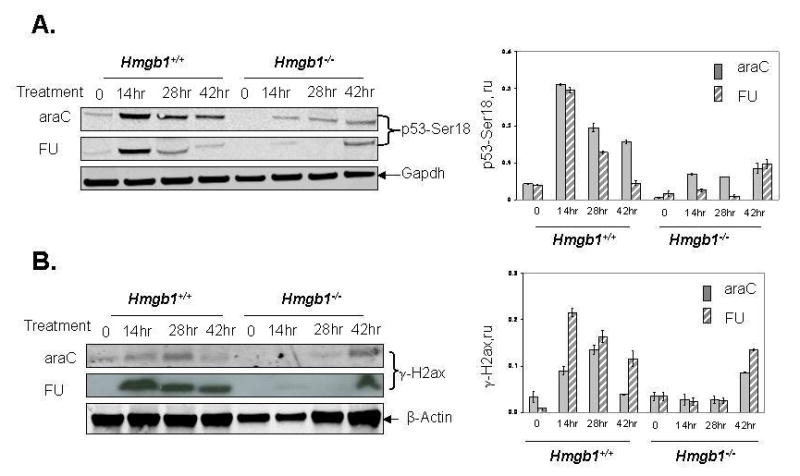
Phosphorylation of p53 and H2ax proteins in MEFs with different Hmgb1 status after araC and FU treatment. Panel A: Western blotting analysis of N-termianl phosphorylation of p53 at Ser 18 (left), and relative levels of p53 phosphorylation normalized vs GAPDH for both drugs (right). Panel B: Western blotting analysis of H2ax phosphorylation at Ser 139 after treatment with araC and FU (left), and relative levels of H2ax phosphorylation normalized vs β-actin for both drugs (right).
Cells were treated with 0.5 μM araC or 10 μM FU for 14–42 hr. Lysates from untreated cells were used as control. Data are expressed as the mean ± S.D. of three independent experiments. Bars, SD. ru, relative units. 0, no drug treatment (control).
B. Accumulation of γ-H2ax
Following the generation of DSB after replication stress, genotoxic insult, or other stimuli, a network of PIKK-like kinases is activated causing rapid phosphorylation of histone H2AX at C-terminal Ser 139 residue (Fernandez-Capetillo et al., 2004). Thus, accumulation of γ-H2AX (a phosphorylated form of H2AX) is a strong indicator of DSB formation. Using antibody specific to H2AX phosphorylated at Ser 139, we evaluated accumulation of DSB after treatment with MP, araC, and FU. Similarly to Ser 12- and Ser 18-phosphorylated forms of p53, accumulation of γ-H2ax following drug treatment was detected after 14 – 42 hr incubation in Hmgb1-proficient MEFs (Fig. 2C and 3B). Importantly, the level of γ-H2ax following MP, araC, and FU treatment was significantly reduced in Hmgb1-deficient cells, indicating a functional role of Hmgb1 in response to drug treatment. The accumulation of γ-H2ax occurred at different rates for the three drugs: following MP treatment of Hmgb1+/+ MEFs, the phosphorylated product was detectable after 14 hr and reached the maximum level after 42 hr incubation. In contrast, the maximum level of γ-H2ax was achieved after 14 hr of incubation with FU and 28 hr incubation with araC, correspondingly. In Hmgb1−/− cells treated with MP, araC and FU, the accumulation of Ser 139-phosphorylated product reached maximal level after 42 hr (the third cycle of cell division) indicating delayed response in comparison with Hmgb1+/+ cells.
At least three PIKK family members, ATM, ATR, and DNA-PK catalyze phosphorylation of Ser 139 in H2AX. There is functional redundancy among the pathways since H2AX phosphorylation remains detectable in ATM−/−, DNA-PK−/− and ATR−/− knockout cells (Fernandez-Capetillo et al., 2004). Using Real-Time PCR technique, we evaluated the relative level of Atm and Atr mRNA in Hmgb1+/+ and Hmgb1−/− MEFs. Our experiments revealed about 2.5-fold higher Atm mRNA expression in Hmgb1+/+ cells compared to Hmgb1−/− cells (p<0.001); Atr mRNA was slightly (1.2 times) overexpressed in Hmgb1+/+ cells compared to Hmgb1−/− cells. This difference did not reach statistical significance (p=0.057). At the protein level, the increase of an active form of ATM phosphorylated at Ser 1981 after treatment with 10 μM MP was not significant (Fig. S3A, B). In contrast, Western analysis revealed increased level of Atr protein in Hmgb1+/+ cells after 28 hr incubation with MP (Fig. S3A, C).
Hmgb1 stimulates trans-activation of p53-regulated pro-apoptotic promoters in cells treated with chemotherapeutic drugs MP and araC
Because chemotherapeutic agents used in this study (FU, araC, and MP) are known to exert their cytotoxic effect via the p53-mediated pathway (Bunz et al., 1999;Zhang et al., 2000;Yin et al., 2006;Zhang et al., 2007), we used luciferase reporter plasmids to assess the role of Hmgb1 in transcriptional activity of p53 following genotoxic stress. Hmgb1+/+ and Hmgb1−/− MEFs were transfected with Luc-expressing reporter plasmids driven by p53-regulated promoters (p21, Bax, Puma, and Noxa) and then treated with anticancer drugs MP and araC. After 28–42 hr incubation with 10 μM MP and 0.5 μM araC, we observed an increased luciferase expression driven by p53-regulated promoters indicative of induction of p53 transcriptional activity (Fig. 4). Specifically, pro-apoptotic promoters Puma and Noxa were 2–3-fold more active in Hmgb1+/+ than in Hmgb1−/− MEFs in response to genotoxic stress after MP treatment. Following treatment with araC, three reporter plasmids (pBax-luc, pPuma-luc, and pNoxa-luc) revealed significantly increased promoter activity (4–8-fold) in Hmgb1+/+ compared with Hmgb1−/− MEFs, though activity of p21 promoter did not differ significantly in these two cell lines (Fig. 4).
Fig. 4.
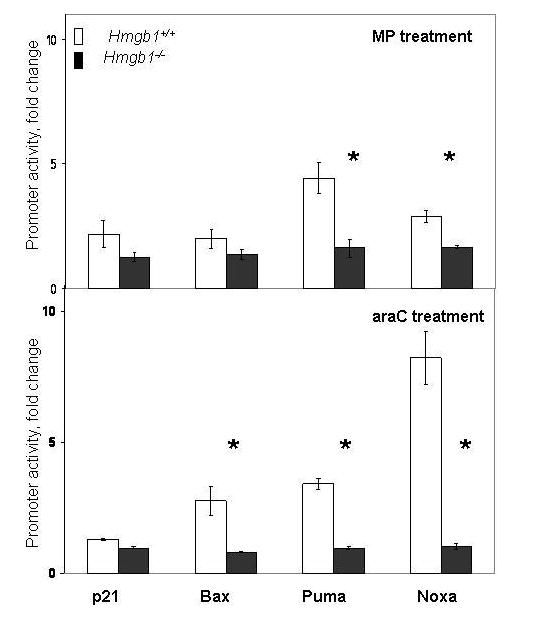
HMGB1 contributes to activation of p53 target genes in response to genotoxic stress. Hmgb1+/+ and Hmgb1−/− MEFs were transfected with luciferase reporter plasmids driven by p21, Bax, Puma, and Noxa promoters, or control plasmid, and treated with 10 μM MP (upper panel) or 0.5 μM araC (lower panel). Trans-activation of p53-regulated promoters p21, Bax, Puma, and Noxa in MEFs was normalized by constitutively expressed Renilla luciferase activity. Activity of Puma and Noxa promoters was significantly different in two cell lines after MP treatment; and activity of Bax, Puma, and Noxa promoters was significantly different after araC treatment (p<0.05). Results of experiments are depicted as fold change of promoter activity relative to cells untreated with genotoxic drugs. Data are expressed as the mean ± S.D. of three independent experiments. Asterisks denote significantly different levels of activity (p<0.05). Bars, SD. ru, relative units.
Transient expression of human HMGB1 rescues phosphorylation of p53 and H2AX in Hmgb1 knockout cells
We examined whether re-expressing HMGB1 in Hmgb1-knockout murine cells restores phosphorylation of p53 and H2ax following genotoxic stress caused by drug treatment. Transfection of Hmgb1−/− MEFs with human HMGB1 cDNA driven by CMV promoter resulted in accumulation of HMGB1 in the cells (Fig. 5). After treatment with 10 μM MP for 28 hr, we observed increased accumulation of p53 phosphorylated at Ser 18, and H2ax phosphorylated at Ser 139 in Hmgb1−/− cells transfected with HMGB1 cDNA, compared with cells transfected with the control plasmid (Fig. 5).
Fig. 5.
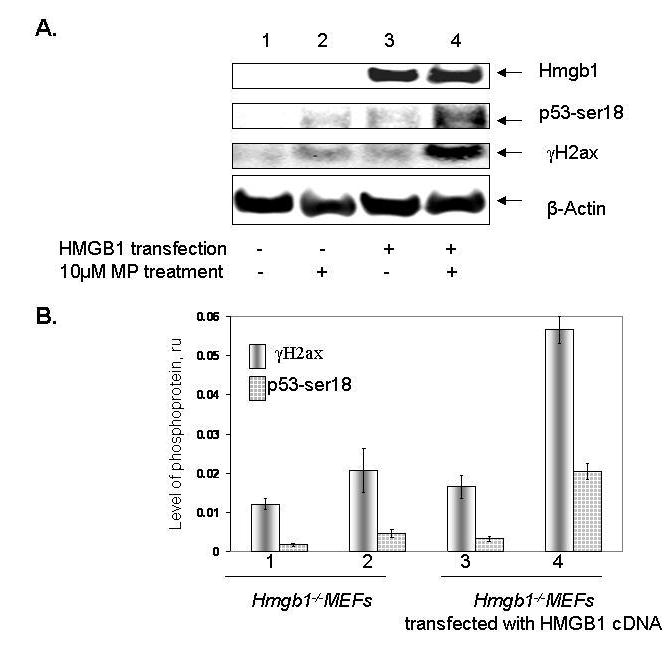
Expression of human HMGB1 restores phosphorylation of p53 and H2ax in Hmgb1 knockout MEFs. Panel A: Accumulation of S18-phsosphorylated p53 and γ-H2ax in Hmgb1−/− cells transfected with human HMGB1-expressing plasmid and treated with 10 μM MP for 28 hr. Expression of HMGB1 in Hmgb1−/− MEFs is shown on the top strip; β-actin, loading control. Panel B: Relative levels of phosphorylation of p53 at Ser 18 and H2ax at Ser 139 after MP treatment of mock- and HMGB1-transfected cells. β-actin was used for normalization of signals.
Microarray experiments reveal distinct gene regulation patterns depending on Hmgb1 status of cells
Using total RNA extracted from Hmgb1+/+ and Hmgb1−/− MEFs in seven independent experiments, we compared gene expression patterns in these two cell lines using DNA microarray technology. All replicates had correlation coefficient value >0.90, which indicated good quality of collected data. Forty-five probe sets representing forty three genes had at least 2-fold higher expression levels in Hmgb1-proficient MEFs compared to Hmgb1-deficient MEFs, and formed tight clusters by hierarchical clustering analysis (Fig. 6A). Hmgb1 gene was identified as one gene having the highest discriminative ratio thus confirming the accuracy of collected data. Identification of gene networks enriched with the selected genes using Ingenuity algorithm elucidated several pathways dependent on the Hmgb1 status of cells (Fig 6B). Specifically, genes involved in intracellular signaling pathways, cell cycle pathway, and apoptosis were differentially expressed in Hmgb1+/+ compared to Hmgb1−/− (p<0.05 by Fisher exact test). This finding parallels our data indicating the role of Hmgb1 in induction of apoptosis. The complete list of genes differentially expressed in Hmgb1+/+ vs Hmgb1−/− cells is provided in Supplementary Table S1. These data suggest that architectural transcription factor Hmgb1 is involved in regulation of several important pathways, and may contribute to modulation of therapeutic efficacy of antimetabolite chemotherapeutic agents.
Fig. 6.
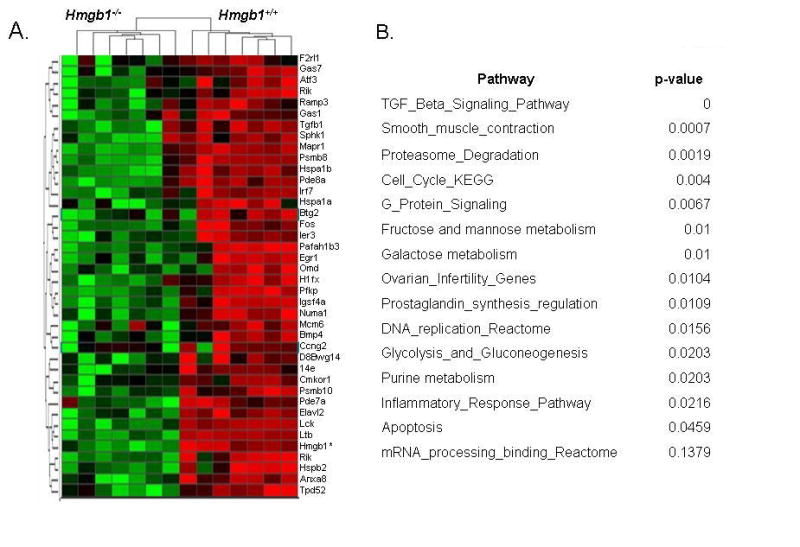
Discriminative genes differently expressed in untreated Hmgb1+/+ vs Hmgb1−/− MEFs. Panel A: Hierarchical clustering analysis of discriminative genes highly expressed in Hmgb1+/+ cells compared with Hmgb1−/− cells (red channel, overexpressed; green channel, underexpressed). Total RNA from two cell lines was extracted and hybridized with Affimetrix microarray chips in seven independent experiments; tight clusters of Hmgb1+/+ vs Hmgb1−/− cells indicate that the gene expression profiles in these two cell lines are essentially different. Supplemental Table S1 contains the complete list of discriminative genes. Panel B: Overrepresented functional categories within gene clusters regulated by Hmgb1. Pathways containing significantly differentially expressed genes in Hmgb1+/+ vs Hmgb1−/− MEFs were identified by Fisher exact test (p value <0.05).
DISCUSSION
Cellular response to DNA damage relies on recognition of structural elements such as double strand breaks, abnormal nucleobases, or apurinic sites, to name just a few. Alternatively, DNA damage sensors may respond to dynamic changes in DNA (e.g., DNA bending) rather than static structural features (Seibert et al., 2005).
In this study, we used a murine fibroblast model system generated from Hmgb1+/+ and Hmgb1−/− mice to assess the role of the DNA-bending protein HMGB1 in cell sensitivity to antimetabolite nucleoside analogs. Our results suggest a mechanistic explanation to increased resistance of Hmgb1-deficient cells to chemotherapeutic agents: induction of p53-mediated apoptosis by chemotherapeutic agents is evidently compromised in Hmgb1-deficient cells. This conclusion is supported by several lines of evidence: first, Hmgb1-deficient cells are more resistant to antimetabolite agents FU, araC, and MP (Table 1) despite comparable incorporation of nucleoside analogs into DNA (Fig. 2S); second, caspase activation is reduced in Hmgb1-deficient MEFs, in comparison with Hmgb1-proficient cells (Fig. 1A); third, Hmgb1-deficient cells manifest a defect in cell cycle arrest after drug treatment (Fig. 1B); fourth, trans-activation experiments demonstrate diminished activation of pro-apoptotic promoters Bax, Puma, and Noxa in Hmgb1-deficient cells (Fig. 2); fifth, phosphorylation of DNA damage markers p53 and H2AX is reduced in Hmgb1-deficient cells (Figs. 3, 4); and sixth, re-expression of HMGB1 in Hmgb1 knockout cells restores phosphorylation of p53 and H2ax (Fig. 5).
The incorporation of nucleoside analogs is similar in Hmgb1+/+ and Hmgb1−/− cells, indicating that metabolic activation of genotoxic drugs occurs at comparable level in these two cell lines. The two drugs used in incorporation experiments (araC and MP) are metabolically activated through two different pathways (correspondingly, nucleoside phosphorylation and purine salvage pathway). Nevertheless, the effect of Hmgb1 abrogation is similar for these drugs suggesting that Hmgb1 exerts its action after the nucleoside analogs are incorporated into DNA.
Because antimetabolites act via several complementary mechanisms, it is difficult to attribute their effects to genotoxicity vs. alternative ways of action (e.g., inhibition of metabolic reactions). The use of gene-targeted cell lines where specific steps in DNA repair or DNA damage response are changed proves to be instrumental in discriminating between metabolic and genotoxic mechanisms of action. Using cells deficient in base excision repair, An and colleagues demonstrated that incorporation of fluorouracil in DNA is the predominant cause of FU cytotoxicity (An et al., 2007). In our experiments, we used murine cells deficient in Hmgb1 protein which is not known to participate in drug metabolism or anabolic activation of drugs. Instead, this protein plays an important role in maintenance of nucleosome structure and regulation of gene transcription (Yamada and Maruyama, 2007). Therefore, changes in chromatine architecture or gene regulation patterns could be important determinants of antimetabolite therapy.
Structural analysis of synthetic DNA showed that incorporation of nucleoside analogs into DNA does not introduce gross changes in DNA geometry (Sahasrabudhe et al., 1996;Somerville et al., 2003). Instead, drastic shifts in DNA dynamics occur; for example, incorporation of deoxythioguanosine makes DNA more flexible (Somerville et al., 2003). While introduction of a kink into perfect DNA is thermodynamically unfavorable, the presence of elements that enhance DNA flexibility facilitates DNA bending (Lorenz et al., 1999). The increased DNA flexibility can be detected by DNA-bending proteins and interpreted as a genotoxic insult, thus providing the primary signal for activation of DNA damage response.
Multiple in vitro and clinical studies provided evidence that antimetabolites act via the p53-mediated apoptotic pathway which includes phosphorylation and transcriptional activation of p53 (Bunz et al., 1999;Kaeser et al., 2004;Zhang et al., 2000;Decker et al., 2003;Longley et al., 2003;Yin et al., 2006). In our experiments, we found that MP, araC, and FU induced Ser12 and Ser 18 phosphorylation in murine p53. Analysis performed over a range of MP concentrations (i.e., at IC50 for wild type and knockout MEFs) demonstrated increased phosphorylation of p53 with increasing drug concentrations. At both drug concentrations phosphorylation of p53-Ser 12 and p53-Ser18 in Hmgb1-deficient cells was significantly lower suggesting that Hmgb1 acts in the beginning of the p53-mediated stress response. In parallel, transcriptional activity of p53-regulated pro-apoptotic promoters was decreased in Hmgb1-knockout cells. Another marker of genotoxic stress used in this study was γ-H2ax which is a strong indicator of DSB formation due to various genotoxic insults, or during the execution phase of apoptosis (Rogakou et al., 2000;Hanasoge and Ljungman, 2007). Similarly to Ser 12-and Ser 18-phosphorylated form of p53, accumulation of γ-H2ax was significantly reduced in Hmgb1-knockout cells.
Ectopic expression of human HMGB1 in murine Hmgb1-knockout cells restored phosphorylation of p53 and H2ax thus confirming functional role of Hmgb1 in genotoxic stress response. Human HMGB1 protein differs in only one amino acid residue from the murine Hmgb1 protein, and evidently can rescue at least some of its functions.
Phosphorylation of Ser 15 in p53, and phosphorylation of Ser 139 in H2AX are catalyzed by one or more of the PIKK-like protein kinases ATM, ATR, or DNA-PK (Meek, 2004). Taken together, the reduced phosphorylation of p53 and H2AX markers in Hmgb1−/− MEFs indicates that the activity of one of these kinases depends on Hmgb1 status of the cells. Gene and protein expression data suggest that this may occur at different levels: transcription of Atm gene was 2.5-fold higher in untreated Hmgb1-proficient compared to Hmgb1-deficient cells, as revealed by RT-PCR. We observed a trend to the increase of activated Atm (phosphorylated at Ser 1981) in Hmgb1+/+ rather than Hmgb1−/− cells after drug treatment, though the difference did not reach significance (Fig. S3A and B). On the other hand, Atr transcription was similar in untreated Hmgb1+/+ and Hmgb1−/− cells. Interestingly, after 28–42 hr exposure to MP we detected the increased level of Atr protein in Hmgb1-proficient but not Hmgb1-deficient cells; this observation suggests that Hmgb1 could augment Atr synthesis or stability under conditions of genotoxic stress (Fig. 3A, C).
Alternatively, abrogation of protein-protein interaction between Hmgb1 and p53 or H2ax in Hmgb1−/− cells could inhibit phosphorylation of p53 and H2ax. Several in vitro studies have examined HMGB1-p53 interaction and demonstrated that HMGB1 enhances p53 sequence-specific DNA binding by a mechanism involving structural changes in the target DNA (Jayaraman et al., 1998;McKinney and Prives, 2002;Imamura et al., 2001). No data on HMGB1-H2AX interactions have been reported, so far. At present, this second scenario cannot be excluded and merits experimental testing.
HMGB1 is an architectural transcription factor involved in regulation of several promoters including human β-globin and Bax (Stros et al., 2002;Bianchi and Agresti, 2005). In unchallenged MEFs, activity of several groups of genes including regulators of cell cycle and apoptosis has been found to differ between Hmgb1-proficient and Hmgb1-deficient cells (Fig. 6A, B). Our experiments with cell viability and cell cycle progression support this notion. Therefore, Hmgb1 could be either directly involved in transcriptional regulation, or act as a co-regulator through interaction with other transcriptional factors, e.g. p53. At present, we expand our studies to other pathways highlighted by DNA microarray analysis.
Since HMGB1 is a DNA bending protein, the role of HMGB1 may consist of active scanning of chromatin, binding to flexible regions of DNA and recruiting other proteins such as p53 and/or regulatory protein kinases. This model does not imply that direct interaction between HMGB1 and p53 is necessary for p53-DNA binding; rather, HMGB1 may change architecture of DNA and facilitate binding of pre-bent DNA by p53 (Imamura et al., 2001).
This hypothetical mechanism predicts that abrogation of Hmgb1 functional activity would not affect cellular response to drugs which induce significant alterations in DNA structure. Indeed, no difference in cytotoxic effect was found between Hmgb1+/+ and Hmgb1−/− cells treated with cisplatin, a chemotherapeutic agent which introduces sharp kinks in DNA (Wei et al., 2003). Because p53 directly binds to cisplatinated DNA in a non-sequence-specific way, HMGB1 probably is not critical for response to this type of DNA lesion.
Our results indicate that abrogation of Hmgb1 in MEFs reduces apoptotic response to nucleoside analogs and precursors, and suggest that decreased Hmgb1 activity due to down-regulated expression or the inactivating mutations can compromise the outcome of chemotherapy. HMGB1 reveals a complex expression pattern in the mouse brain during development, with a number of cells showing undetectable level of HMGB1. Moreover, its intracellular localization also varies depending on yet uncharacterized factors (Guazzi et al., 2003). High resolution mapping of genome imbalance and gene expression profiles of 26 serous epithelial ovarian tumors identified HMGB1 as a gene associated with resistance to chemotherapy (Bernardini et al., 2005). In addition to the regulation of HMGB1 gene expression, the genetic variations in the HMGB1 gene can contribute to variability in drug response. In a recent study of 103 healthy blood donors, six genetic polymorphisms were identified in HMGB1 locus which may have a regulating role in expression of this protein (Kornblit et al., 2007). Therefore, HMGB1 is a potential target for modulating activity of chemotherapeutic nucleoside analogs and precursors. Identification of proteins sensitive to DNA lesions which occur without the loss of DNA integrity provides new insights into the determinants of drug sensitivity in cancer cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are grateful to Dr. M. Bianchi for Hmgb1+/+ and Hmgb1−/− cell lines, Drs. K. Helin, M. Oren, T. Taniguchi, B. Vogelstein for their generous gift of plasmids; Ashley Miller, Jennifer Smith, Bettyann Rogers, Hiren Patel, and Manali Phadke for their excellent technical assistance.
This work is supported by grant R01 CA104729 from the National Cancer Institute (EK), and a grant with the Pennsylvania Department of Health (ZO).
Abbreviations
- HMGB1
high mobility group protein B1
- ATM
ataxia-telangiectasia mutated protein kinase
- ATR
ataxia-telangiectasia and Rad3-related protein kinase
- DNA-PK
DNA-dependent protein kinase
- AMC
7-amino-4-methylcoumarin
- araC
cytosine arabinoside, 4-amino-1-[(2R,3S,4R,5R)-3,4-dihydroxy-5- (hydroxymethyl)oxolan-2-yl] pyrimidin-2-one
- cladribine
5-(6-amino-2-chloro-purin-9-yl)-2-(hydroxymethyl)oxolan-3-ol
- clofarabine
5-(6-amino-2-chloro-purin-9-yl) -4-fluoro-2- (hydroxymethyl)oxolan-3-ol
- fludarabine
[(2R,3R,4S,5R)-5-(6-amino-2-fluoro-purin-9-yl)- 3,4-dihydroxy-oxolan-2-yl]methoxyphosphonic acid
- FU
5-fluorouracil, 5-fluoro-1H-pyrimidine-2,4-dione
- gemcitabine
4-amino-1-[3,3-difluoro-4-hydroxy-5- (hydroxymethyl) tetrahydrofuran-2-yl]-1H-pyrimidin- 2-one
- MP
6-mercaptopurine, 3,7-dihydropurine-6-thione
- thioguanine
2-amino-7H-purine-6-thiol
- MTT 3
(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
References
- An Q, Robins P, Lindahl T, Barnes DmE. 5-Fluorouracil Incorporated into DNA Is Excised by the Smug1 DNA Glycosylase to Reduce Drug Cytotoxicity. Cancer Res. 2007;67:940–945. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA Damage Activates ATM Through Intermolecular Autophosphorylation and Dimer Dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Ben-Shaul Y, Bergman H, Soreq H. Identifying Subtle Interrelated Changes in Functional Gene Categories Using Continuous Measures of Gene Expression. Bioinformatics. 2005;21:1129–1137. doi: 10.1093/bioinformatics/bti149. [DOI] [PubMed] [Google Scholar]
- Bernardini M, Lee CH, Beheshti B, Prasad M, Albert M, Marrano P, Begley H, Shaw P, Covens A, Murphy J, Rosen B, Minkin S, Squire JA, Macgregor PF. High-Resolution Mapping of Genomic Imbalance and Identification of Gene Expression Profiles Associated With Differential Chemotherapy Response in Serous Epithelial Ovarian Cancer. Neoplasia. 2005;7:603–613. doi: 10.1593/neo.04760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi ME, Agresti A. HMG Proteins: Dynamic Players in Gene Regulation and Differentiation. Curr Opin Genet Dev. 2005;15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW, Vogelstein B. Disruption of P53 in Human Cancer Cells Alters the Responses to Therapeutic Agents. J Clin Invest. 1999;104:263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calogero S, Grassi F, Aguzzi A, Voigtlander T, Ferrier P, Ferrari S, Bianchi ME. The Lack of Chromosomal Protein Hmg1 Does Not Disrupt Cell Growth but Causes Lethal Hypoglycaemia in Newborn Mice. Nat Genet. 1999;22:276–280. doi: 10.1038/10338. [DOI] [PubMed] [Google Scholar]
- Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a Tetrazolium-Based Semiautomated Colorimetric Assay: Assessment of Chemosensitivity Testing. Cancer Res. 1987;47:936–942. [PubMed] [Google Scholar]
- Chao C, Hergenhahn M, Kaeser MD, Wu Z, Saito S, Iggo R, Hollstein M, Appella E, Xu Y. Cell Type- and Promoter-Specific Roles of Ser18 Phosphorylation in Regulating P53 Responses. J Biol Chem. 2003;278:41028–41033. doi: 10.1074/jbc.M306938200. [DOI] [PubMed] [Google Scholar]
- Decker RH, Levin J, Kramer LB, Dai Y, Grant S. Enforced Expression of the Tumor Suppressor P53 Renders Human Leukemia Cells (U937) More Sensitive to 1-[Beta-D-Arabinofuranosyl]Cytosine (Ara-C)-Induced Apoptosis. Biochem Pharmacol. 2003;65:1997–2008. doi: 10.1016/s0006-2952(03)00149-7. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the Histone Guardian of the Genome. DNA Repair (Amst) 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Guazzi S, Strangio A, Franzi AT, Bianchi ME. HMGB1, an Architectural Chromatin Protein and Extracellular Signalling Factor, Has a Spatially and Temporally Restricted Expression Pattern in Mouse Brain. Gene Expr Patterns. 2003;3:29–33. doi: 10.1016/s1567-133x(02)00093-5. [DOI] [PubMed] [Google Scholar]
- Hanasoge S, Ljungman M. H2AX Phosphorylation After UV-Irradiation Is Triggered by DNA Repair Intermediates and Is Mediated by the ATR Kinase. Carcinogenesis. 2007 doi: 10.1093/carcin/bgm157. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Hock R, Furusawa T, Ueda T, Bustin M. HMG Chromosomal Proteins in Development and Disease. Trends Cell Biol. 2007;17:72–79. doi: 10.1016/j.tcb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T, Izumi H, Nagatani G, Ise T, Nomoto M, Iwamoto Y, Kohno K. Interaction With P53 Enhances Binding of Cisplatin-Modified DNA by High Mobility Group 1 Protein. J Biol Chem. 2001;276:7534–7540. doi: 10.1074/jbc.M008143200. [DOI] [PubMed] [Google Scholar]
- Jayaraman L, Moorthy NC, Murthy KG, Manley JL, Bustin M, Prives C. High Mobility Group Protein-1 (HMG-1) Is a Unique Activator of P53. Genes Dev. 1998;12:462–472. doi: 10.1101/gad.12.4.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser MD, Pebernard S, Iggo RD. Regulation of P53 Stability and Function in HCT116 Colon Cancer Cells. J Biol Chem. 2004;279:7598–7605. doi: 10.1074/jbc.M311732200. [DOI] [PubMed] [Google Scholar]
- Kornblit B, Munthe-Fog L, Petersen SL, Madsen HO, Vindelov L, Garred P. The Genetic Variation of the Human HMGB1 Gene. Tissue Antigens. 2007;70:151–156. doi: 10.1111/j.1399-0039.2007.00854.x. [DOI] [PubMed] [Google Scholar]
- Krynetskaia NF, Cai X, Nitiss JL, Krynetski EY, Relling MV. Thioguanine Substitution Alters DNA Cleavage Mediated by Topoisomerase II. FASEB J. 2000;14:2339–2344. doi: 10.1096/fj.00-0089com. [DOI] [PubMed] [Google Scholar]
- Krynetski EY, Krynetskaia NF, Bianchi ME, Evans WE. A Nuclear Protein Complex Containing High Mobility Group Proteins B1 and B2, Heat Shock Cognate Protein 70, ERp60, and Glyceraldehyde-3-Phosphate Dehydrogenase Is Involved in the Cytotoxic Response to DNA Modified by Incorporation of Anticancer Nucleoside Analogues. Cancer Res. 2003;63:100–106. [PubMed] [Google Scholar]
- Krynetski EY, Krynetskaia NF, Gallo AE, Murti KG, Evans WE. A Novel Protein Complex Distinct From Mismatch Repair Binds Thioguanylated DNA. Mol Pharmacol. 2001;59:367–374. doi: 10.1124/mol.59.2.367. [DOI] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Hillisch A, Payet D, Buttinelli M, Travers A, Diekmann S. DNA Bending Induced by High Mobility Group Proteins Studied by Fluorescence Resonance Energy Transfer. Biochemistry. 1999;38:12150–12158. doi: 10.1021/bi990459+. [DOI] [PubMed] [Google Scholar]
- Maybaum J, Mandel HG. Unilateral Chromatid Damage: a New Basis for 6-Thioguanine Cytotoxicity. Cancer Res. 1983;43:3852–3856. [PubMed] [Google Scholar]
- McKinney K, Prives C. Efficient Specific DNA Binding by P53 Requires Both Its Central and C-Terminal Domains As Revealed by Studies With High-Mobility Group 1 Protein. Mol Cell Biol. 2002;22:6797–6808. doi: 10.1128/MCB.22.19.6797-6808.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek DW. The P53 Response to DNA Damage. DNA Repair (Amst) 2004;3:1049–1056. doi: 10.1016/j.dnarep.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Rich TA, Shepard RC, Mosley ST. Four Decades of Continuing Innovation With Fluorouracil: Current and Future Approaches to Fluorouracil Chemoradiation Therapy. J Clin Oncol. 2004;22:2214–2232. doi: 10.1200/JCO.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA Fragmentation During Apoptosis Induces Phosphorylation of H2AX Histone at Serine 139. J Biol Chem. 2000;275:9390–9395. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- Sahasrabudhe PV, Pon RT, Gmeiner WH. Solution Structures of 5-Fluorouracil-Substituted DNA and RNA Decamer Duplexes. Biochemistry. 1996;35:13597–13608. doi: 10.1021/bi960535y. [DOI] [PubMed] [Google Scholar]
- Seibert E, Osman R, Ross JBA. Dynamics of DNA Damage Recognition. In: Siede W, Kow YW, Doetsch PW, editors. DNA Damage Recognition. Taylor and Francis; New York London: 2005. pp. 3–19. [Google Scholar]
- Somerville L, Krynetski EY, Krynetskaia NF, Beger RD, Zhang W, Marhefka CA, Evans WE, Kriwacki RW. Structure and Dynamics of Thioguanine-Modified Duplex DNA. J Biol Chem. 2003;278:1005–1011. doi: 10.1074/jbc.M204243200. [DOI] [PubMed] [Google Scholar]
- Soubeyrand S, Schild-Poulter C, Hache RJ. Structured DNA Promotes Phosphorylation of P53 by DNA-Dependent Protein Kinase at Serine 9 and Threonine. Eur J Biochem. 2004;271:3776–3784. doi: 10.1111/j.1432-1033.2004.04319.x. [DOI] [PubMed] [Google Scholar]
- Stros M, Ozaki T, Bacikova A, Kageyama H, Nakagawara A. HMGB1 and HMGB2 Cell-Specifically Down-Regulate the P53- and P73-Dependent Sequence-Specific Transactivation From the Human Bax Gene Promoter. J Biol Chem. 2002;277:7157–7164. doi: 10.1074/jbc.M110233200. [DOI] [PubMed] [Google Scholar]
- Wei M, Burenkova O, Lippard SJ. Cisplatin Sensitivity in Hmbg1−/− and Hmbg1+/+ Mouse Cells. J Biol Chem. 2003;278:1769–1773. doi: 10.1074/jbc.M210562200. [DOI] [PubMed] [Google Scholar]
- Xie H, Midic U, Vucetic S, Obradovic Z. Handbook of Applied Algorithms. Wiley; 2007. [Google Scholar]
- Yamada S, Maruyama I. HMGB1, a Novel Inflammatory Cytokine. Clin Chim Acta. 2007;375:36–42. doi: 10.1016/j.cca.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Yin B, Kogan SC, Dickins RA, Lowe SW, Largaespada DA. Trp53 Loss During in Vitro Selection Contributes to Acquired Ara-C Resistance in Acute Myeloid Leukemia. Exp Hematol. 2006;34:631–641. doi: 10.1016/j.exphem.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the Apoptotic Response to Anticancer Agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jeffs G, Ren X, O’Donovan P, Montaner B, Perrett CM, Karran P, Xu YZ. Novel DNA Lesions Generated by the Interaction Between Therapeutic Thiopurines and UVA Light. DNA Repair (Amst) 2007;6:344–354. doi: 10.1016/j.dnarep.2006.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.