

Abstract
The present study shows that rat liver and brain mitochondrial nitric oxide synthase (mtNOS) are functionally associated with mitochondrial respiratory chain complex I. When complex I is activated, mtNOS exerts high activity and generates nitric oxide, whereas inactivation of complex I leads mtNOS to abandon its NOS activity. Functional association of mtNOS with complex I is potentially important in regulating mtNOS activity and mitochondrial functions.
Keywords: Mitochondrial nitric oxide synthase, nitric oxide, respiratory chain complex I, mitochondrial respiration
Introduction
Mitochondrial nitric oxide (NO) synthase (mtNOS) regulates various functions of mitochondria in liver [1–5], heart [6–8], brain [2, 9, 10], kidney [11, 12], as well as cells including endothelial [13], kidney [14], neuronal [15], cardiac myocytes [16], and breast cancer [17]. mtNOS-derived NO competes with oxygen and regulates mitochondrial primary functions including respiration and transmembrane potential (ΔΨ) [1,5,6,9]. mtNOS-derived NO also produces oxidative nitrogen oxide species that cause oxidative damage to mitochondria and cells and play a key role in apoptosis [1–3,8,12,15–17]. Despite significance of the regulation by mtNOS of mitochondrial functions, it is not fully understood how mitochondria regulate mtNOS activity. The rationale for the present study was to delineate whether mitochondria exert regulatory function over mtNOS activity. The present study shows that in isolated rat liver and brain mitochondria, mtNOS activity is associated with mitochondrial respiratory chain complex I (RCC-I) and suggest RCC-I regulates mtNOS activity.
Material and Methods
Purification of mitochondria
Liver mitochondria were isolated from Sprague-Dawley rats by differential centrifugation and purified by Percoll purification as described [18]. Brain mitochondria were purified as described [19]. Brains were removed, washed, and homogenized in Medium A (sucrose 320 mM, potassium EDTA 1 mM, 0.1% fatty acid–free bovine serum albumin [FAF-BSA], Tris-HCl 10 mM, pH 7.40). The homogenate was centrifuged at 1,100 x g for 10 min, and the supernatant was re-centrifuged at 19,000 x g for 20 min to obtain crude mitochondrial fraction. The pellet was resuspended in Medium A, laid on 6% Ficoll solution, and centrifuged at 19,000 x g for 30 min. Supernatant and the fluffy layer above the mitochondria pellet were discarded. Each 10 mg of the brown pellet was resuspended in 70 mg of the Purification Medium (2.56 g of 20% [w/w] dextran T500; 1.28 g of 40% [w/w] polyethylene glycol (PEG) 4000; 2.24 g of 1 M sorbitol; 70 mg of 10 mM potassium EDTA; 700 mg of 1% FAF-BSA; 200 mg of 200 mM potassium phosphate pH 7.80; H2O to 7.00 g) and centrifuged at 600 x g for 2 min in a swing-bucket centrifuge. The upper phase was discarded, 300 μl of the Washing Medium (96 mg of 20% [w/w] dextran T500; 480 mg of 40% [w/w] PEG 4000; 960 mg of 1 M sorbitol; 75 mg of 200 mM potassium phosphate pH 7.80; 30 mg of 10 mM potassium EDTA; 300 mg 1% FAF-BSA; H2O to 3.00 g) was added to the lower part, mixed and centrifuged at 600 x g for 2 min. The upper part was discarded and the lower part was mixed with 3 ml of Medium B (sorbitol 320 mM, potassium EDTA 1 mM, 0.1% FAF-BSA, potassium phosphate 5 mM, pH 7.80).
The suspension was centrifuged at 19,000 x g for 20 min, the supernatant was discarded, and the mitochondrial pellet was collected. All steps were carried out at 4 °C. Purity of the isolated mitochondria was assessed by measuring cytochrome a using ε605–630nm 12 mM−1 cm−1 as described [18]. Only mitochondria with less than 5% impurity were used. Broken mitochondria were prepared by 5 times freezing the intact mitochondria in liquid nitrogen followed by thawing as described [1, 17]. Submitochondrial particles (SMP) were prepared from mitoplasts as described [20]. Briefly, the outer membrane was removed by treating the intact mitochondria with digitonin (0.12 mg/mg of mitochondrial protein) for 15 min at 4 °C followed by 2 times wash. Mitoplasts were frozen-thawed as above and the unbroken mitoplasts were removed by 2 times spinning at 9,500 x g for 10 min. The supernatant of the second centrifugation was centrifuged at 100,000 x g for 30 min. The pellet referred to as SMP and supernatant referred as to mitochondrial matrix were collected.
Activation or inactivation of RCCs
The following experimental conditions were used in freshly isolated mitochondria: A) No respiratory substrate (No RS) refers to when respiratory chain was not altered by exogenous substrates; B) RCC-I activation refers to when RCC-I was activated by pyruvate/malate (10/5 mM); C) RCC-I inactivation refers to when RCC-I was inactivated by rotenone (5 μM); D) RCC-II activation refers to when RCC-I was inactivated by rotenone (5 μM) and mitochondria were energized from RCC-II by succinate (2.5 mM). In SMP, RCC-I was activated by NADH (100 μM).
Determination of mtNOS activity
mtNOS activity was determined using the following assays
Oxyhemoglobin assay
mtNOS activity was detected in broken mitochondria at 401–420 nm in the presence of oxyhemoglobin (4 μM) using ε401–420nm 100 mM−1 cm−1 as described [18].
Citrulline radioassay
Mitochondria were supplemented with L-[3H]arginine (30,000 – 50,000 cpm). Radio-labeled L-citrulline was separated from L-arginine using Dowex columns and the radioactivity was measured as described [3, 17].
Chemiluminescence assay
Mitochondria (50 μg) were injected into the purge vessel containing vanadium chloride (0.8 % in 1M HCl) thermostated at 93 °C, and NO was measured using chemiluminescence analyzer (Sievers 280i, General Electric, Boulder, CO) as described [17].
Amperometric assay
mtNOS activity in SMP was measured by NO-sensitive electrode (Apollo 4000; World Precision Instruments, FL) using a 7 μm tip micro-sensor (ISO-NO-007) placed in the chamber containing SMP (200 μg).
Oxygen consumption
Oxygen consumption was measured using a Clark-type oxygen electrode (Harvard Apparatus, Holliston, MA) as described [18, 21].
Intramitochondrial ionized calcium ([Ca2+]m)
The [Ca2+]m was measured by using dual-wavelength excitation fluorometery as described [8, 17]. Mitochondria were loaded with fura-2/AM for 20 min at room temperature followed by twice wash. Fura-2/AM loaded mitochondria were excited at 352–362 nm and emission was collected at 510 nm.
Statistical analysis
Barograms are Mean ± SEM of n = 4–6. Differences were tested by ANOVA and considered significant at p < 0.05. All computations were performed using PlotiT (Scientific Programming).
Results
mtNOS activity
Figs. 1A–F show mtNOS activity of liver and brain mitochondria as determined by three NOS assays. As shown, mtNOS activity was significantly higher when mitochondria were respiring on complex I, compared to when mitochondria were not supplied with exogenous respiratory substrates, RCC-I was inactivated, or RCC-II was activated. As shown, mtNOS activity in all samples was inhibited by mtNOS inhibitor, L-NG-monomethyl arginine (L-NMMA; Ref 3, 18).
Fig. 1. mtNOS activity and mitochondrial respiratory chain complexes.
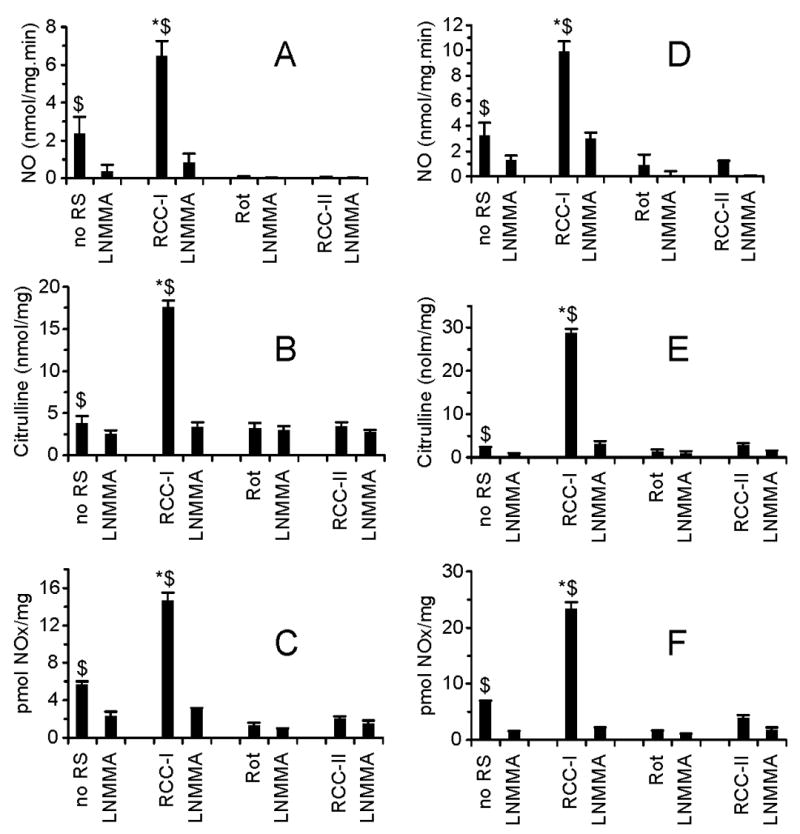
mtNOS activity in isolated liver (A, B, C) and brain (D, E, F) mitochondria were measured using oxyhemoglobin (A, D; expressed), citrulline radioassay (B, E), and chemiluminescence assay (C, F). Measurements were performed under the conditions that mitochondrial respiratory chain activity was not altered by exogenous respiratory substrates (no RS), RCC-I was activated (RCC-I), RCC-I was inactivated (Rot), or RCC-I was inactivated and mitochondria were energized from RCC-II (RCC-II). Where indicated mtNOS was inhibited by L-NMMA. Results are expresses as nmol NO per mg mitochondrial protein minute (A, D); nmol citrulline per mg mitochondrial protein (B, E), and pmol NOx per mg mitochondrial protein (C, F). *P < 0.05 RCC-I vs. no RS, Rot, RCC-II; $P < 0.05 vs. LNMMA.
Mitochondrial respiration
Figs. 2A, B show respiration of liver and brain mitochondria supported by activation of RCC-I, or when RCC-I was inactivated and respiration was supported from RCC-II. As shown, respiration supported by RCC-I was decreased by loading mitochondria with Ca2+, and that Ca2+-induced decrease of respiration was prevented by L-NMMA. However, when RCC-I was inactivated and respiration was supported from RCC-II, Ca2+ did not decrease the respiration.
Fig. 2. Oxygen consumption.
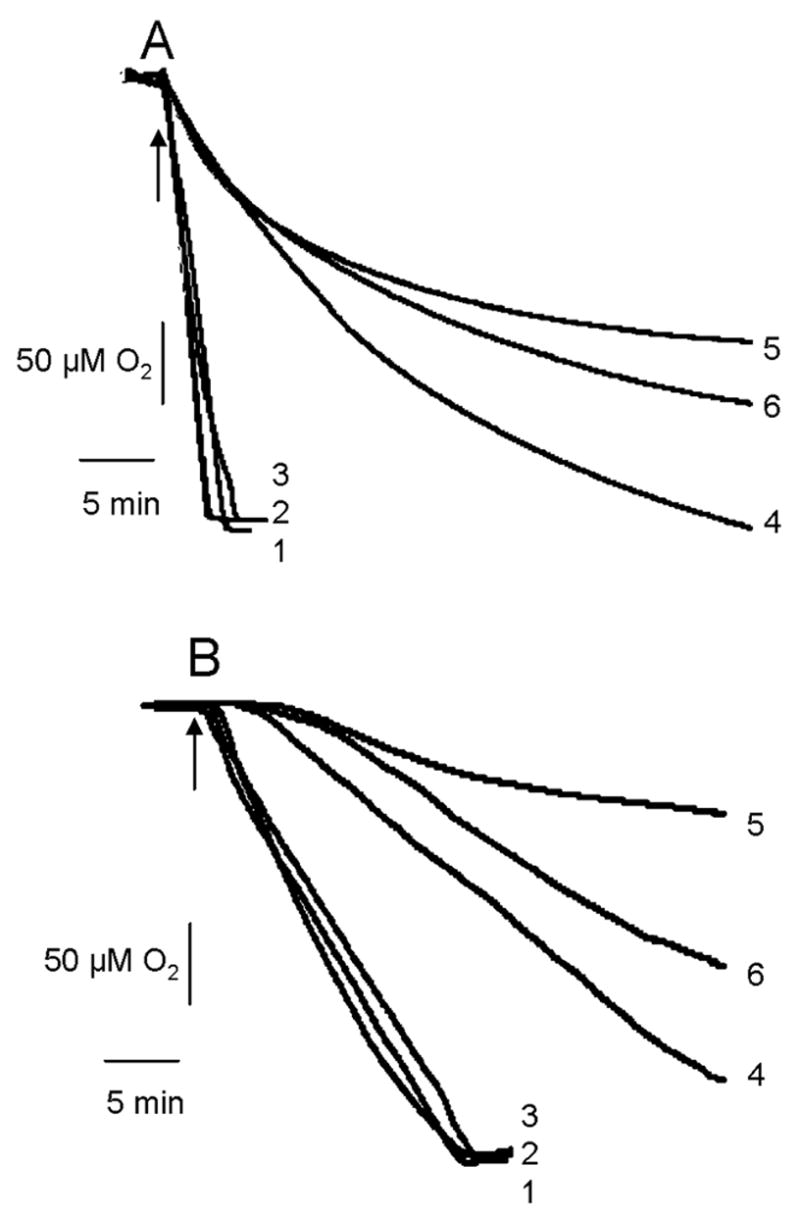
Oxygen consumption of liver (A) and brain mitochondria (B) are shown. Trace 1 represents respiration supported from RCC-II. Trace 2 is as trace 1 except Ca2+ was present in the medium. Trace 3 is as trace 2 except mitochondria were pre-treated with L-NMMA. Trace 4 represents respiration supported from RCC-I. Trace 5 is as trace 4, except Ca2+ was present in the medium. Trace 6 is as trace 5 except mitochondria were treated with L-NMMA. Arrows indicates addition of RCC-I or RCC-II substrates to the chamber.
Intramitochondrial calcium ([Ca2+]m)
mtNOS is Ca2+-dependent [1,7,9,13] and elevation of [Ca2+]m per se is sufficient to increases mtNOS activity [3,8,9,13,16,17]. Thus, the present study determined whether RCC-I activation increases [Ca2+]m. Fig. 3 shows that [Ca2+]m was not increased when RCC-I was activated compared to when RCC-I was inactivated and RCC-II was activated.
Fig. 3. Mitochondrial calcium ([Ca2+]m).
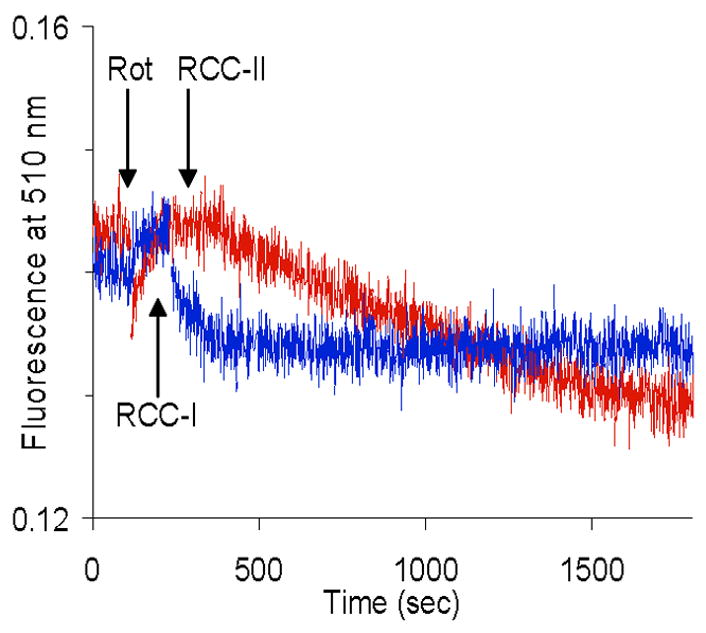
Intramitochondrial ionized calcium was measured under the conditions that RCC-I was activated (RCC-1) or RCC-I was inactivated by rotenone (Rot) and RCC-II was activated by succinate (RCC-II).
mtNOS activity of submitochondrial particle (SMP)
Figs. 4A, B show mtNOS activity of SMP. As shown, activation of SMP RCC-I by NADH stimulates mtNOS in an L-NMMA-sensitive manner. Increased mtNOS activity was inhibited when RCC-I was inactivated by rotenone. Replacing NADH with NADPH did not support mtNOS activity.
Fig. 4. mtNOS activity of submitochondrial particles.
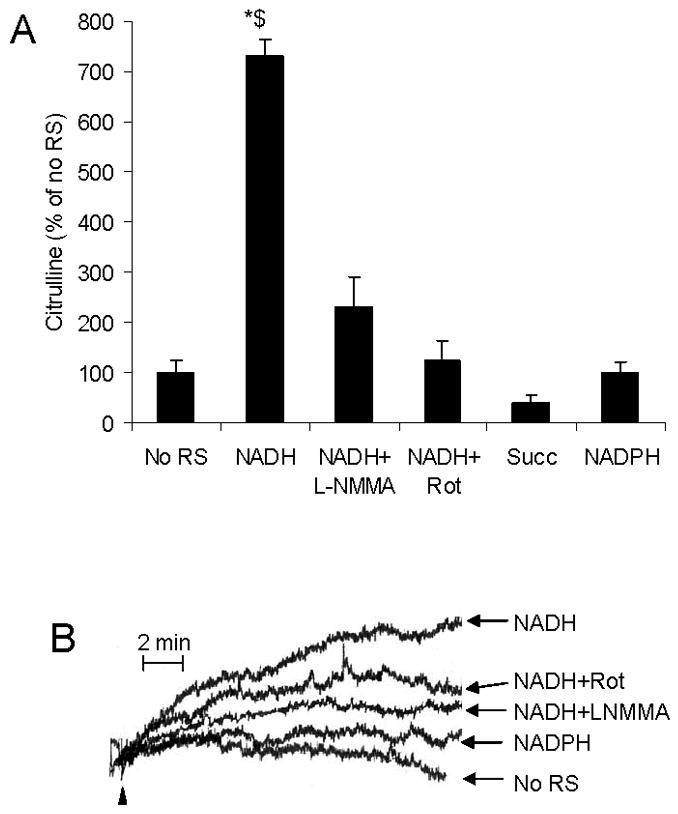
A) mtNOS activity was measured in submitochondrial particles by using citrulline assay and expressed as percentage of when submitochondrial particles were not supplied with exogenous respiratory substrates (No RS). Bars represent when RCC-I was activated by NADH in the absence (NADH) or presence of L-NMMA (NADH + LNMMA) or rotenone (NADH + Rot), when RCC-I was inactivated by rotenone and RCC-II was activated by succinate (Succ), or NADPH (NADPH) was present in place of NADH. B) mtNOS activity was measured by using NO-sensitive electrode under the conditions that submitochondrial particles were not supported with respiratory substrates (No RS), or RCC-I was activated by NADH in the absence (NADH) or presence of L-NMMA (NADH + LNMMA) or rotenone (NADH + Rot), or NADH was replaced with NADPH (NADPH). *P < 0.05 NADH vs. no RS, NADH + Rot, Succ, NADPH; $P < 0.05 vs. LNMMA.
Discussion
The present study provides evidence strongly suggesting that mtNOS activity in isolated rat liver and brain mitochondria is associated with RCC-I. Using 3 well-established NOS detection assays, Fig. 1 shows mtNOS activity as determined by all NOS assays is significantly higher when RCC-I is activated, compared to when mitochondria were not supplied with exogenous respiratory substrates, RCC-I was inactivated, or when mitochondria were energized from RCC-II. mtNOS is Ca2+-sensitive and increased [Ca2+]m stimulates mtNOS activity [1, 3, 7, 8, 13, 17]. mtNOS-derived NO decreases mitochondrial respiration [22] by competing with O2 for the binding site of cytochrome oxidase [23]. Figs. 2A, B confirm findings of Fig. 1 by showing that mitochondrial respiration supported from RCC-I was decreased by loading mitochondria with Ca2+ and that this effect of Ca2+ was prevented when mtNOS was inhibited. However, when RCC-I was inactivated and mitochondrial respiration was supported from RCC-II, Ca2+ did not decrease mitochondrial respiration. Findings presented under Figs. 1 and 2 strongly support the hypothesis that mtNOS activity in brain and liver mitochondria is associated with RCC-I activity.
Intramitochondrial calcium homeostasis is governed by continuous precipitation of [Ca2+]m to non-ionized calcium stores known as matrix electron-dense granules, and release of [Ca2+]m from the granules [8, 24]. Since mtNOS is [Ca2+]m-sensitive [8, 13, 17] and RCC-I activation stimulated mtNOS activity, we tested the effect of RCC-I activation on [Ca2+]m. As shown in Fig. 3, RCC-I activation did not increase [Ca2+]m, even [Ca2+]m was slightly decreased compared to when RCC-I was inactivated and mitochondria were energized from RCC-II. These findings rule out elevation of [Ca2+]m as a mechanism underling stimulation of mtNOS upon RCC-I activation. To further study mechanisms responsible for association of mtNOS activity with RCC-I, we measured mtNOS activity in SMP. mtNOS is located at the inner mitochondrial membrane [1, 3, 8, 10] and determination of mtNOS in SMP allows excluding potential interference of mitochondrial matrix, intermembrane space, outer membrane, or other components of mitochondria with RCC-I-mtNOS association. Figs. 4A, B show that NADH that activates RCC-I significantly increased mtNOS activity in an L-NMMA sensitive manner. NOS isozymes utilize L-arginine to produce NO and L-citrulline. For cytoplasmic NOS isozymes, this reaction utilizes electrons donated by NADPH rendering a redox potential of −280 mV [25]. Figs. 4A, B show that NADPH that serves native electron donor to cytoplasmic NOS isozymes [26, 27] fails to support mtNOS activity of SMP. These figures also show that NADH-increased mtNOS activity was inhibited when RCC-I was inactivated by rotenone. This finding rules out possible direct interaction of NADH with mtNOS. These findings also explain lack of NO formation when SMP were supplied NADPH, as well as inhibition of mtNOS by rotenone.
NOS isozymes are oxidoreductase proteins comprising of an oxygenase and a reductase domain [25]. Each NOS monomer homodimerizes in a configuration that allows the reductase domain of each monomer couples with the oxygenase domain of the coupling monomer. Electrons enter the NADPH-binding site at the reductase domain of a monomer and transverse to the arginine-binding site at the oxygenase domain of the coupling monomer where NO is generated (Diagram 1). Coupling is essential for NOS isozymes to retain their NOS activity and uncoupling NOS isozymes abolishes their NOS activity [25,26]. mtNOS is located at the inner mitochondrial membrane [1,18] where respiratory chain complexes are embedded. Mitochondrial respiratory chain complexes provide a range of redox potentials from −280 mV for RCC-I to +250 mV for cytochrome c oxidase [28]. Electrons derived from oxidation of NADH enter RCC-I and flow through the chain following the redox hierarchy of the respiratory complexes. Our results show that when RCC-I is active, mtNOS produces NO, while inactivation of RCC-I abolishes mtNOS activity. These findings suggest a functional association between mtNOS and RCC-I whereby mtNOS utilizes RCC-I as its source of electrons (Diagram 1), and describe a reciprocal regulation between mtNOS and mitochondrial respiratory chain.
Diagram 1. Functional association of mtNOS with RCC-I.
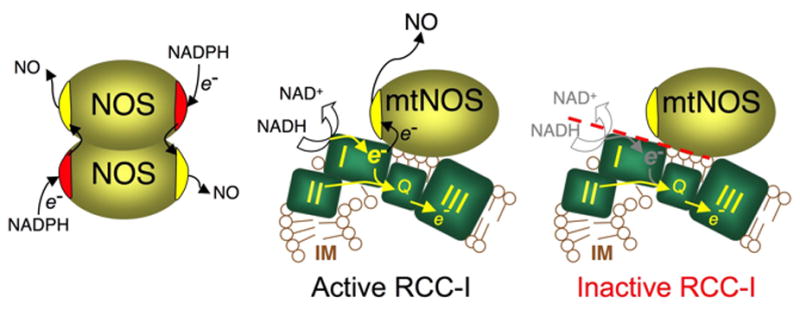
Cytoplasmic NOS isozymes consist of a reductase and an oxygenase domain. Each monomer homodimerizes in a configuration that allows electrons flow from the reductase domain of one monomer to the oxygenase domain of the coupling monomer where NO is produced. mtNOS that is localized at mitochondrial inner membrane (IM) where respiratory chain complexes are located. Electrons enter the chain from respiratory complex I (I) or RCC-II (II) and flow down to ubiquinone (Q) and downstream complexes (only RCC-III is shown). mtNOS functionally associates with RCC-I and utilizes RCC-I as source of electrons. When RCC-I is active, mtNOS receives electrons enabling the enzyme to produces NO. Inactivation of RCC-I ceases mtNOS reducing equivalents and abolishes its NOS activity.
Taken together, the present study demonstrates that liver and brain mtNOS are functionally associated with RCC-I. When RCC-I is activated mtNOS generates NO, whereas RCC-I inactivation leads mtNOS to abandon NO production. Exact molecular mechanism underlying association of mtNOS with RCC-I needs further investigations.
Acknowledgments
Financial support was provided by the National Institute on Aging (award AG023264-02) and American Heart Association (award 0565221B).
Abbreviations used
- RCC-I
respiratory chain complex I
- RCC-II
respiratory chain complex II
- NO
nitric oxide
- mtNOS
mitochondrial nitric oxide synthase
- L-NMMA
NG-monomethyl-L-arginine
- Rot
rotenone, Succ, succinate, Pyr, Pyruvate
- Mal
malate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 2.Navarro A, Boveris A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1244–R1249. doi: 10.1152/ajpregu.00226.2004. [DOI] [PubMed] [Google Scholar]
- 3.Ghafourifar P, Schenk U, Klein SD, Richter C. Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria, Evidence for intramitochondrial peroxynitrite formation. J Biol Chem. 1999;274:31185–31188. doi: 10.1074/jbc.274.44.31185. [DOI] [PubMed] [Google Scholar]
- 4.Navarro A, Sanchez-Pino MJ, Gomez C, Bandez MJ, Cadenas E, Boveris A. Dietary thioproline decreases spontaneous food intake and increases survival and neurological function in mice. Antioxid Redox Signal. 2007;9:131–141. doi: 10.1089/ars.2007.9.131. [DOI] [PubMed] [Google Scholar]
- 5.Valdez LB, Zaobornyj T, Boveris A. Mitochondrial metabolic states and membrane potential modulate mtNOS activity. Biochim Biophys Acta. 2006;1757:166–172. doi: 10.1016/j.bbabio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Gonzales GF, Chung FA, Miranda S, Valdez LB, Zaobornyj T, Bustamante J, Boveris A. Heart mitochondrial nitric oxide synthase is upregulated in male rats exposed to high altitude (4,340 m) Am J Physiol Heart Circ Physiol. 2005;288:H2568–H2573. doi: 10.1152/ajpheart.00812.2004. [DOI] [PubMed] [Google Scholar]
- 7.Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc Natl Acad Sci USA. 2001;98:14126–14131. doi: 10.1073/pnas.241380298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zenebe WJ, Nazarewicz RR, Parihar MS, Ghafourifar P. Hypoxia/Reoxygenation of isolated rat heart mitochondria causes cytochrome c release and oxidative stress; evidence for involvement of mitochondrial nitric oxide synthase. J Mol Cell Cardiol. 2007;43:411–419. doi: 10.1016/j.yjmcc.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lores-Arnaiz S, D'Amico G, Czerniczyniec A, Bustamante J, Boveris A. Brain mitochondrial nitric oxide synthase: in vitro and in vivo inhibition by chlorpromazine. Arch Biochem Biophys. 2004;430:170–177. doi: 10.1016/j.abb.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 10.Riobo NA, Melani M, Sanjuan N, Fiszman ML, Gravielle MC, Carreras MC, Cadenas E, Poderoso JJ. The modulation of mitochondrial nitric-oxide synthase activity in rat brain development. J Biol Chem. 2002;277:42447–42455. doi: 10.1074/jbc.M204580200. [DOI] [PubMed] [Google Scholar]
- 11.Boveris A, Valdez LB, Alvarez S, Zaobornyj T, Boveris AD, Navarro A. Kidney mitochondrial nitric oxide synthase. Antioxid Redox Signal. 2003;5:265–271. doi: 10.1089/152308603322110841. [DOI] [PubMed] [Google Scholar]
- 12.Vinas JL, Sola A, Hotter G. Mitochondrial NOS upregulation during renal I/R causes apoptosis in a peroxynitrite-dependent manner. Kidney Int. 2006;69:1403–1409. doi: 10.1038/sj.ki.5000361. [DOI] [PubMed] [Google Scholar]
- 13.Dedkova EN, Ji X, Lipsius SL, Blatter LA. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am J Physiol Cell Physiol. 2004;286:C406–C415. doi: 10.1152/ajpcell.00155.2003. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Figueroa MO, Caamano C, Marin R, Guerra B, Alonso R, Morano MI, Akil H, Watson SJ. Characterization of basal nitric oxide production in living cells. Biochim Biophys Acta. 2001;1540:253–264. doi: 10.1016/s0167-4889(01)00138-0. [DOI] [PubMed] [Google Scholar]
- 15.Dennis J, Bennett JP., Jr Interactions among nitric oxide and Bcl-family proteins after MPP+ exposure of SH-SY5Y neural cells I: MPP+ increases mitochondrial NO and Bax protein. J Neurosci Res. 2003;72:76–88. doi: 10.1002/jnr.10539. [DOI] [PubMed] [Google Scholar]
- 16.Nazarewicz RR, Zenebe WJ, Parihar A, Parihar MS, Ghafourifar P. 12(S)-Hydroperoxyeicosatetraenoic acid (12-HETE) increases mitochondrial nitric oxide by increasing intramitochondrial calcium. Arch Biochem Biophys. 2007 doi: 10.1016/j.abb.2007.09.018. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nazarewicz RR, Zenebe WJ, Parihar A, Larson SK, Alidema E, Choi J, Ghafourifar P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res. 2007;67:1282–1290. doi: 10.1158/0008-5472.CAN-06-3099. [DOI] [PubMed] [Google Scholar]
- 18.Ghafourifar P, Asbury ML, Joshi SS, Kincaid ED. Determination of mitochondrial nitric oxide synthase activity. Methods Enzymol. 2005;396:424–444. doi: 10.1016/S0076-6879(05)96036-9. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Perez MJ. Preparation of synaptosomes and mitochondria from mammalian brain. Methods Enzymol. 1994;228:403–411. doi: 10.1016/0076-6879(94)28040-1. [DOI] [PubMed] [Google Scholar]
- 20.Ghafourifar P. Characterization of mitochondrial nitric oxide synthase. Methods Enzymol. 2002;359:339–350. doi: 10.1016/s0076-6879(02)59197-7. [DOI] [PubMed] [Google Scholar]
- 21.Ghafourifar P, Klein SD, Schucht O, Schenk U, Pruschy M, Rocha S, Richter C. Ceramide induces cytochrome c release from isolated mitochondria. Importance of mitochondrial redox state. J Biol Chem. 1999;274:6080–6084. doi: 10.1074/jbc.274.10.6080. [DOI] [PubMed] [Google Scholar]
- 22.Peralta JG, Finocchietto PV, Converso D, Schopfer F, Carreras MC, Poderoso JJ. Modulation of mitochondrial nitric oxide synthase and energy expenditure in rats during cold acclimation. Am J Physiol Heart Circ Physiol. 2003;284:H2375–H2383. doi: 10.1152/ajpheart.00785.2002. [DOI] [PubMed] [Google Scholar]
- 23.Boveris DL, Boveris A. Oxygen delivery to the tissues and mitochondrial respiration. Front Biosci. 2007;12:1014–1023. doi: 10.2741/2121. [DOI] [PubMed] [Google Scholar]
- 24.Ashraf M, Bloor CM. X-ray microanalysis of mitochondrial deposits in ischemic myocardium. Virchows Arch B Cell Pathol. 1976;22:287–297. doi: 10.1007/BF02889222. [DOI] [PubMed] [Google Scholar]
- 25.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ortiz de Montellano PR, Nishida C, Rodriguez-Crespo I, Gerber N. Nitric oxide synthase structure and electron transfer. Drug Metab Dispos. 1998;26:1185–1189. [PubMed] [Google Scholar]
- 27.Ghosh DK, Holliday MA, Thomas C, Weinberg JB, Smith SM, Salerno JC. Nitric-oxide synthase output state. Design and properties of nitric-oxide synthase oxygenase/FMN domain constructs. J Biol Chem. 2006;281:14173–14183. doi: 10.1074/jbc.M509937200. [DOI] [PubMed] [Google Scholar]
- 28.Tyler DD. The mitochondrion in health and disease. VCH Publishers Inc.; New York: 1992. [Google Scholar]