

Abstract
Summary
Transcriptional and post-translational signals are known mechanisms which promote efficient responses to DNA damage. We have identified Saccharomyces cerevisiae tRNA methyltransferase 9 (Trm9) as an enzyme that prevents cell death via translational enhancement of DNA damage response proteins. Trm9 methylates the uridine wobble base of tRNAARG(UCU) and tRNAGLU(UUC). We used computational and molecular approaches to predict that Trm9 enhances the translation of some transcripts over-represented with specific arginine and glutamic acid codons. We found that translation elongation factor 3 (YEF3) and the ribonucleotide reductase (RNR1 and RNR3) large subunits are over-represented with specific arginine and glutamic acid codons, and demonstrated that Trm9 significantly enhances Yef3, Rnr1, and Rnr3 protein levels. Additionally, we identified 425 genes, which included YEF3, RNR1, and RNR3, with a unique codon usage pattern linked to Trm9. We propose that Trm9-specific tRNA modifications enhance codon-specific translation elongation and promote increased levels of key damage response proteins.
Introduction
Previous studies have shown that hundreds to thousands of S. cerevisiae transcripts are regulated in a dose and time dependent manner after methyl methanesulfonate (MMS) exposure (Gasch et al., 2000; Gasch et al., 2001; Gasch and Werner-Washburne, 2002; Jelinsky and Samson, 1999; Jelinsky et al., 2000; Workman et al., 2006). The magnitude and complexity of transcription-based responses to MMS-induced alkylation damage suggest that downstream mechanisms are used to fine-tune cellular pathways after transcription. In support of this hypothesis, post-transcriptional regulation of DNA damage response pathways has been demonstrated by targeted studies (Yao et al., 2003 Zhao and Rothstein, 2002). For example, the ribonucleotide reductase (Rnr) transcripts RNR1, RNR2, RNR3, and RNR4 are all induced after DNA alkylation damage, with the Rnr complex being the rate-limiting step in the production of dNTPs for use in DNA synthesis. Rnr regulation continues after transcription; Rnr2 and Rnr4 proteins are re-localized from the nucleus to the cytoplasm after damage (Yao et al., 2003), and the Rnr inhibitor Sml1 is inactivated via phosphorylation (Zhao and Rothstein, 2002). Thus post-transcriptional regulatory mechanisms promote Rnr activity, which in turn influence cell viability post-damage (Chabes et al., 2003), highlighting the role of post-transcriptional events in the DNA damage response.
In previous studies we used high-throughput screening of S. cerevisiae gene deletion libraries to identify proteins that modulate the toxicity of alkylating agents (Begley et al., 2002; Begley et al., 2004). These screens identified many known components of DNA repair and damage signaling pathways, and identified many proteins with the potential to participate in the cellular DNA damage response. In particular, S. cerevisiae tRNA methyltransferase 9 (Trm9) was identified as a potential enhancer of the DNA damage response; a trm9Δ allele increases cell sensitivity to MMS and γ-irradiation (Begley et al., 2002; Begley et al., 2004; Bennett et al., 2001). Trm9 uses the methyl donor S-adenosyl methionine (SAM) to catalyze the last step in the formation of 5-methylcarbonylmethyluridine (mcm5U) and 5-methylcarbonylmethyl-2-thiouridine (mcm5s2U) in specific arginine and glutamic acid charged tRNAs (Kalhor and Clarke, 2003) (Fig. 1), thus suggesting a role for Trm9 in translation. Trm9 deficient cells have a damage-induced cell cycle phenotype similar to that of members of the Rad52 epistasis group (Bennett et al., 2001). Rad52 members are involved in the repair of double-strand breaks, and the phenotypic overlap with Trm9 suggests that tRNA modifications help coordinate DNA repair. Trm9 is conserved in humans and the human gene (KIAA1456) is found on the end of chromosome 8 (8p22), a region commonly lost in colorectal tumors (Ilyas et al., 1999). Importantly, Flanagan and colleagues have used microcell mediated chromosome transfer and cell culture based assays to implicate human Trm9 as a potential tumor-suppressor gene for colorectal cancer (Flanagan et al., 2004).
Figure 1. Trm9 Catalyzed tRNA Modifications.

Trm9 completes the formation of mcm5U and mcm5s2U at position 34 of tRNAARG(UCU) and tRNAGLU(UUC), respectively.
There are 16 tRNA methyltransferase (Trm) genes in S. cerevisiae and they promote the formation of over twenty different methyl-based modifications in the anticodon and other loops of tRNA, varying in nucleoside position and modification type (Cherry et al., 1998). In addition to mcm5U and mcm5s2U, methyl modifications in tRNA include N2, N2-dimethylguanosine, 5-methyluridine, 5-methylcytidine, 3-methylcytidine, 1-methylguanosine, 1-methyladenosine, 7-methylguanosine, 2-methylguanosine, wybutosine, and 2'-O-methylated nucleosides. Generally, methyl modifications play some role in tRNA stability, translation, and cell growth, thus theoretically affecting translational efficiency, frameshifting or amino acid substitution rates (Agris, 2004; Urbonavicius et al., 2001). The exact biological role of each modification is hard to identify because of the proposed functional redundancy of some methyl based modifications, and the lack of strong phenotypes for some of the corresponding tRNA methytransferase deficient strains. However some tRNA methyltransferases (Trm6 and Trm61) are essential proteins supporting an important role for these enzymes in the cell. In addition, mild growth or stress-based defects have been observed in strains deficient in Trm1, Trm4, Trm7, and Trm8, and Trm9 has been implicated in damage and stress signaling (Cherry et al., 1998; Bennett et al., 2001; Kalhor and Clarke, 2003; Alexandrov et al., 2005).
The modification catalyzed by S. cerevisiae Trm9 occurs in the wobble base of arginine and glutamic acid charged tRNAs that have UCU and UUC anticodons, respectively (Kalhor and Clarke, 2003; Lu et al., 2005). [Note: all codon and anticodon wobble bases are underlined]. Further, cells deficient in Trm9 were sensitive to the translation inhibitor paromomycin, further supporting a role for Trm9 in translation. The mcm5U wobble base produced by Trm9 has been reported to modulate tRNA-mRNA pairing and enhance binding with the arginine AGA codon (Weissenbach and Dirheimer, 1978). This observation suggests that the mcm5U base modification affects translation through modulation of the codon-anticodon interface, and suggests a codon specific role for Trm9.
The potential for Trm9 to modulate specific codon-anticodon interactions would make it an ideal enhancer of codon-specific translation. In the following study, we have used computational approaches to identify 425 genes with skewed AGA codon usage patterns and whose corresponding protein levels could be enhanced by Trm9-specific tRNA modifications. We determined that Trm9 specifically increased the protein levels of Yef3, Rnr1 and Rnr3, all of which have AGA rich codon usage patterns, without affecting transcription. Further, we found that general translation is intact in trm9Δ cells, and have defined a codon-specific role for Trm9-catalyzed tRNA modifications in translation.
Results
Trm9 Enhances the Translation of AGA and AGG Reporters
To validate our high throughput result that the trm9Δ allele conferred MMS sensitivity (Begley et al., 2002; Begley et al., 2004), we performed plate based sensitivity assays in haploid and diploid S. cerevisiae cells. We demonstrated that TRM9 can complement the MMS-sensitive phenotype of trm9Δ cells, in both the By4741 and CenPK2−1C backgrounds, and that TRM9 gene dosage is inversely related to MMS sensitivity in diploid By4743 cells (Supplemental Figure S1). To explore the molecular mechanism by which Trm9 provided its protective effect, we developed an S. cerevisiae codon-specific reporter system to analyze the importance of Trm9 in the translation of the arginine codons AGA and AGG. We used PCR based methods to add an internal codon run near the N-terminus and in frame with the lacZ gene (Supplemental Figure S2A), to generate a pair of reporters specific to synonymous codons for arginine (AGA-lacZ and AGG-lacZ). Importantly the amino acid matched pair of codon reporters differs with respect to the wobble bases in the codon run, but the resulting proteins are the same at the level of amino acid sequence. As a result of the identical protein sequences, the design allows the reporter to reflect the effects of translation while removing protein degradation or enzyme activity as a potential mechanism of altered activity.
We ligated the constructs into a pYES-based plasmid to place the lacZ reporter in a high copy plasmid under the control of a strong galactose-inducible promoter. Each of the reporter systems was transformed into wild-type and trm9Δ S. cerevisiae. We grew isolates under inducing conditions for 3 to 6 hours and then harvested the resulting cells and prepared total extracts for lacZ reporter analysis. Total protein levels were similar in wild-type and trm9Δ cells and equal amounts of proteins were used for β-galactosidase assays. We measured the activity of lacZ, 10× AGA, 10× AGG, and 5× AGG reporters in wild-type and trm9Δ cells. The control reporter behaved similarly in both wild-type and trm9Δ cells (Table 1). The ratio of β-galactosidase activity in wild-type and trm9Δ cells was nearly 1, indicating that general translation was similar in both cell types. Next we analyzed 10× AGA reporter activity in wild-type and trm9Δ cells. We determined that wild-type cells had a ∼6-fold greater 10× AGA reporter activity than trm9Δ cells, supporting that Trm9-catalyzed tRNA modification dramatically increases anticodon pairing with the AGA codon. We also attempted to monitor the activity of a 10× AGG reporter in wild-type cells, but determined that we could not consistently observe activity over background levels. We reasoned that the long run of AGG codons either prohibited translation or made the plasmid unstable, so we switched to a 5× AGG reporter format. We observed a small yet detectable amount of reporter activity using the 5× AGG reporter, and determined that wild-type cells have ∼10-fold higher activity than trm9Δ cells. Combined, our results from wild-type and trm9Δ cells indicate that Trm9 enhances the translation of reporters with either 10× AGA or 5× AGG codon runs.
Table 1.
Codon Reporter Activities in Wild-type and trm9Δ Cells
| wild-type (Activity units) | trm9 Δ (Activity units) | |
|---|---|---|
| IacZ | 46.1 +/− 1.9 | 50.1 +/− 7.0 |
| AGA | 29.9 +/− 9.1 | 4.3 +/− 0.54 |
| AGG | 0.23 +/− 0.07 | 0.023 +/− 0.004 |
Gene-Specific Codon Usage Patterns Identify Potential Trm9 Targets
In the S. cerevisiae genome AGA are found at higher frequency than AGG codons, with arginine codon frequencies of 0.48 and 0.21 respectively. In addition, there is an alternative tRNA species containing a CCU anticodon that is a perfect Watson-Crick match for AGG codons (Cherry et al., 1998). These observations suggested that AGA codons would be an easier signal to specifically key to mcm5U modifications, and we decided to initially analyze the signaling potential of Trm9 on the translation of AGA codons. We hypothesized that Trm9 could affect the translation of endogenous transcripts containing long runs of AGA codons or transcripts globally enriched for AGA codons. To identify potential downstream targets, we developed a computational approach to compile and help visualize gene-specific codon usage (GSCU) trends for all 5,783 S. cerevisiae genes. We surveyed all S. cerevisiae genes and did not identify any transcripts with more than three AGA codons in a row, suggesting that an overall enrichment of AGA codons throughout a specific gene would be the physiological signal keyed to Trm9-specific tRNA modifications. The developed GSCU algorithm compiled codon usage data for all codons and this data was used to calculate gene-specific codon Z-scores for all S. cerevisiae genes (Supplemental Table S1). The gene-specific codon usage data are an integrated measure of codon usage in an individual gene, with over-represented codons correlated to short codon runs. The gene-specific codon Z-scores, which utilize frequency data and detail whether a gene is over- or under-represented with a given codon relative to genome averages, were imported into the program CLUSTER (Eisen et al., 1998), subjected to hierarchical clustering analysis, and then visualized using a heat map in the program TREEVIEW (Eisen et al., 1998) (Fig. 2A). It should be noted that the heat map shown in Figure 2 does not describe transcriptional data. Rather, it represents an integrated signal representative of gene-specific codon usage. All positive and negative deviations from genome averages are shown, with statistically significant deviations shown in Supplemental Figure S3.
Figure 2. Gene-Specific Codon Usage Patterns Identify Codon-Skewed Genes.

(A) Hierarchical clustering and heat map analysis of all GSCU data for 5,783 S. cerevisiae genes. Gene-specific over-represented codons are displayed in yellow, under-represented codons displayed in purple, codons with gene-specific codon usage patterns showing no deviation from genome average values displayed as black. Supplemental Figure S3 contains the x-axis key. (B) Northern and Tap tagged western blots for genes and proteins that are over-represented with AGA and GAA codons (i.e., Group 1), and (C) genes outside of Group 1 which contain average codon values. Cells were grown to 5 × 106 cells/ml in YPD and either mock or 0.0125% MMS treated for 1-hour. The loading control (LC) for northern blots was ACT1 while for western blots it is β-tubulin.
We analyzed the gene and codon clustered data (Fig 2A) and determined that there were 425 genes that clustered together (Group 1), and as group were skewed to contain more AGA codons than average genes. We performed 10,000 random samplings of 425 genes to assemble group codon statistics and to determine the significance of the codon usage pattern identified in Group 1 genes. Notably, Group 1 genes were significantly (p < 10−10) over-represented with the arginine AGA codon and under-represented with the AGG codon. Group 1 genes were also over-represented 24 other codons and under-represented with 35 other codons, many of which were synonymous pairs (Supplemental Table S2). It should be noted that we did not identify any group of genes over-represented with AGG codons, but a general trend observed for Group 1 genes in Figure 2A was that synonymous codons are split in the heat map, with one being significantly over-represented (yellow) in Group 1 and the other being under-represented (purple).
Importantly, Group 1 genes were significantly over-represented (p < 10−10) with the glutamic acid GAA codon. Trm9 has been shown to modify the wobble base of tRNAGLU(UUC) (Kalhor and Clarke, 2003; Jablonskowski et al., 2006) and this tRNA anticodon can pair with GAA codons. Together with our finding that Group 1 genes are over-represented with GAA codons, the published Trm9 activity data suggests that GAA codons will more efficiently pair with an anticodon wobble base that has been Trm9-modified. We subsequently performed 10× GAA-lacZ reporter experiments and determined that wild-type cells had 7-fold more activity than trm9Δ cells, thus supporting a role for Trm9 in the efficient translation of GAA codons (Supplemental Figure S2B).
Codon bias has previously been correlated to expression levels (Bennetzen and Hall, 1982). It is important to note however, that Group 1 genes were not only over-represented with codons that are normally classified as high usage. While both AGA and GAA codons are considered high usage, the Group 1 genes were also over-represented with some codons that are considered low usage codons, including aspartic acid (GAC), isoleucine (ATC), tyrosine (TAC), lysine (AAG) and phenylalanine (TTC) specific codons. The large number of over-represented codons in Group 1 genes suggests that Trm9 or other tRNA modifying enzymes will affect the translation of other codons or codon combinations, a possibility we are investigating.
Biological Themes Associated with Group 1 Proteins
To identify regulatory and functional themes in our 425 identified Group 1 genes, we compared them to other global and functional lists. First we compared Group 1 genes to ∼900 transcripts belonging to the transcription-specific environmental stress response (ESR) (Gasch et al., 2000), to determine if Group 1 genes were over-represented with ESR transcripts. We identified 185 ESR transcripts in Group 1, which represents a 3.6 fold enrichment over the expected value (p < 9.9 × 10−10). The transcription-based ESR identified in S. cerevisiae encompasses ∼900 transcripts, all of which are regulated in response to ten different environmental stresses. The significant overlap with ESR genes suggests that the Group 1 genes may be similarly regulated and involved in stress-response pathways.
To identify functional themes associated with Group 1 we used FunSpec (Robinson et al., 2002) analysis of all 425 corresponding proteins. FunSpec identifies MIPS functional categories that are significantly enriched in a list of proteins, relative to a random list of the same size (Supplemental Table S3). The Group 1 proteins are over-represented in the functional categories of protein synthesis, energy and metabolism, stress and damage responses. The protein syntheses category contained the most significantly enriched AGA and GAA containing transcript YEF3 (Supplemental Table S4), thus suggesting that YEF3 translation would be keyed to Trm9-catalyzed tRNA modifications. Because we and others have demonstrated that trm9Δ cells have a DNA damage phenotype, we were particularly interested in DNA damage-response genes with skewed codon usage patterns. FunSpec analysis demonstrated that Group 1 protein were over-represented with stress and damage response activities, specifically activities involved in deoxyribonucleotide metabolism (Rnr1, Rnr2, Rnr3, and Rnr4). All four RNR genes contain a high number of AGA and GAA codons (Supplemental Table S4) and RNR1−4 collectively contained ∼8× more AGA codons than expected. Importantly, Rnr1−4 are all key members of the DNA damage response.
Trm9 Enhances Yef3, Rnr1 and Rnr3 Protein Levels
The identification of YEF3, and the ribonucleotide reductase genes in Group 1 revealed the potential for the corresponding Yef3, Rnr1, Rnr2, Rnr3, and Rnr4 protein levels to be perturbed in trm9Δ cells. We analyzed the basal and damaged-induced levels of Yef3, Rnr1, Rnr3, and Rnr4 in wild-type and trm9Δ cells. We used genome based C-terminal TAP-tagged versions (Ghaemmaghami et al., 2003) and determined that Yef3-TAP, Rnr1-TAP and Rnr3-TAP protein levels were all notably decreased in trm9Δ cells (Fig. 2B). The most dramatic reduction in protein level was observed for Yef3 in trm9Δ cells relative to wild-type, and this correlates to the high number (132) of AGA and GAA codons. Northern blot analysis indicated that transcriptional regulation of YEF3, RNR1, and RNR3 was similar between wild-type and trm9Δ cells, under both basal and MMS-induced conditions, and the observed decrease of specific protein levels in trm9Δ cells supports a post-transcriptional role for Trm9.
Transcriptional induction by MMS occurs for RNR1 and RNR3 in wild-type and trm9Δ cells, and the transcript levels are similar in both cell types (Fig. 2B and Supplemental Figure S4). Under basal conditions, Rnr1 protein levels were much higher in wild-type cells compared to trm9Δ. Similarly, after MMS-treatment Rnr1 protein levels were higher in wild-type compared to trm9Δ cells, but the level of the Rnr1 protein was lower in both wild-type and trm9Δ cells when comparing MMS to untreated samples, suggesting Rnr1 is targeted for degradation after DNA damage. We also determined that Rnr3 protein levels are higher in wild-type cells relative to trm9Δ, under basal conditions; this increase in wild-type cells is maintained as Rnr3 protein levels go up in both cell types after MMS-treatment. Rnr3 is considered the damage-inducible large subunit of the ribonucleotide reductase complex, and our results support this observation.
Endogenous and damage-induced levels of Rnr4 showed no difference between wild type and trm9Δ cells (Supplemental Figure S5), and indicated that Rnr4 protein levels were not affected by Trm9 under normal conditions. Although we have been unable to detect endogenous Rnr2-TAP in wild-type cells (not shown), we have determined that PAB1, encoding Poly(A) binding protein, is found in Group 1 genes and have demonstrated that the levels of Pab1-TAP are modestly decreased in the trm9Δ background (Supplemental Figure S5). While not as dramatic as the results observed for Yef3-TAP, Rnr1-TAP, and Rnr3-TAP, the Pab1-Tap result further supports that the Group 1 codon signature is linked to Trm9. Clearly though, systems level analysis encompassing hundreds of matched transcript and protein levels measurements will be needed to determine the precise codon signature keyed to Trm9 and to ascertain the predictive nature of the Group 1 classification.
We also analyzed protein levels for codon usage patterns found outside of the Group 1 genes. Nap1 (6 AGA and 4 AGG) and Pih1 (5 AGA and 4 AGG) have small but similar numbers of AGA and AGG codons, and have average codon usage values that fall outside of Group 1. Similar to above we used TAP-tagged proteins, western, and northern blots to analyze transcript and protein levels in wild-type and trm9Δ cells (Fig. 2C). Transcript levels for both NAP1 and PIH1 were very similar in the wild-type and trm9Δ cells. Likewise, protein levels for Nap1 and Pih1 were similar when wild-type and trm9Δ cells were compared, further supporting that Trm9-initiated translational enhancement is keyed to gene-specific codon usage patterns. The RAD53 gene also contains average codon usage values (12 AGA and 12 AGG) and falls outside of the Group 1 classification. We have analyzed Rad53 levels in wild-type and trm9Δ cells and demonstrated they are at similar levels, further supporting that average transcripts are not affected by Trm9 (Supplemental Figure S5). Importantly, the Rad53 result demonstrates that only select DNA damage response proteins have their levels keyed to Trm9.
Yef3 and Rnr1 protein levels were markedly reduced in trm9Δ cells, yet transcript levels were similar when wild-type and mutant cells were compared. We verified that both YEF3 and RNR1 were in the process of being translated in trm9Δ cells by analyzing polysome profiles using cells grown under basal conditions. We determined that both YEF3 and RNR1 mRNA were predominantly found in the polysomes of both wild-type and trm9Δ cells (Fig. 3A-B). We observed that the ACT1 mRNA loading control was found at similar levels in fraction 1−6, comparing wild-type to trm9Δ cells. In contrast, both the YEF3 and RNR1 transcripts were represented at higher levels in matched polysome fractions of trm9Δ samples, when compared to wild-type, further indicating that translation was perturbed in a codon specific fashion in the mutant cells.
Figure 3. YEF3 and RNR1 transcripts are Associated with Translational Machinery in trm9Δ Cells.

(A) Polysome profiles of YEF3 in wild-type and trm9Δ cells. RNA was extracted from the gradient fractions and subjected to northern blot analysis, probing with ACT1 and YEF3 RNA as indicated. (B) Polysome profiles of RNR1 in wild-type and trm9Δ cells. The RNR1 and ACT1 transcripts were detected using northern blot analysis. Nonspecific signal was detected in lanes 10, 13, and 14 and was due to the low RNR1 transcript levels and bleeding from 25S and 18S pools. For all polysome profiles By4741 and trm9Δ cells were grown in YPD to ∼5×106 cells/ml and ethidium bromide staining was used to indicate the relative amounts of 25 and 18S rRNA in each fraction.
The DNA Damage Phenotype in trm9Δ Cells is Due to Rnr1 and Rnr3 Deficiencies
Hydroxyurea (HU) is an inhibitor of ribonucleotide reductase activity, and acts to quench the active site and prevent the formation of deoxyribonucleotide diphosphates (Eklund et al., 2001). We postulated that the Rnr1 and Rnr3 protein deficiencies identified in trm9Δ cells should result in a HU-induced growth phenotype. Therefore we performed HU sensitivity assays to determine if the observed Rnr1 and Rnr3 protein deficiencies were physiologically significant. Wild-type (By4741), trm9Δ, and trm9Δ cells over-expressing TRM9 were grown over-night in SD-URA and serially diluted onto SD-URA + galactose plates with and without HU. Plates were imaged after four days and demonstrated that trm9Δ cells are growth impaired in the presence of HU, a phenotype that can be complemented by the over-expression of TRM9 (Fig. 4A). Analysis of our plate based phenotypic data supported the hypothesis that ribonucleotide reductase activity was reduced in trm9Δ cells.
Figure 4. Decreased Protein Levels of Rnr1 and Rnr3 Result in a DNA Damage Phenotype.
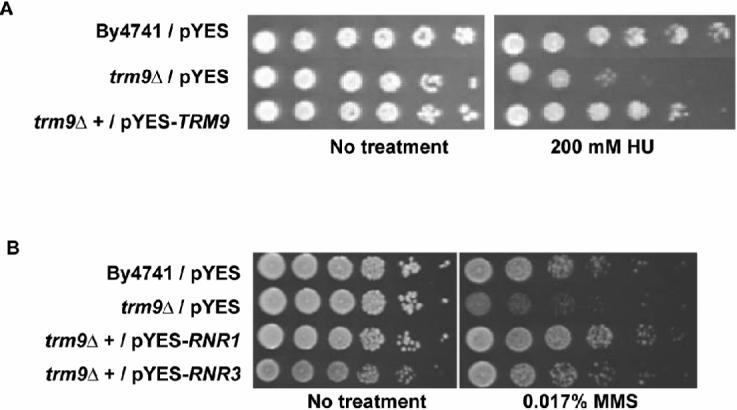
(A) The HU sensitivity of trm9Δ cells was complemented by over-expression of TRM9 from pYES-TRM9. (B) Over-expression of RNR1 and RNR3, from pYES-RNR1 and pYES-RNR3, rescued the MMS sensitive phenotype of trm9Δ cells. For both experiments cell cultures were grown overnight in SD-URA and serially diluted onto plates with HU or MMS, respectively. SD-URA + galactose plates were used for HU assays. YP-galactose plates were used in MMS experiments, because the MMS sensitive phenotype identified in trm9Δ cells is stronger under rich media conditions, compared to defined media.
Given the central role of the Rnr complex in the DNA damage response, we reasoned that transcriptional over-expression of RNR1 or RNR3 might rescue the originally observed MMS sensitive phenotype of trm9Δ cells by compensating for the reduced translational efficiency. We put RNR1 and RNR3 under the control of a strong galactose promoter and performed MMS rescue experiments. Our plate based assays demonstrated that over-expression of RNR1 or RNR3 can rescue the MMS sensitive phenotype of trm9Δ cells (Fig. 4B). Taken together, our western blot data, HU assays, and RNR1 and RNR3 over-expression results indicate that low Rnr levels caused the observed DNA damage phenotype in Trm9 deficient cells.
S. cerevisiae Trm9 Levels Are Unchanged After MMS Damage
After showing that Trm9 enhances the levels of DNA damage response proteins, we investigated whether Trm9 activity changed in response to DNA damage. We used northern and western blots to characterize transcript and protein levels for Trm9 before and after 0.0125% MMS treatment for one hour. The dose of MMS was chosen because this treatment will increase the levels of the DNA damage inducible RNR1, RNR3, and MAG1 transcripts and the Mag1 and Rnr3 proteins (Fig. 2B and Fig 5A-B). The TRM9 transcript levels did not change after MMS exposure, relative to untreated controls. Also, we analyzed the level of the Trm9 protein in wild-type cells, before and after MMS damage, using an endogenous TAP-tagged protein (Fig. 5B). We monitored protein levels from 0 to 60 minutes, and found that Trm9 protein levels were relatively unchanged after MMS treatment.
Figure 5. Trm9 Transcript and Protein Levels Are Unchanged after MMS Damage.
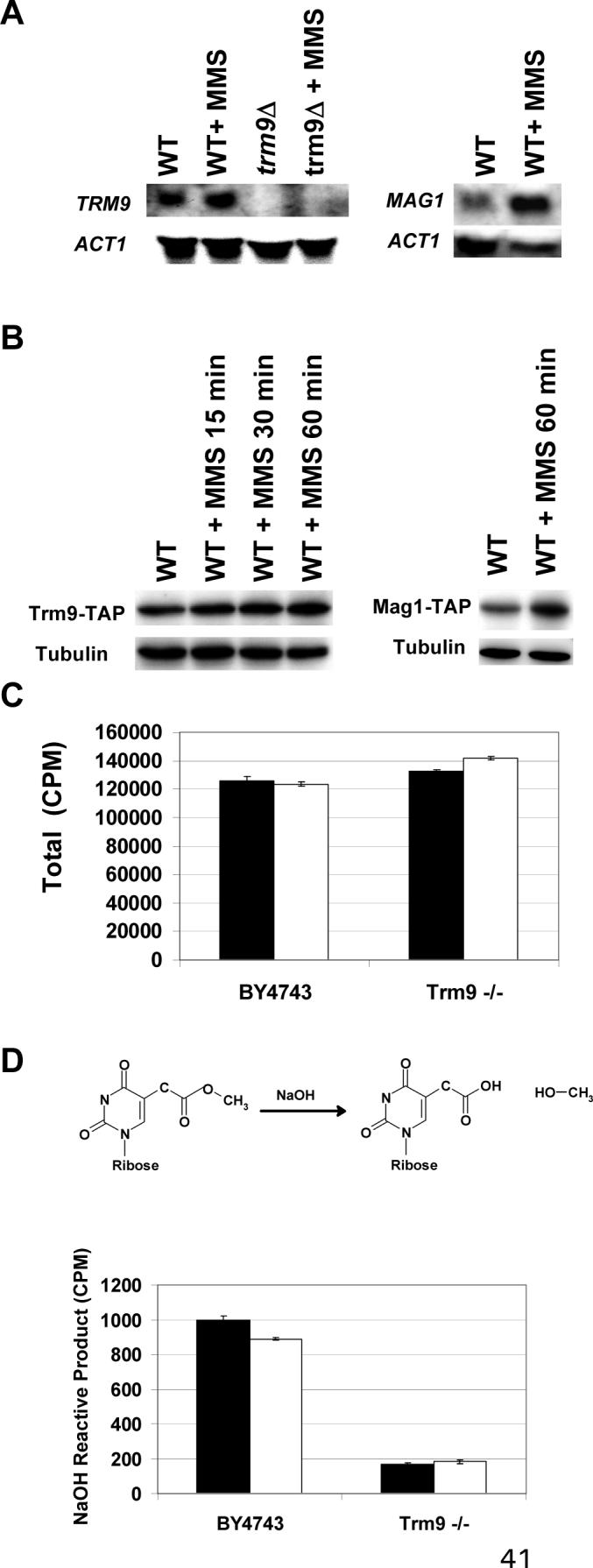
(A) The transcription of TRM9 in wild-type (BY4741) and trm9Δ cells was analyzed before and after MMS treatment using northern blots. The induction of the MAG1 transcript, which encodes an MMS-inducible DNA glycosylase used to repair 3-methyladenine lesions in DNA, serves as a positive control for DNA damage. (B) Western blots against endogenous TAP tagged Trm9 (ATCC201388 background) before and after MMS treatment. Mag1-TAP (ATCC201388 background) levels increase under these exposure conditions, and serve as a positive control for DNA damage. Cells used for northern and western blots were grown to 5 × 106 cells/ml in YPD and either mock or MMS treated as indicated. (C) Total radioactivity of tRNA purified from wild-type (By4743) and trm9−/− cells labeled with [3H]-methionine before (black bars) and after (white bars) MMS treatment. (D) NaOH derivatization of mcm5U nucleosides will generate labeled methanol. NaOH reactive methylesters in tRNA from wild-type (BY4743) and trm9−/− cells labeled with [3H]-methionine, before (black bars) and after (white bars) MMS treatment. Error bars represent the standard deviation of two replicate experiments in panels C and D.
To analyze Trm9 activity in vivo after MMS treatment, we used radioactive labeling of tRNA, followed by tRNA purification and NaOH derivatization. Trm9 uses the biological methyl donor SAM to generate a methylester to complete the synthesis of mcm5U and mcm5s2U wobble bases. Similar to MMS, SAM is a SN2 alkylating agent, and the high concentrations used in labeling experiments can damage DNA (Kalhor and Clarke, 2003; Barrows and Magee, 1982). Therefore we used methionine, which is a precursor to SAM, and were able to generate physiological pools of [3H]-SAM from [3H]-methionine. We determined that the total amount of radioactivity in tRNA from wild-type and trm9Δ cells was very similar, indicating that label incorporation in total tRNA in trm9Δ cells was comparable to that of wild-type (Fig. 5C). The total radioactivity in each sample represents all methyl-based modifications, of which Trm9 specific tRNA modifications were a small part. Next, we used NaOH derivatization of total tRNA to identify Trm9 specific tRNA modifications. We compared tRNA from untreated wild-type and untreated trm9Δ cells, and determined that the wild-type cells have an approximate 4-fold increase in NaOH reactive radiolabeled methanol compared to trm9Δ cells (Fig. 5D). Next, we determined that the levels of Trm9-specifc tRNA modifications in wild-type cells before and after MMS treatment were similar (Fig. 5D), suggesting that Trm9 activity is unchanged after DNA damage. While our transcript and protein data support that Trm9 levels are constitutive, more precise activity assays are needed to determine if Trm9 specific tRNA modifications increase after DNA damage.
Discussion
Trm9 Catalyzed tRNA Modifications Enhance Translation
We have shown that Trm9 specific tRNA modifications can enhance some protein levels in a codon-predictable fashion. Initially, Trm9-specific tRNA modifications promote high basal levels of codon-specific proteins and can work coordinately with transcription to enhance damage-induced protein levels. Our codon reporter data points to translation rather than protein degradation as the step at which protein levels are affected. The decreased protein levels of Yef3, Rnr1, and Rnr3, with no difference in transcription, in trm9Δ cells further implicated Trm9 as a transcript-specific enhancer of translation. Similarly, our observation that YEF3 and RNR1 are overly engaged with the polysomes in trm9Δ cells supports the role of Trm9 in enhancing translation. While the exact mechanism of translational enhancement is unknown, modulation of translational elongation would appear to be a viable method to enhance codon-specific translation.
Our data support a basic model of codon-specific translational enhancement by Trm9 (Fig. 6). For simplicity, we only depict the AGA codon, but additionally our reporter data supports that Trm9 enhances the translations of AGG and GAA codons, while or computational results predicate that other codons will be enhanced by Trm9 catalyzed tRNA modifications. In our model cells with functional Trm9 activity and corresponding tRNA modifications are primed for the enhanced production of proteins corresponding to AGA rich transcripts. In effect, the Trm9-specific tRNA modifications allow for a more efficient use of the existing AGA-rich transcripts. Trm9-specifc tRNA modifications are proposed to have no influence on the translation of average transcripts which contain a normal codon usage pattern. Ultimately, our model requires mcm5U to be a translational signals that promote anticodon interactions with AGA codons, or with other codon combinations. Biochemical studies of thiolated wobble and modified uridines in anticodon stem loops have shown that these modifications differentially affect interactions with synonymous codons (Yarian et al., 2002 ; Krueger et al., 1998), thus providing precedence for our translational enhancement model. It is important to note that our model of translational enhancement is predicted to work at the level of translation elongation, and one of the targets (Yef3) identified in this study is a translation elongation factor. The exact function of Yef3 is unclear, but it has a proposed role of removing deacylated tRNA from the ribosome E-site during translation elongation (Anand et al., 2003). It is possible that Yef3 plays a role in codon specific translation, either directly or indirectly, and in theory could work coordinately with Trm9 to recruit tRNAs after modification, but this is speculative.
Figure 6. Model for Codon-Specific Translational Enhancement.

Our model has (panel A, left) Trm9 dependent tRNA modifications enhancing the translation of transcripts over-represented with AGA codons while not affecting average transcripts. Translational enhancement is most likely facilitated (panel A, right) by mcm5U (*) at the tRNA wobble base promoting interactions with the AGA in mRNA. It is important to note that the up- and down-stream codons surrounding AGA most likely play an important yet still undefined role in this model. The lack of Trm9-dependent tRNA modifications (panel B, left) slows the translation of transcripts over-represented with AGA codons. Codon-specific translation is predicted to be slowed due to (panel B, right) inefficient interactions between unmodified uridine in the anticodon and adenine in the codon wobble position. AGA specific transcripts and proteins are shown in yellow, while average transcripts and proteins are shown in gray.
Translational Enhancement is Linked to Gene-Specific Codon Usage Patterns
We have used computational approaches to determine that the Trm9 associated codon signatures are found in 425 transcripts whose corresponding proteins are involved in stress response and other biological processes. We have demonstrated that Yef3, Rnr1, and Rnr3 protein levels are specifically linked to Trm9 activity, and their regulation fits into our model for enhanced translation. The AGA and GAA codon skew has been analyzed from the perspective of codon frequency, but it is more likely that codon totals or grouped codon runs will be the mRNA specific regulatory sequence keyed to Trm9 activity. The previous codon total and codon run statement is supported by our 10× AGA reporter data and by the observation that while RNR4 has the distinctive codon usage pattern found in Group 1 genes, its levels were not affected by Trm9 activity. Rnr4 is approximately half the size of Yef3, Rnr1, and Rnr3. Rnr4 contains ∼50% less AGA and GAA codons and in theory does not have the same frequency of codon runs or combinations.
The exact parameters of the codon sequence keyed to Trm9 have yet to be determined. Our heat map results demonstrated that many codons are over-represented in Group 1 genes, suggesting a number of different codon runs or codon combinations could be keyed to Trm9. For example, in addition to the arginine (AGA) and (GAA) codons, the glutamine codon CAA was significantly over-represented in Group 1 genes. Recent studies have shown that Trm9 modifies the corresponding glutamine tRNAGLN(UUG) (Lu et., al. 2005), suggesting that Trm9 could be enhancing the translation of these codons as well. The large number of over-represented codons in Group 1 suggests that not only homogenous codon runs (i.e., AGA-AGA-AGA) are keyed to Trm9, but also distinct combinations of codons (i.e., AGA-GAA-CAA). In addition, our reporter data supports that the translation of AGG codons are enhanced by Trm9. While AGG codons are not over-represented in Group 1 genes, their presence in distinct codon runs should be factored into any codon signal keyed to Trm9. Thus a complicated signal based on multiple codon combinations may be associated with Trm9, and investigations on the role of Trm9 in the translation of all 64 codon runs and codon combinations are underway.
Translational Signals are Coupled to the DNA Damage Response
The regulation of S. cerevisiae Rnr activity has been widely studied and shown to occur at the levels of transcription, sub-cellular localization, and post-translational modification. Each of these regulatory mechanisms is used to either control the level of individual Rnr subunits or overall activity, with deficiencies corrupting the DNA damage response (Chabes et al., 2003; Elledge and Davis, 1990). We report that the DNA damage phenotype in trm9Δ cells is due to a deficiency in Rnr1 and Rnr3 proteins, thus implicating tRNA modifications in the coordination of the DNA damage response. Our plates based HU assays, showing trm9Δ cells are growth impaired, reinforce this point. Complementation of HU and MMS sensitive phenotypes in trm9Δ cells was observed by over-expression of TRM9. The failure to fully compliment each phenotype may be due to over-expression of the tRNA methyltransferase promoting toxicity in the trm9Δ cells, by off-target methylation, but this is speculative. Over-expression of either RNR1 or RNR3 will rescue the MMS-sensitive phenotype of trm9Δ cells, which is most likely because either large subunit can substitute for the other in the final Rnr complex. Rnr activity is an important component of the cellular DNA damage response, and deficiencies or dysregulation of this activity can affect survival after DNA damage (Chabes et al., 2003; Elledge and Davis, 1990). Our study demonstrates that translation based signals play an important role in enhancing the levels of proteins that participate in the DNA damage response, and enhanced translation should be applicable to other codon-specific transcripts.
Major findings of our study are that Trm9-catalyzed tRNA modifications enhance the translation of arginine and glutamic acid codons in specific gene sequences, and that the levels of some DNA damage response proteins are linked to tRNA modifications. One can imagine sophisticated programs of translational regulation keyed to dynamic increases or decreases in tRNA modifications. The translational enhancement by Trm9 appears to work during elongation, and this system could be exploited to dynamically regulate translation elongation in other organisms. Ultimately the proposed model provides a unique mechanism to enhance protein levels in a gene and codon specific fashion, and due to Trm9's conservation, we suggest that a similar type of codon-specific translational enhancement will be found in humans.
Experimental Procedures
Strains, Plasmids, Media, and Plate Based Assays
A table of strains and oligonucleotides used is provided in supplementary material (Supplemental Table 5). All TAP tagged strains were obtained from Open Biosystems and were made trm9Δ using the G418 knockout cassette derived from Yml014w. Transformants for the deletion alleles were obtained on YPD (Guthrie and Fink, 2002) plates supplemented with G418 (200 μg/ml); trm9Δ deletions were confirmed by PCR. Plasmids were made using PCR based approaches and included pYES-TRM9, pYES-RNR1, and pYES-RNR3, derivatives of pYES2 (Invitrogen, Carlsbad, CA). Codon reporter plasmids were derived from pYES2/NT/lacZ (Invitrogen) and included pYES-10×AGA-lacZ, pYES-10×AGG-lacZ, pYES-10×GAA-lacZ, pYES-5×AGG-lacZ. YPD, YP-galactose, SD-URA (synthetic defined minus uracil), SD-TRP (synthetic defined minus tryptophan), SD-MET (synthetic defined minus methionine) and SD-URA + galactose (synthetic defined minus uracil + galacotose) media were made using reagents from QBiogene (Irvine, CA) as described (Guthrie and Fink, 2002). Plate-based assays were performed by spotting 5 μl of an over-night culture, with a corresponding 10-fold dilution series on YPD, YP-galactose, SD-TRP or SD-URA + galactose agar plates supplemented with MMS or HU.
Computational Analysis of Gene-Specific Codon Usage Patterns
We downloaded 5,783 S. cerevisiae gene sequences from the Saccharomyces Genome Database (SGD) and analyzed each gene using our GSCU algorithm. The GSCU algorithm interrogated a gene from the start to stop codon, counted the number of times a specific codon was used in a gene, and determined gene-specific codon frequencies for all 64 codons. Next, the genome-average codon frequency for all codons was determined using gene-specific codon frequency values for all 5,783 S. cerevisiae genes. The genome average codon frequency values were nearly identical to codon usage frequencies previously published for S. cerevisiae (Cherry et al., 1998), which is expected. In contrast, individual gene-specific codon frequencies can deviate significantly from genome averages. The gene-specific codon frequency for each S. cerevisiae gene was analyzed to compute a gene-specific codon Z-score.
The Z-score measures whether a codon is over- or under-represented in a gene relative to the average genome value; it is also a measure of significance. Clustering and heat map analyses were performed using CLUSTER and TREEVIEW. Group codon statistics were compiled using random sampling and a normal curve approximation, similar to that described (Begley et al., 2004). Briefly, the set of GSCU statistics representing 5,783 S. cerevisiae genes was randomly sampled to pick N = 425 genes. Group codon usage statistics were compiled for M = 10,000 iterations and group average values and standard deviations were then generated for statistical analysis, with Z-scores compiled for each gene-specific codon using the following formula:
Corresponding p-values were determined for all over-represented or under-represented codons using a one-tailed test and normal approximation.
β-galactosidase Assays, Transcript and Protein Measurements, and Polysome Profiles
Codon reporter plasmids were transformed into wild-type (By4741) and trm9Δ cells and individual colonies were used to seed overnight cultures, in SD-URA media. Overnight cultures were diluted into fresh SD-URA+GAL media and grown for 3 to 6 hours. Cells were harvested and then analyzed for β-galactosidase activity (Rupp et al., 2002). The specific activity (SA) measure was utilized to account for galactose induction time (I), exposure time (E) for assay development, and total protein used, with SA = 1000 * Absorbance / I * E * total protein. lacZ reporter assays to quantitate RNR1-RNR4 transcript levels were performed as described (Klinkenberg et al., 2006).
For the analysis of transcript and protein levels, By4741 and ATCC201385 derived cells were grown to ∼5 × 106 cells/ml in YPD and either untreated or MMS treated (0.0125% MMS) for 60 minutes. RNA was purified using an RNeasy Mini Kit (Qiagen) and analyzed as described (Jelinsky et al., 2000). Detection was facilitated using the Chemiluminescent Nucleic Acid Detection Module (Pierce, Rockford, IL). Protein extracts were prepared as described (Ghaemmaghami et al., 2003) and protein concentrations were measured using a BCA™ Protein Assay Kit (Pierce). Western blots were performed as described (Ghaemmaghami et al., 2003). Polysome profiles were performed similar as described (Mangus and Jacobson, 1999).
Trm9 Specific tRNA Methylation Assays
[3H]–methionine labeling was initiated by inoculating overnight cultures in SD-MET for 12-huours. Next cells were inoculated at ∼0.5 × 106 cells/ml into SD-MET media supplemented with 12.5 uC/ml [3H]–methionine (PerkinElmer, Waltham, MA). Cells were incubated with shaking for three days, followed by an additional three days with fresh SD-MET + [3H]–methionine labeling media. Pre-labeled cells were then inoculated at ∼1.0 × 106 cells/ml and incubated in fresh labeling media to ∼4 × 106 cells/ml, mock or MMS-treated for 1-hour, after which cells were harvested and stored at −80°C. tRNA was specifically isolated from labeled wild-type and trm9Δ samples using the Purelink miRNA isolation kit from Invitrogen. We assayed aliquots of tRNA from wild-type and trm9Δ cells for NaOH-reactive [3H]-methylesters, as described (Kalhor and Clarke, 2003).
Acknowledgments
This work was supported by grants to TJB from the NIH (1K22ES01225101 and 1R01ES015037) and NYSTAR. Special thanks to Dr. Steven Clarke (UCLA) for strains.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
References
- Agris PF. Decoding the genome: a modified view. Nucleic Acids Res. 2004;32:223–238. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A, Grayhack EJ, Phizicky EM. tRNA m7G methyltransferase Trm8p/Trm82p: Evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA. 2005;11:821–30. doi: 10.1261/rna.2030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell. 2006;21:87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Anand M, Chakraburtty K, Marton MJ, Hinnebusch AG, Kinzy TG. Functional interactions between yeast translation eukaryotic elongation factor (eEF) 1A and eEF3. J Biol Chem. 2003;278:6985–6991. doi: 10.1074/jbc.M209224200. [DOI] [PubMed] [Google Scholar]
- Barrows LR, Magee PN. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis. 1982;3:349–351. doi: 10.1093/carcin/3.3.349. [DOI] [PubMed] [Google Scholar]
- Begley TJ, Rosenbach AS, Ideker T, Samson LD. Recovery Pathways in S. cerevisiae Revealed by Genomic Phenotyping and Interactome Mapping. Mol Canc Res. 2002;1 [PubMed] [Google Scholar]
- Begley TJ, Rosenbach AS, Ideker T, Samson LD. Hot spots for modulating toxicity identified by genomic phenotyping and localization mapping. Mol Cell. 2004;16:117–125. doi: 10.1016/j.molcel.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Bennett CB, Lewis LK, Karthikeyan G, Lobachev KS, Jin YH, Sterling JF, Snipe JR, Resnick MA. Genes required for ionizing radiation resistance in yeast. Nat Genet. 2001;29:426–434. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Hall BD. Codon Selection in Yeast. J Biol Chem. 1982;257:3026–3031. [PubMed] [Google Scholar]
- Cartlidge RA, Knebel A, Peggie M, Alexandrov A, Phizicky EM, Cohen P. The tRNA methylase METTL1 is phosphorylated and inactivated by PKB and RSK in vitro and in cells. Embo J. 2005;24:1696–1705. doi: 10.1038/sj.emboj.7600648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- Chan MM, Lu X, Merchant FM, Iglehart JD, Miron PL. Gene expression profiling of NMU-induced rat mammary tumors: cross species comparison with human breast cancer. Carcinogenesis. 2005;26:1343–1353. doi: 10.1093/carcin/bgi100. [DOI] [PubMed] [Google Scholar]
- Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund H, Uhlin U, Farnegardh M, Logan DT, Nordlund P. Structure and function of the radical enzyme ribonucleotide reductase. Prog Biophys Mol Biol. 2001;77:177–268. doi: 10.1016/s0079-6107(01)00014-1. [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Davis RW. Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Genes Dev. 1990;4:740–751. doi: 10.1101/gad.4.5.740. [DOI] [PubMed] [Google Scholar]
- Flanagan JM, Healey S, Young J, Whitehall V, Trott DA, Newbold RF, Chenevix-Trench G. Mapping of a candidate colorectal cancer tumor-suppressor gene to a 900-kilobase region on the short arm of chromosome 8. Genes Chromosomes Cancer. 2004;40:247–260. doi: 10.1002/gcc.20039. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Huang M, Metzner S, Botstein D, Elledge SJ, Brown PO. Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol Biol Cell. 2001;12:2987–3003. doi: 10.1091/mbc.12.10.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GM. Guide to Yeast Genetics and Molecular Biology. Academic Press; 2002. [Google Scholar]
- Ilyas M, Straub J, Tomlinson IP, Bodmer WF. Genetic pathways in colorectal and other cancers. Eur J Cancer. 1999;35:1986–2002. doi: 10.1016/s0959-8049(99)00298-1. [DOI] [PubMed] [Google Scholar]
- Jablonowski D, Zink S, Mehlgarten C, Daum G, Schaffrath R. tRNAGlu wobble uridine methylation by Trm9 identifies Elongator's key role for zymocin-induced cell death in yeast. Mol Microbiol. 2006;59:677–688. doi: 10.1111/j.1365-2958.2005.04972.x. [DOI] [PubMed] [Google Scholar]
- Jelinsky SA, Estep P, Church GM, Samson LD. Regulatory networks revealed by transcriptional profiling of damaged saccharomyces cerevisiae cells: rpn4 links base excision repair with proteasomes. Mol Cell Biol. 2000;20:8157–8167. doi: 10.1128/mcb.20.21.8157-8167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinsky SA, Samson LD. Global response of Saccharomyces cerevisiae to an alkylating agent. Proc Natl Acad Sci U S A. 1999;96:1486–1491. doi: 10.1073/pnas.96.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhor HR, Clarke S. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol Cell Biol. 2003;23:9283–9292. doi: 10.1128/MCB.23.24.9283-9292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkenberg LG, Webb T, Zitomer RS. Synergy among differentially regulated repressors of the ribonucleotide diphosphate reductase genes of Saccharomyces cerevisiae. Eukaryot Cell. 2006;5:1007–1017. doi: 10.1128/EC.00045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger MK, Pedersen S, Hagervall TG, Sùrensen MA. The modifcation of the wobble base of tRNAGlu modulates the translation rate of glutamic acid codons in vivo. J. Mol. Biol. 1998;284:621–631. doi: 10.1006/jmbi.1998.2196. [DOI] [PubMed] [Google Scholar]
- Lu J, Huang B, Esberg A, Johansson MJ, Bystrom AS. The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. Rna. 2005;11:1648–1654. doi: 10.1261/rna.2172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangus DA, Jacobson A. Linking mRNA turnover and translation: assessing the polyribosomal association of mRNA decay factors and degradative intermediates. Methods. 1999;17:28–37. doi: 10.1006/meth.1998.0704. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Grigull J, Mohammad N, Hughes TR. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics. 2002;3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp S, Guthrie Christine, Gerald RF. Methods in Enzymology. Vol. 112. Academic Press; 2002. [Google Scholar]
- Urbonavicius J, Qian Q, Durand JM, Hagervall TG, Bjork GR. Improvement of reading frame maintenance is a common function for several tRNA modifications. Embo J. 2001;20:4863–4873. doi: 10.1093/emboj/20.17.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl GM, Carr AM. The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat Cell Biol. 2001;3:E277–286. doi: 10.1038/ncb1201-e277. [DOI] [PubMed] [Google Scholar]
- Weissenbach J, Dirheimer G. Pairing properties of the methylester of 5-carboxymethyl uridine in the wobble position of yeast tRNA3Arg. Biochim Biophys Acta. 1978;518:530–534. doi: 10.1016/0005-2787(78)90171-5. [DOI] [PubMed] [Google Scholar]
- Workman CT, Mak HC, McCuine S, Tagne JB, Agarwal M, Ozier O, Begley TJ, Samson LD, Ideker T. A systems approach to mapping DNA damage response pathways. Science. 2006;312:1054–1059. doi: 10.1126/science.1122088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R, Zhang Z, An X, Bucci B, Perlstein DL, Stubbe J, Huang M. Subcellular localization of yeast ribonucleotide reductase regulated by the DNA replication and damage checkpoint pathways. Proc Natl Acad Sci U S A. 2003;100:6628–6633. doi: 10.1073/pnas.1131932100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarian C, Marszalek M, Sochacka E, Malkiewicz A, Guenther R, Miskiewicz A, Agris PF. Modified nucleoside dependent Watson-Crick and wobble codon binding by tRNALysUUU species. Biochemistry. 2000;39:13390–13395. doi: 10.1021/bi001302g. [DOI] [PubMed] [Google Scholar]
- Yarian C, Townsend H, Czestkowski W, Sochacka E, Malkiewicz AJ, Guenther R, Miskiewicz A, Agris PF. Accurate translation of the genetic code depends on tRNA modified nucleosides. J Biol Chem. 2002;277:16391–16395. doi: 10.1074/jbc.M200253200. [DOI] [PubMed] [Google Scholar]
- Zhao X, Rothstein R. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc Natl Acad Sci U S A. 2002;99:3746–3751. doi: 10.1073/pnas.062502299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Elledge SJ. DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell. 1993;75:1119–1127. doi: 10.1016/0092-8674(93)90321-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.