

Summary
The effect of protein kinase C (PKC) on rapid N-type inactivation of K+ channels has not been reported previously. We found that PKC specifically eliminates rapid inactivation of a cloned human A-type K+ channel (hKv3.4), converting this channel from a rapidly inactivating A type to a noninactivating delayed rectifier type. Biochemical analysis showed that the N-terminal domain of hKv3.4 is phosphorylated in vitro by PKC, and mutagenesis experiments revealed that two serines within the inactivation gate at the N-terminus are sites of direct PKC action. Moreover, mutating one of these serines to aspartic acid mimics the action of PKC. Serine phosphorylation may thus prevent rapid inactivation by shielding basic residues known to be critical to the function of the inactivation gate. The regulatory mechanism reported here may have substantial effects on signal coding in the nervous system.
Introduction
Protein phosphorylation is a key regulatory mechanism of membrane excitability and ion channel function (Levitan and Kaczmareck, 1991). Various studies have shown that protein kinases regulate ligand-gated and voltage-gated ion channels (e.g., Shuster et al., 1985; Arm strong and Eckert, 1987; Sigel and Baur, 1988; Shearman et al., 1989; Levitan et al., 1990; Chung et al., 1991; Busch et al., 1992; Swope et al., 1992; Sculptoreanu et al., 1993; Krishek et al., 1994). For instance, it has been found that protein kinase C (PKC) and cyclic AMP-dependent protein kinase (PKA) directly regulate gating of voltage-gated Na+ channels (Numann et al., 1991; West et al., 1991; Li et al., 1993). These studies suggested that phosphorylation of a single serine within the inactivation domain of the Na+ channel slowed inactivation and regulated the ability of PKA to phosphorylate additional sites that affected peak Na+ current. Voltage-gated K+ channels may also be regulated by protein kinases (Walsh and Kass, 1988; Augustine and Bezanilla, 1990; Moran et al., 1991; Perozo and Bezanilla, 1990; Covarrubias et al., 1992, Soc. Neurosci., abstract), but the molecular mechanisms are not well understood. In a recent study (Drain et al., 1994), it was found that phosphatase slows inactivation of Drosophila melanogaster Shaker K+ channels and that PKA reverses this action. It was deduced that this effect is most likely mediated by PKA-dependent phosphorylation of serine residues at the cytoplasmic C-terminal region of the channel polypeptide. However, the mechanism by which phosphorylation may affect K+ channel inactivation was not elucidated.
Inactivation of voltage-gated K+ channels affects their ability to control spike repolarization and interspike interval (Hille, 1992). Rapid N-type inactivation in several Shaker-related K+ channels is mediated by a cytoplasmic domain at the N-terminus of a channel subunit (Zagotta et al., 1990; Hoshi et al., 1990; Murrell-Lagnado and Aldrich, 1993a, 1993b). This region has been identified as the inactivation gate, which acts as a blocking particle tethered to the channel (Demo and Yellen, 1991). These findings support the “ball and chain” hypothesis of channel inactivation originally proposed to explain rapid inactivation of voltage-gated Na+channels (Bezanilla and Armstrong, 1977). Although voltage-gated K+ channels are tetrameric, a single gate seems sufficient to inactivate Shaker K+ channels (MacKinnon, 1991; MacKinnon et al., 1993), and basic and hydrophobic amino acids within the N-terminal domain are critical to the interaction of the inactivation gate with the internal mouth of the channel pore (Murrell-Lagnado and Aldrich, 1993a, 1993b).
We examined whether rapid N-type inactivation of a voltage-gated K+ channel from human brain can be regulated directly by PKC-dependent protein phosphorylation and investigated the mechanism of this action. This channel is encoded by hKv3.4, a member of the Shaw subfamily of K+ channel genes related to Shaker (Wei et al., 1990; Rudy et al., 1991; Schroter et al., 1991). Comparing the configuration of basic residues at the N-terminus of several inactivating K+ channels, we were struck by their close correspondence with unique serine residues in hKv3.4. We report evidence which strongly suggests that phosphorylation of the inactivation gate modulates N-type inactivation in this K+ channel.
Results
Rapid Inactivation of hKv3.4
In Xenopus laevis oocytes, hKv3.4 expresses a voltage-dependent outward K+ current which, in response to a step depolarization, inactivates rapidly and almost completely within approximately 100 ms (Figure 1A). Current decay was well described, assuming an exponential time course (Figure 1A, inset), and the time constant of such decay was independent of whole-cell current amplitude (Figure 1B). Also, the recording configuration did not affect inactivation significantly. Currents inactivated similarly when recorded from whole oocytes and cell-attached or inside-out patches (Figure 1C; see Figure 3). Rapid inactivation is an inherent property of hKv3.4, since removal of the first 28 amino acids at the N-terminus (Rettig et al., 1992) resulted in complete elimination of current inactivation (Figure 1D). Thus, as in other rapidly inactivating K+ channels, the N-terminal region in hKv3.4 controls rapid inactivation. Contrary to Shaker B K+ channels, the N-terminal deletion mutant did not reveal evidence of slow C-type inactivation in hKv3.4.
Figure 1. Rapid Inactivation of hKv3.4 K+ Channels Expressed in Xenopus Oocytes.

(A) Whole-oocyte outward K+ currents were elicited by 112 ms step depolarizations between −50 and +50 mV in 10 mV increments from a holding potential of −100 mV. Inset shows that current inactivation is well described as an exponential decay (decaying solid line) with a time constant (τ) of 9.6 ms at +50 mV. When whole-oocyte currents were >10 μA (at +50 mV), a second small component (<10%) with slower time constant (~50 ms) was needed to describe current decay.
(B) τ of current inactivation at +50 mV is plotted as a function of peak current (to represent level of expression; n = 20 cells from four separate batches of oocytes). Dotted line represents the average value.
(C) Macroscopic outward K+ current recorded from a cell-attached macropatch. Response was elicited by a 220 ms step depolarization to +30 mV from a holding potential of −90 mV. The smooth line across the current trace represents the best exponential fit with a τ of 16 ms. This compares with a τ of 13 ms determined from whole-oocyte currents at +30 mV.
(D) Whole-oocyte outward currents elicited by 900 ms step depolarizations between −30 and +50 mV in 20 mV increments, from a holding potential of −100 mV for wild-type (WT) and ΔN28 (deletion mutant removing the first 28 amino acids; see Figure 5).
Figure 3. Effect of PKC on Single-Channel Outward Currents in an Inside-Out Patch Expressing a Single hKv3.4 K+ Channel.
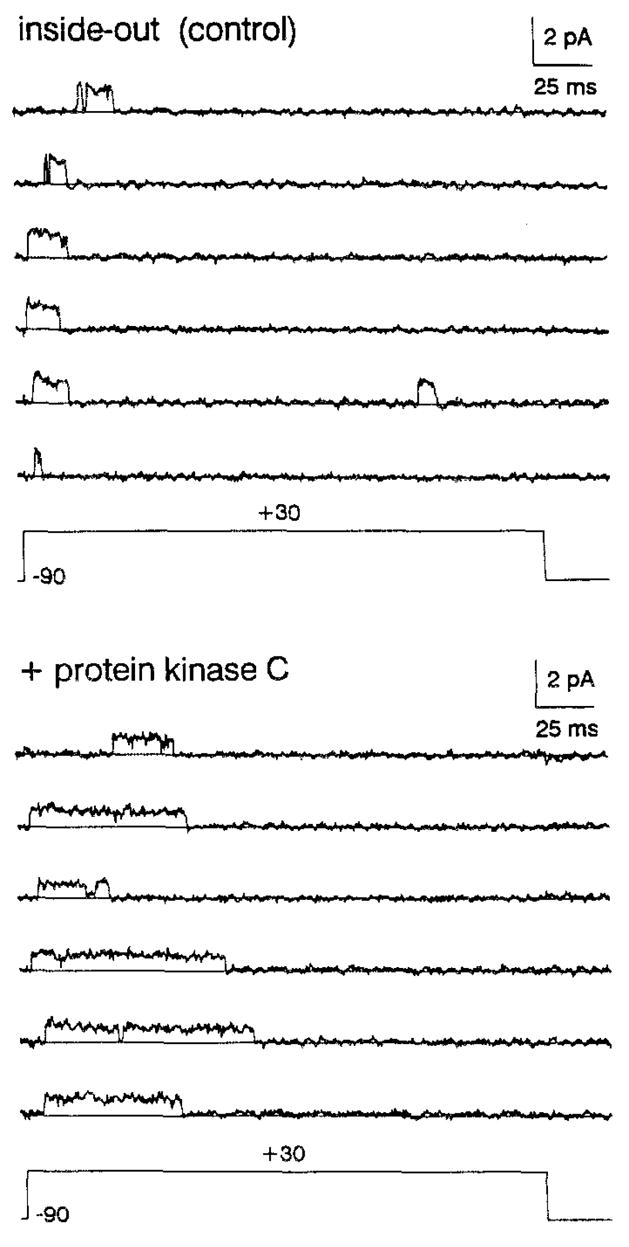
Currents were elicited by 250 ms step depolarizations from −90 to +30 mV at intervals of 5 s. Records were low-pass filtered at 1 kHz and sampled at 250 μs per point. In a total of 32 consecutive records (including cell-attached period), no overlapping events were seen. To test the action of PKC, the cytoplasmic side of the patch was perfused with high calcium intracellular solution supplemented with 1 μM brain phosphatidylserine, 1 μM diacyl-glycerol, 1 mM ATP, and ~10 pM purified brain PKC. PKC recording was taken ~3 min after application of the enzyme (the latency of PKC action in this patch). Pipette solution contained 140 mM NaCl, 6 mM MgCl2, and 5 mM HEPES (pH 7.2}. Intracellular bath solution contained 140 mM KCl, 2 mM MgCl2, 1 mM CaCl2, 11 mM EGTA, and 10 mM HEPES (pH 7.2). High calcium intracellular solution contained 140 mM KCl, 2 mM MgCl2, 1.5 mM CaCl2, 1 mM EGTA, and 10 mM HEPES (pH 7.2). Intracelluar solutions were supplemented with 5 mM glutathioneto prevent inhibition of inactivation owing to cysteine oxidation (see Experimental Procedures). All were titrated to the indicated pH with N-methyl-D-glucamine.
PKC Selectively Regulates Rapid Inactivation of hKv3.4
To examine whether PKC modulates K+ channels encoded by hKv3.4, we exposed Xenopus oocytes expressing these channels to two typical PKC activators. Whole-cell currents were recorded using two-microelectrode voltage clamp. Both phorbol 12-myristate 13-acetate (PMA) and 1-oleoyl-2-acetyl-rac-glycerol (OAG) substantially reduced inactivation of the current in a time-dependent manner (Figures 2A and 2B). This was particularly striking in the presence of PMA. In all instances, it induced elimination of rapid current inactivation. As generally observed (Shearman et al., 1989), PMA was more potent than OAG. In a parallel experiment, the current integral over a 112 ms depolarization was enhanced 3.1-fold by 10 nM PMA and 1.7- and 1.9-fold by 20 and 60 μM OAG, respectively. To measure the time course of the effect, the relative amplitude of the steady-state current (at the end of a 112 ms depolarization) was measured at intervals of 30 s, before and after PMA or OAG (Figure 2C). The action of PKC activators was relatively slow, as expected for a second messenger-mediated effect. The response had a latency of 1.5–2 min and reached a maximum in approximately 10–12 min. A response similar to that obtained with PMA was seen in all attempts (n = 28). This action seems specific (Figure 2D), since calphostin C (a specific PKC inhibitor) significantly reduced the action of PMA, and α-PMA (an inactive phorbol ester analog) produced no effect.
Figure 2. Effect of PKC Activators on Inactivation of Whole-Oocyte K+ Currents Expressed by hKv3.4.
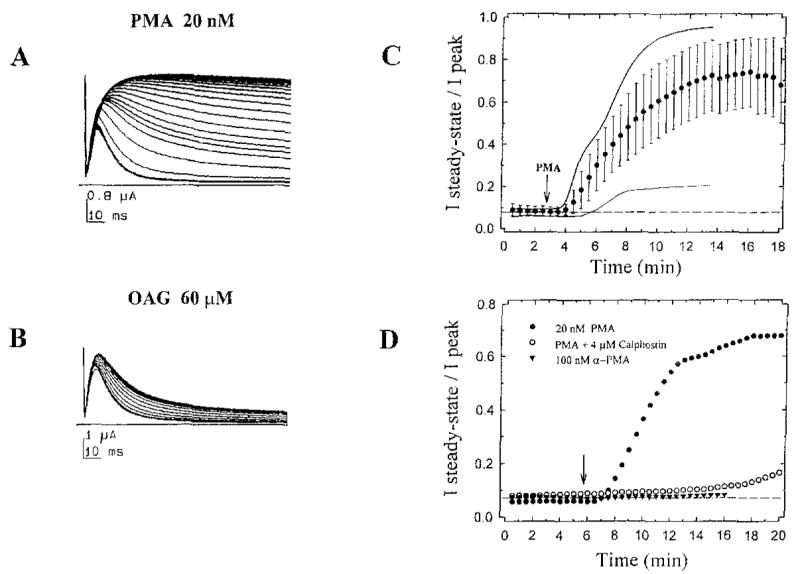
(A) Consecutive whole-oocyte current responses (32) elicited by repetitive 112 ms step depolarizations to +30 rnV from a holding potential of −100 mV, at 30 s intervals. After 10 control responses in normal bath solution (under continuous bath perfusion), the oocyte was bathed in normal bath solution plus 20 nM PMA.
(B) Same as in (A) but adding 60 μM OAG, Both compounds induced a time-dependent inhibition of current inactivation. To apply activators, the chamber (volume ~ 350 μl) was perfused at a rate of 3–4 ml/min. PMA and OAG dilutions were prepared immediately before the experiments from concentrated stock solutions (dimethylsulfoxide solutions stored at −20°C).
(C) Time course of the effect of PKC activators. From each current trace, we measured peak (Ipeak) and steady-state (Isteady-state) amplitudes (the apparent steady-state current was measured as the average of the last 10 points in a trace). Thus, Isteady-state/Ipeak is a measure of the fraction of current that does not inactivate at the end of a 112 ms step depolarization. This variable was then plotted against recording time. Graph shows the average time course from 12 independent experiments (each point [closed circles] and corresponding bars represent the mean ± SD). Dashed line represents the average Isteady-state/Ipeak before PMA (<10 % ). For comparison, the time courses of experiments shown in (A) and (B) are plotted here too (PMA, thick solid line; OAG, thin solid line).
(D) As described above, experiments were conducted in three different conditions (additions at the arrow): 20 nM PMA alone (closed circles), 20 nM PMA plus 4 μM calphostin C (open circles), and 100 nM α-PMA alone (closed triangles). Calphostin C was illuminated under standard fluorescent light for ~1 hr before the experiment and incubated for 7.5 min with the oocyte, before PMA application (inhibitor was present throughout the rest of the experiment). Calphostin C by itself did not affect kinetics or amplitude of currents.
At the single-channel level, direct application of purified brain PKC (Huang et al., 1986) to the cytoplasmic side of an inside-out patch also reduced channel inactivation (Figure 3). In control, openings occur in isolation near the beginning of the pulse and are short in duration owing to rapid inactivation. In the presence of PKC and necessary cofactors (Figure 3, legend), openings become longer, since inactivation is partially removed. The delay of this response was ~ 3 min after PKC application. Records did not have sufficient events (<50) to construct dwell time histograms; however, the average open time increased from 11 ± 8 ms in control to 48 ± 23 ms in the presence of PKC.
To test further the specificity and selectivity of PKC activators on hKv3.4, we examined oocytes expressing other subtypes of inactivating K+ channels (Wei et al., 1990; Pak et al., 1991). Figure 4 shows the effect of PMA on Drosophila Shal2, mouse mKv4.1 (or mShal1), and Drosophila Shaker H37. It can be seen that PKC activation significantly enhanced Shal2 and mKv4.1 and slightly inhibited Shaker H37. For Shal2 and mKv4.1, peak currents were enhanced 92% (n = 2) and 53% ± 32% (n = 7), respectively, and this was inhibited by calphostin C. The slow time course of these effects resembles the action of PMA on inactivation of hKv3.4 (Figure 4A). In all three cases, however, inactivation kinetics was virtually unaltered (Figures 4B–4D, insets). Another study has also shown that PKC inhibits Shaker B peak currents, without affecting inactivation kinetics (Moran et al, 1991). Thus, PKC selectively regulates rapid inactivation of hKv3.4.
Figure 4. Effect of PKC Activation on A-Type K+ Currents Expressed in Xenopus Oocytes.

(B–D) Whole-oocyte outward K+ currents elicited by 450 ms step depolarizations to +40 mV from a holding potential of −100 mV. Currents are shown before and after bath application of 20 nM PMA. The effect of PMA reached a maximum within 5–10 min (see below). To investigate an effect on current kinetics, traces were scaled to match their peak current (insets)
(A) Time course of experiments shown in (B), (C), and (D). This was measured as described in Figure 2, except that only peak current was measured. For comparison, this variable was normalized to the averaged peak amplitude before PMA.
PKC Phosphorylates the N-Terminal Region of hKv3.4
Since deletion of the first 28 amino acids of hKv3.4 removes rapid inactivation (Figure 1D), we assumed that target sites of PKC were located within the N-terminal region. The first 40 N-terminal amino acids of three A-type K+ channels are compared in Figure 5. For these K+ channels, strong evidence has suggested that the N-terminal region controls rapid inactivation (Zagotta et al., 1990; Hoshi et al., 1990; Ruppersberg et al., 1991b;Rettig etal., 1992). However, a close correspondence between positively charged amino acids and putative PKC sites is clearly apparent in hKv3.4 only. Basic residues flanking putative phosphate acceptors are found in known PKC consensus sequences (Marais et al., 1990; Kennelly and Krebs, 1991). To test directly whether the N-terminal region of hKv3.4 is phosphorylated by PKC in vitro, we used purified brain PKC and a synthetic peptide that matches the first 28 amino acids in hKv3.4 as the substrate. We found that diacylglycerol (DAG) stimulated phosphorylation of the N-terminal peptide >10-fold: 521 ± 5 and 7537 ± 227 nanomoles per minute per microgram of PKC (n = 3), in the absence and presence of DAG, respectively. Thus, residues within the inactivation gate of hKv3.4 act as specific phosphate acceptors in the presence of PKC.
Figure 5. Comparison of the Amino Acid Sequence at the N-Terminus of Three A-Type K+ Channels.
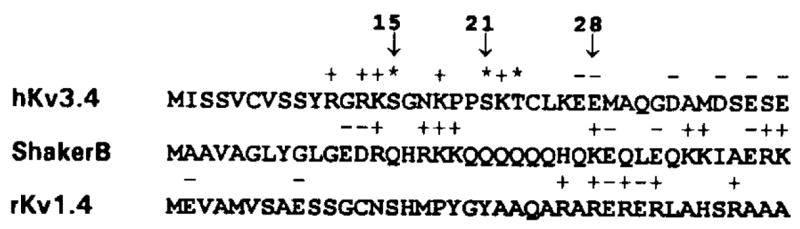
To compare the primary structures of inactivation gates of three A-type K+ channels, we show the first 40 amino acids at the N-termini of hKv3.4, Shaker B, and rKv4.1. Basic and acidic amino acids are indicated above the corresponding sequence by a positive or negative sign, respectively. Potential PKC phosphorylation sites in hKv3.4 are indicated by asterisks. Arrows mark residues 15, 21, and 28. A; Ala; R, Arg; N, Asn; D, Asp; C, Cys; E, Glu; Q, Gln; G, Gly; H, His; I, Ile; L, Leu; K, Lys; M, Met; F, Phe; P, Pro; S, Ser; T, Thr; W, Trp; Y, Tyr; V, Val.
Serine Residues at the N-Terminus of hKv3.4 Are Targets of PKC
We focused our attention on residues S15, S21, and T23. To test whether these sites are PKC targets associated with regulation of channel inactivation, we created a series of point mutations in which alanine (A) was substituted for serine (S) or for threonine (T). These mutations are listed in Table 1. In unstimulated oocytes, there was little or no difference between wild-type (WT) and mutant channels. For instance, the time constants of inactivation at +50 mV were 9.4 ± 0.7, 8.4 ± 0.6, 8.0 ± 0.5, 7.3 ± 0.4, and 7.7 ± 0.9 ms for WT, S15A, S21A, S15A/S21A, and S15A/S21A/T23A, respectively (n = 4–9). To test the ability of PKC to remove inactivation from WT and mutant channels, whole-oocyte currents were recorded before and after 20 nM PMA (Figure 6). Long step depolarizations revealed a slow current decay in the presence of PMA. Since the time course of this decay was variable (compare WT responses in Figure 6), we did not attempt to characterize it as a sum of exponentials. Thus, to quantitate the results, we integrated WT and mutant currents (before PMA and after PMA) over the length of a 900 ms depolarization (Figure 6; Table 1). This is a simple model-independent method to study the effect of point mutations on regulation of hKv3.4 inactivation by PKC.
Table 1.
Effect of PKC on Inactivation of WT and Mutant hKv3.4 K+ Channels
| K+ Channel | Qba (nC/μA) | Qsa (nC/μA) | (Qb−Qs)ac (nC/μA) | Qfb | n |
|---|---|---|---|---|---|
| WT | 60 ± 29 | 615 ± 81 | 552 ± 93 | 1.0 | 15 |
| S15A | 71 ± 44 | 400 ± 20 | 329 ± 42 | 0.58 | 4 |
| S21A | 36 ± 5 | 308 ± 12 | 266 ± 76 | 0.48 | 3 |
| S15A/S21A | 45 ± 13 | 240 ± 15 | 195 ± 9 | 0.35 | 4 |
| S21AAT23A | 54 ± 9 | 235 ± 30 | 272 ± 9 | 0.49 | 4 |
| S15A/S21A/T23A | 44 ± 4 | 310 ± 65 | 181 ± 42 | 0.33 | 3 |
All values are mean ± SD. Current integral (nC) over the length of a 900 ms pulse to ±50 mV was normalized to its corresponding peak amplitude (μA). Qb is basal current integral, before PMA. Qs is stimulated current integral, after PMA. Thus, (Qs − Qb) represents PKC-dependent increment in current integral.
Qf = (Qs − Qb)M/(Qs − Qb)WT (i.e., fractional increment), where (Qs − Qb)M and (Qs − Qb)WT are PKC-dependent current integrals for mutant and wild-type channels, respectively. This analysis assumes that Qf for wild-type is 1.0.
All mutant values were significantly different from wild type at p < .001 (two-tailed Student’s t test). By the same test, values for S15A, S21A, and S21AAT23A were not significantly different, and values for S15A/S21A and S15A/S21A/T23A were also not significantly different. This ruled out a role of T23 on PKC-dependent removal of inactivation. Differences between S21A and S15A/S21A or S21A/T23A and S15A/S21A/T23A were not evaluated further, since it was clear that the effects of S15A and S21A were not additive (see Figure 6).
Figure 6. Whole-Oocyte Outward K+ Currents Expressed by WT and Mutant hKv3.4 in the Absence and Presence of PMA.

Currents evoked by 900ms step depolarizations from −100 mV to +50 mV were recorded before and after exposure to 20 nM PMA. In the presence of PMA, currents were recorded after reduction of inactivation had reached steady state (10–15 min; Figure 2). For comparison, all currents were normalized to their corresponding peak and overlaid. WT peak currents were 2.1, 2.6, 3.5, 2.4, and 1.9 μA. The corresponding mutant peak currents were 0.9,2.5, 3.5,5.6, and 1.2 μA.
We found that S15 and S21 were especially important. Compared with WT, inhibition of inactivation by PKC was intermediate in S15A and S21A (Figure 6). Individually, these mutations affected ~50% of the response to PMA (Qf in Table 1). This clearly showed that S15 and S21 are sites that can affect rapid inactivation when phosphorylated by PKC. Furthermore, the double mutant (S15A/S21A) produced additional inhibition, but ~30% of PKC action remained (Qf in Table 1). This indicated that the effect of S15A and S21A is only partially additive and suggested that secondary PKC sites may also be involved (see Discussion). Two more mutations were created to test the role of T23. We found that, compared with S21A or S15A/S21A, another double mutation (S21A/T23A) and a triple mutation (S15A/S21A/T23A) did not produce additional inhibition (Figure 6; Table 1). This ruled out T23 as an important PKC target that affects channel inactivation. Thus, S15 and S21 are key sites involved in regulation of hKv3.4 inactivation by PKC.
Phosphorylation of the inactivation Gate May Disrupt Critical Electrostatic Interactions
The inactivation gate of rapidly inactivating A-type K+ channels acts as an open-channel blocker (Demo and Yellen, 1991). Long-range electrostatic interactions control the on-rate of channel blockage, and hydrophobic interactions stabilize binding of the inactivation domain to a receptor near the inner mouth of the pore (Isacoff et al., 1991; Murrell-Lagnado and Aldrich, 1993a, 1993b). Such electrostatic interactions are determined by multiple basic residues at the N-terminal region of the channel polypeptide, and the rate of inactivation is affected by substitutions that alter the net positive charge of the inactivation domain (Zagotta et al., 1990; Murrell-Lagnado and Aldrich, 1993a). We propose that phosphorylation of S15 and S21 in hKv3.4 K+ channels reduces the net positive charge of the inactivation gate (each phosphate added may contribute one or two negative charges at physiological pH). To test this hypothesis, we also replaced a phosphorylation site (S15) with an acidic amino acid (aspartate), to introduce a single constitutive negative charge, and found that this mutant (S15D) inactivated significantly more slowly than WT, without affecting the rising phase of the current (Figure 7). The time constant of inactivation at +50 mV increased from 10 ± 1 ms (n = 11) to 16 ± 2 ms (n = 9) for WT and S15D, respectively. Thus, slower inactivation of phosphorylated channels can result from shielding of critical positive charges, which reduces the affinity of the inactivation gate for its receptor near the pore.
Figure 7. Whole-Oocyte Outward K+ Currents Expressed by hKv3.4 (S15D) and the Corresponding WT Controls.
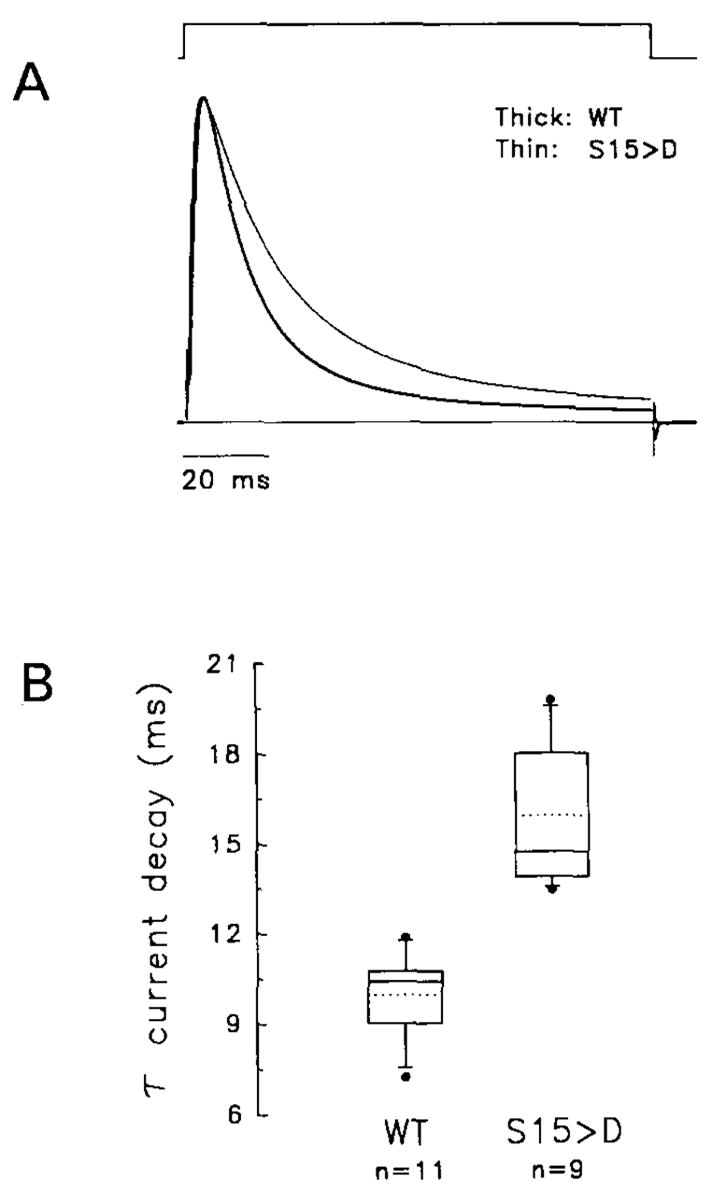
(A) Currents were evoked by a 112 ms step depolarization from −100 to +70 mV. For comparison, currents were normalized to their peak and overlaid (thick trace, WT; thin trace, S15D). Peak currents were 20 and 12 μA for WT and S15D, respectively.
(B) Tukey box plot summarizing the effect of S15D on the time constant of current inactivation (τ at +50 mV). The lower border, middle line, and upper border of the box represent the 25th, 50th, and 75th percentiles, respectively. The dotted line indicates the arithmetic mean (WT, 10 ± 1 ms [n = 11]; S15D, 16 ± 2 ms [n = 9]). The upper and lower whiskers represent the 90th and 10th percentiles, respectively, and outliers are shown as closed circles.
Discussion
The results of our experiments demonstrate that the inactivation gate of a mammalian A-type K+ channel is regulated by direct action of PKC. The main findings are summarized here: PKC specifically inhibits N-type inactivation of a human K+ channel encoded by hKv3.4; a peptide corresponding to the inactivation gate at the N-terminus of hKv3.4 is phosphorylated by PKC in vitro; specific serine residues within this region are important PKC targets mediating the action of this kinase on N-type inactivation; and substitution of one of these serines for an acidic amino acid mimics PKC-dependent regulation of N-type inactivation.
The Effect of PKC on N-Type Inactivation Is Specific and Selective
PKC is activated physiologically by the coordinate action of phosphatidylserine, DAG, and Ca2+ (Shearman et al., 1989). This can be mimicked by tumor-promoting phorbol esters like PMA and synthetic DAG analogs like OAG. In our study, elimination of rapid inactivation by PKC activators (Figure 2) is probably mediated by action of PKC because PMA and OAG produced a similar effect, specific inhibition of PKC by calphostin C blocked PMA action, the inactive phorbol ester α-PMA failed to produce any effect, and the action of PKC activators can be mimicked by direct cytoplasmic application of active PKC to an inside-out patch expressing a single channel (Figure 3). Comparing the magnitude of the effect, it was clear that PMA produced the largest inhibition of current inactivation. This is to be expected because, compared with DAG derivatives, phorbol esters are very potent and effective activators of PKC (Shearman et al., 1989). The magnitude of PMA action also suggests that the pool of PKC available for activation is relatively large and sufficient to modify the majority of hKv3.4 channels expressed in the membrane. The fact that direct cytoplasmic application of PKC produced a partial effect on inactivation of single channels may also be explained. hKv3.4 channels expressed in Xenopus oocytes are homotetrameric, and each subunit contributes one inactivation domain with at least two phosphorylation sites each (see below). Under conditions that may cause submaximal phosphorylation, we expect to find at least three classes of subunits in a channel: fully phosphorylated forms, several partially phosphorylated forms, and nonphosphorylated forms. This could cause the slow and variable decay of whole-cell currents revealed by long depolarizations in the presence of PMA (Figure 6). To produce maximum reduction of current inactivation, all sites should be phosphorylated. Thus, if phosphorylation of a single channel is incomplete, this leads to partial removal of channel inactivation. This could happen if the amount of PKC that reaches the vicinity of a channel were limited. Since the intracellular side of an inside-out membrane patch may have a complex structure (Ruknudin et al., 1991), diffusion of an exogenous enzyme into the cytoplasmic compartment of the patch may be rate limiting (Drain et al., 1994).
Among four distinct A-type K+ channels expressed in Xenopus oocytes (Figure 4), only hKv3.4 current kinetics was affected by PMA. This probably reflects specific differences in the primary structure of these channels. In general, PKC consensus sequences have been defined as a serine or threonine flanked by basic residues (Marais et al., 1990; Kennelly and Krebs, 1991). This is clearly apparent at the N-terminus of hKv3.4. In fact, residues in this region are phosphate acceptors, since brain PKC can phosphorylate the N-terminal region of hKv3.4 (see Results).
Identification of PKC Target Sites That Regulate N-Type Inactivation
Mutagenesis experiments provided further evidence in favor of a direct action of PKC. Regulation of N-type inactivation was sensitive to mutations that affected serines at positions 15 and 21 (Table 1). Each residue may be responsible for approximately 50% of PKC action. However, the effect of a double mutation (S15A/S21A) was not fully additive, suggesting that PKC may also act through additional auxiliary sites in the absence of S15 and S21. PKC recognition sequences are diverse, and a broad range of substrate specificity has been reported (Marais et al., 1990; Kennelly and Krebs, 1991). In addition, potential PKC sites in close proximity may not be equivalent or independent. S15 and S21 are clearly important targets of PKC, but phosphorylation of these sites may actually affect recognition of neighboring potential sites by PKC or vice versa. Less typical potential sites like S8 and S9 may also be phosphorylated by PKC. In a pilot experiment, we found that a double mutant, S8A/S9A, significantly affected the action of PKC on rapid inactivation. Thus, to regulate rapid N-type inactivation, PKC may phosphorylate multiple nonindependent sites within the inactivation gate of hKv3.4.
Related Studies
Previous reports examined the effects of PKC on gating of rat brain Na+ channels (Numann et al., 1991; West et al., 1991). PKC reduces peak Na+ current and slows Na+ current inactivation. These studies concluded that phosphorylation of one or more sites between homologous domains I and II in the channel polypeptide caused inhibition of peak current, and phosphorylation of serine 1506 in the inactivation gate was responsible for slower inactivation. The inactivation gate of voltage-gated Na+ channels is located between homologous domains III and IV (Vassilev et al., 1988; Stühmer et al., 1989; Patton et al., 1992; West et al., 1992). In a subsequent study, it was also found that PKA can cause inhibition of peak Na+ current, but this effect was dependent on phosphorylation of serine 1506 by PKC or the presence of a negatively charged amino acid at this position (Li et al., 1993). Thus, it seems that PKC slows inactivation of rat brain Na+ channels and hKv3.4 K+ channels by a direct action involving phosphorylation of sites within the inactivation domain of these channels. For hKv3.4 K+ channels, however, we did not observe inhibition of peak current by PKC. Another study examined the action of phosphatase and PKA on rapid N-type inactivation of Drosophila Shaker K+ channels (Drain et al., 1994). Phosphatase slows inactivation, and PKA reverses this action. Mutagenesis revealed that this effect was associated with serines 507 and 508 at the cytoplasmic C-terminal region of the Shaker polypeptide. Mutations at the N-terminus did not affect phosphatase action. Thus, it was concluded that PKA-dependent phosphorylation of serines at the C-terminus of Shaker may regulate N-type inactivation allostericaliy. By contrast, PKC can eliminate N-type inactivation of hKv3.4 K+ channels, and this is in most part due to phosphorylation of serine residues within the inactivation gate. We have not yet examined the effect of PKA or phosphatase on gating of hKv3.4 K+ channels.
Model
In a general model of K+ channel gating (Zagotta and Aldrich, 1990; Hoshi et al., 1994), voltage-dependent activation is followed by a voltage-independent transition that opens the channel, and subsequent inactivation. According to a modern view of the “ball and chain” hypothesis (Bezanilla and Armstrong, 1977; Zagotta et al., 1990; Hoshi et al., 1990), the cytoplasmic N-terminus of the channel polypeptide is the inactivation gate (“ball” domain) tethered to the rest of the molecule by a hydrophilic arm. Multiple basic amino acids are essential for the function of the inactivation gate (Zagotta et al., 1990; Murrell-Lagnado and Aldrich, 1993a). Thus, the N-terminal region of A-type K+ channels acts as a positively charged particle that, following channel opening, occludes the inner mouth of the channel, causing inactivation (Demo and Yellen, 1991; Ruppersberg et al., 1991b). Our results with hKv3.4 are consistent with this model. Phosphorylation of S15 and S21 may reduce the net positive charge of the inactivation gate and in this manner disrupt long-range electrostatic interactions that control the on-rate of channel blockage (Murrell-Lagnado and Aldrich, 1993a). Within the first 27 amino acids of hKv3.4, the net charge (z) under basal conditions is +4. If S15 and S21 are phosphorylated (i.e., two negative charges per phosphate group), z may be as low as 0. Supporting the electrostatic mechanism presented here, we showed that an acidic amino acid at position 15 (S15D) mimicked the effect of PKC-dependent phosphorylation on N-type inactivation (Figure 7). In this case, a novel aspartate residue introduces a single constitutive negative charge, reducing z to +3. This explains why the effect of this mutation on current inactivation is smaller compared with the effect of PMA on WT channels. The electrostatic hypothesis to explain the effects of phosphorylation on channel function was also supported in a previous study (Perozo and Bezanilla, 1990). This revealed that phosphorylation affects voltage-dependent gating of a delayed rectifier K+ channel by electrostatic interactions.
Significance
PKC phosphorylation of hKv3.4 K+ channels can eliminate rapid channel inactivation, converting these channels from rapidly inactivating A type to noninactivating delayed rectifier type. Physiologically, neurotransmitters acting on specific receptors trigger a second messenger cascade that induces activation of PKC (Nicoll, 1988). The effect of PKC on inactivation of hKv3.4 may thus have significant physiological repercussions. Although there is evidence for the localization of hKv3.4 K+ channels in muscle and in discrete regions of the brain (Weiser et al., 1994), the function of these channels is still unknown. Since these channels activate at membrane potentials more positive than −20 mV, they may not normally regulate sub-threshold depolarizations, and therefore, spike frequency. However, Kv3.4 channels reopen during recovery from inactivation, and it has been proposed that they may control firing frequency via current flow at hyperpolarized membrane potentials (Ruppersberg et al., 1991 b). Another possibility is that these channels regulate excitability by controlling spike duration (Hochner and Kandel, 1992; Goldsmith and Abrams, 1992). If hKv3.4 channels inactivate rapidly, a depolarizing spike may be prolonged. On the other hand, if hKv3.4 channels inactivate slowly or do not inactivate, the spike may be shortened.
Experimental Procedures
Oocyte Expression
hKv3.4 was subcloned into a hybrid plasmid derived from pBluescript II KS (Stratagene) and pMXT (a gift from Dr. M. White, Medical College of Pennsylvania). This novel plasmid includes the 5′ and 3′ untranslated regions of Xenopus (β-globin that flank the hKv3.4 insert. For expression in Xenopus oocytes, the linearized plasmid was used as template to produce capped mRNA by in vitro transcription driven by T3 RNA polymerase (Megascript, Ambion). Harvesting of oocytes and microinjection was done by standard procedures (Goldin and Sumikawa, 1992).
Electrophysiological Recording
Electrophysiological recording from Xenopus oocytes was done using a TEV-200 two microelectrode voltage-clamp amplifier (Dagan, Minneapolis, MN), to record whole-oocyte currents, or an Axopatch 200 patch-clamp amplifier (Axon Instruments, Burlingame, CA), to record single-channel currents (Stühmer, 1992). For whole-oocyte recording, the bath solution contained 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES (pH 7.4; titrated with NaOH). PMA and OAG were purchased from Sigma Chemical Corp. (St. Louis, MO) and Calbiochem Corp. (San Diego, CA), respectively. Calphostin C (Kobayashi et al., 1989) and α-PMA were purchased from LC Services Corp. (Woburn, MA). Whole-oocyte currents were low-pass filtered at 2 kHz using an internal 4 pole Bessel filter and digitized at 4 kHz. Macroscopic leak and capacitive currents were subtracted digitally using scaled template sweeps with no active current. For patch-clamp recording, the patch pipette contained 140 mM NaCl, 6 mM MgCl2, and 5 mM HEPES (pH 7.2; titrated with N-methyl-D-glucamine). In the cell-attached configuration, to zero the oocyte membrane potential, the cell was bathed in 140 mM K-aspartate, 10 mM KCl, 1.8 mM CaCl2, 10 mM HEPES (pH 7.4; titrated with KOH). For inside-out patches, the internal solution contained 140 mM KCl, 2 mM MgCl2, 1 mM CaCl2, 11 mM EGTA, and 10 mM HEPES (pH 7.2). Internal solutions were supplemented with 5 mM glutathione to prevent inhibition of inactivation owing to cysteine oxidation (Ruppersberg et al., 1991a). Single-channel currents were low-pass filtered at 1 or 2 kHz using an 8 pole Bessel filter (Frequency Devices, Haverhill, MA) and digitized at 4 or 8 kHz, respectively. Capacitive currents were electronically compensated. Any residual capacitive transient and leak current were digitally subtracted using averaged blank sweeps. All currents were recorded at room temperature (21.5°C–22.5°C). Data acquisition and analysis were done using pCLAMP 5.5 or 6.0 (Axon Instruments, Burlingame, CA). For further analysis, we used Peakfit and Sigmaplot (Jandel, San Rafael, CA). All results are reported as means ± SD.
Phosphorylation In Vitro
A crude (~40% pure) preparation of Raw3 N-terminal peptide (Schroter et al., 1991) was provided by Dr. R. W. Aldrich (Stanford University). The amino acid sequence of this peptide is identical to the first 28 residues of hKv3.4. This peptide was purified (>95%) using a C18 column and reverse phase fast-protein liquid chromatography (Pharmacia, Piscataway, NJ). PKC phosphorylation in vitro was carried out using a filter assay as described previously (Slater et al., 1993). PKC was purified from rat brain as described earlier (Huang et al., 1986). This material was provided by Dr. C. Stubbs (Jefferson Medical College).
Mutagenesis
Site-directed mutagenesis was performed using either pALTER (Altered Sites, Promega, Madison, WI) or an overlap polymerase chain reaction (PCR) strategy. The 563 bp HindIII-Sacl fragment encoding the 5′ end of hKv3.4 was subcloned into pALTER. Mutagenesis was performed on this fragment, which was then substituted into the full length construct as a HindIII-Sacl cassette. S15A was generated by annealing and extension with an oligonucleotide encoding this mutation (using single-stranded DNA template generated from the pALTER subclone). All other mutations were generated by overlap PCR employing vector primers paired with internal primers incorporating the desired mutations. Mutations were confirmed by sequencing through the entire cassette. ΔN28 was recovered as a clone with a fortuitous point mutation introduced by Taq polymerase in the process of generating overlap PCR fragments. This point mutation (ATG → GTG) resulted in loss of the initiation codon. Translation thus started at the next initiation methionine at position 29.
Acknowledgments
We thank Mr. C. Choe for valuable technical assistance and Drs. P. De Weer and R. Horn for critical comments on earlier versions of this manuscript. We also thank Drs. S. Slater and C. Stubbs (Jefferson Medical College) for providing purified brain PKC and for testing phosphorylation in vitro of the Raw3 peptide and Dr. R. Sorensen (Jefferson Medical College) for purifying Raw3 N-terminal peptide. hKv3.4 clone was provided by Dr. B. Rudy (New York University Medical Center), and Raw3 N-terminal peptide was a gift from Dr. R. W. Aldrich (Stanford University). We thank them for supplying these materials. This work was supported by NIH grants NS32337 (M. C.) and NS24785 (L. S.) and in part by NIAAA program project grant AA07186. T. B. V. was supported by PHS training grant AA07463. All correspondence should be addressed to M. C.
References
- Armstrong D, Eckert R. Voltage-activated calcium channels that must be phosphorylated to respond to membrane depolarization. Proc Natl Acad Sci USA. 1987;84:2518–2522. doi: 10.1073/pnas.84.8.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine CK, Bezanilla F. Phosphorylation modulates potassium conductance and gating current of perfused giant axons of squid. J Gen Physiol. 1990;95:245–271. doi: 10.1085/jgp.95.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel I: sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Varnum MD, North RA, Adelman JP. An amino acid mutation in a potassium channel that prevents inhibition by protein kinase C. Science. 1992;255:1705–1707. doi: 10.1126/science.1553557. [DOI] [PubMed] [Google Scholar]
- Chung SK, Reinhart PH, Martin BL, Brautigan D, Levitan IB. Protein kinase activity closely associated with a reconstituted calcium-activated potassium channel. Science. 1991;253:560–562. doi: 10.1126/science.1857986. [DOI] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+ channel behaves like an open-channel biocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Drain P, Dubin AE, Aldrich RW. Regulation of Shaker K+ channel inactivation gating by cAMP-dependent protein kinase. Neuron. 1994;12:1097–1109. doi: 10.1016/0896-6273(94)90317-4. [DOI] [PubMed] [Google Scholar]
- Goldin AL, Sumikawa K. Preparation of RNA for injection into Xenopus oocytes. Meth Enzymol. 1992;207:279–297. doi: 10.1016/0076-6879(92)07018-j. [DOI] [PubMed] [Google Scholar]
- Goldsmith BA, Abrams TW. cAMP modulates multiple K+ currents, increasing spike duration and excitability in Aplysia sensory neurons. Proc Natl Acad Sci USA. 1992;89:11481–11485. doi: 10.1073/pnas.89.23.11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, Massachusetts; Sinauer Associates Inc: 1992. [Google Scholar]
- Hochner B, Kandel ER. Modulation of a transient K+ current in the pleural sensory neurons of Aplysia by serotonin and cAMP: implications for spike broadening. Proc Natl Acad Sci USA. 1992;89:11476–11480. doi: 10.1073/pnas.89.23.11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aid rich R. Shaker potassium channel gating I: transitions near the open state. J Gen Physiol. 1994;103:249–278. doi: 10.1085/jgp.103.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KP, Chan KF, Singh TJ, Nakabayashi H, Huang FL. Autophosphorylation of rat brain Ca2+-activated and phospholipid-dependent protein kinase. J Biol Chem. 1986;261:12134–12140. [PubMed] [Google Scholar]
- Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+ channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem. 1991;266:15555–15558. [PubMed] [Google Scholar]
- Krishek Bj, Xie X, Blackstone C, Huganir RL, Moss SJ, Smart TG. Regulation of GABAA receptor function by protein kinase C phosphorylation. Neuron. 1994;12:1081–1095. doi: 10.1016/0896-6273(94)90316-6. [DOI] [PubMed] [Google Scholar]
- Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1989;759:548–553. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- Levitan IB, Kaczmareck LK. The Neuron: Cell and Molecular Biology. New York: Oxford University Press; 1991. [Google Scholar]
- Levitan IB, Chung S, Reinhart PH. Modulation of a single ion channel by several different protein kinases. Adv Second Messenger Phosphoprotein Res. 1990;24:36–40. [PubMed] [Google Scholar]
- Li M, West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- MacKinnon R, Aldrich RW, Lee AW. Functional stoichiometry of Shaker potassium channel inactivation. Science. 1993;262:757–759. doi: 10.1126/science.7694359. [DOI] [PubMed] [Google Scholar]
- Marais RM, Nguyen O, Wodgett JR, Parker P. Studies on the primary sequence requirements for PKC-α, -β1 and -γ peptide substrates. FEBS Lett. 1990;277:151–155. doi: 10.1016/0014-5793(90)80831-3. [DOI] [PubMed] [Google Scholar]
- Moran O, Dascal N, Lotan I. Modulation of a Shaker potassium A-channel by protein kinase C activation. FEBS Lett. 1991;279:256–260. doi: 10.1016/0014-5793(91)80162-v. [DOI] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Interactions of amino terminal domains of Shaker K+ channels with a pore blocking site studied with synthetic peptides. J Gen Physiol. 1993a;102:949–975. doi: 10.1085/jgp.102.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Energetics of Shaker K+ channels blocked by inactivation peptides. J Gen Physiol. 1993b;102:977–1003. doi: 10.1085/jgp.102.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA. The coupling of neurotransmitter receptors to ion channels in the brain. Science. 1988;241:545–551. doi: 10.1126/science.2456612. [DOI] [PubMed] [Google Scholar]
- Numann R, Catterall WA, Scheuer T. Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science. 1991;254:115–118. doi: 10.1126/science.1656525. [DOI] [PubMed] [Google Scholar]
- Pak MD, Baker K, Covarrubias M, Butler A, Ratcliffe A, Salkoff L. mShal, a subfamily of A-type K+ channel cloned from mammalian brain. Proc Natl Acad Sci USA. 1991;88:4386–4390. doi: 10.1073/pnas.88.10.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton DE, West JW, Catterall WA, Goldin AL. Amino acid residues required for fast Na+ channel inactivation: charge neutralizations and deletions in the III–IV linker. Proc Natl Acad Sci USA. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E, Bezanilla F. Phosphorylation affects voltage gating of the delayed rectifier K+ channel by electrostatic interactions. Neuron. 1990;5:685–690. doi: 10.1016/0896-6273(90)90222-2. [DOI] [PubMed] [Google Scholar]
- Rettig J, Wunder F, Stocker M, Lichtinghagen R, Mastiaux F, Beckh S, Kues W, Pedarzani P, Schröter KH, Ruppersberg JP, Veh R, Pongs O. Characterization of a Shaw-related potassium channel family in rat brain. EMBO J. 1992;11:2473–2486. doi: 10.1002/j.1460-2075.1992.tb05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, Sen K, Vega-Saenz de Meira EC, Lau E, Reid T, Ward D. Cloning of a human cDNA expressing a high voltage-activating TEA-sensitive, type A K+ channel which maps to human chromosome 1 band p21. J Neurosci Res. 1991;29:401–412. doi: 10.1002/jnr.490290316. [DOI] [PubMed] [Google Scholar]
- Ruknudin A, Song MJ, Sachs F. The ultrastructure of patch-clamped membranes: a study using high voltage electron microscopy. J Cell Biol. 1991;112:125–134. doi: 10.1083/jcb.112.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991a;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Frank R, Pongs O, Stocker M. Cloned neuronal IK(A) channels reopen during recovery from inactivation. Nature. 1991b;353:657–660. doi: 10.1038/353657a0. [DOI] [PubMed] [Google Scholar]
- Schroter KH, Ruppersberg JP, Wunder F, Rettig J, Stocker M, Pongs O. Cloning and functional expression of a TEA-sensitive A-type potassium channel from rat brain. FEBS Lett. 1991;278:211–216. doi: 10.1016/0014-5793(91)80119-n. [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A, Scheuer T, Catterall WA. Voltage-dependent potentiation of L-type Ca2+ channels due to phosphorylation by cAMP-dependent protein kinase. Nature. 1993;364:240–243. doi: 10.1038/364240a0. [DOI] [PubMed] [Google Scholar]
- Shearman MS, Sekiguchi K, Nishizuka Y. Modulation of ion channel activity: a key function of the protein kinase C enzyme family. Pharmacol Rev. 1989;41:211–237. [PubMed] [Google Scholar]
- Shuster MJ, Camardo JS, Siegelbaum SA, Kandel ER. Cyclic AMP-dependent protein kinase closes the serotonin-sensitive K channels of Aplysia sensory neurones in cell-free membrane patches. Nature. 1985;313:392–395. doi: 10.1038/313392a0. [DOI] [PubMed] [Google Scholar]
- Sigel E, Baur R. Activation of protein kinase C differently modulates neuronal Na+, Ca+, and γ-aminobutyrate type A channels. Proc Natl Acad Sci USA. 1988;85:6192–6196. doi: 10.1073/pnas.85.16.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater SJ, Cox KJA, Lombardi JV, Ho C, Kelly MB, Rubin E, Stubbs CD. Inhibition of protein kinase C by alcohols and anaesthetics. Nature. 1993;364:82–84. doi: 10.1038/364082a0. [DOI] [PubMed] [Google Scholar]
- Stühmer W. Electrophysiological recording from Xenopus oocytes. Meth Enzymol. 1992;207:319–339. doi: 10.1016/0076-6879(92)07021-f. [DOI] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Blackstone CD, Huganir RL. Phosphorylation of ligand-gated ion channels: a possible mode of synaptic plasticity. FASEB J. 1992;6:2514–2523. [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;247:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Walsh KA, Kass RS. Regulation of a heart potassium channel by protein kinase A and C. Science. 1988;242:67–69. doi: 10.1126/science.2845575. [DOI] [PubMed] [Google Scholar]
- Wei A, Covarrubias M, Butler A, Baker K, Pak M, Salkoff L. K+ current diversity is produced by an extended gene family conserved in Drosophila and mouse. Science. 1990;248:599–603. doi: 10.1126/science.2333511. [DOI] [PubMed] [Google Scholar]
- Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA. A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science. 1991;254:866–868. doi: 10.1126/science.1658937. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Sheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Aldrich RW. Voltage-dependent gating of Shaker A-type potassium channels in Drosophila muscle. J Gen Physiol. 1990;95:29–60. doi: 10.1085/jgp.95.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]