

Abstract
Following certain patterns of electrical activity the strength of conventional chemical synapses in many areas of the mammalian brain can be subject to long-term modifications. Such modifications have been extensively characterised and are hypothesised to form the basis of learning and memory. A recent study in Science now shows that activity-dependent long-term modifications may also occur in the strength of mammalian electrical synapses. This raises the enticing possibility that electrical synapses might also contribute to neural plasticity and challenges the notion that in the mammalian CNS they are a simple mechanism for ‘hardwiring’ discrete neuronal populations.
Introduction
A longstanding challenge of neuroscience research is to understand the neural basis of learning and memory and to identify the mechanisms which allow the brain to adapt in response to different types of experience. For a long time, neuroscientists had proposed that such mechanisms would most likely involve modifications in the strength of chemical synapses. The discovery of long term potentiation (LTP) just over 30 years ago gave this idea a sound molecular/cellular basis [1, 2], and LTP has since emerged as the prime candidate mechanism for explaining experience-dependent plasticity and certain types of memory [3]. LTP refers to the sustained strengthening of a chemical synapse following a brief tetanic stimulation of afferent pathways and occurs in numerous brain areas and by various mechanisms. It was later discovered that following a different stimulation protocol the inverse of LTP, i.e. long-term depression (LTD), can occur [4-6].
Recently, there has been an explosion of interest in a different type of synapse in the mammalian brain: the electrical synapse. Electrical synapses are formed by the direct connection of neurons via gap junctions (GJs) which are specialized cell-to-cell contacts consisting of a collection of intercellular channels [reviewed in 7-11]. These channels provide electrical and chemical continuity by allowing the ‘passive’ diffusion of ions and low molecular weight proteins. Each channel is composed of two hemichannels, or connexons, each of which is comprised of six connexins. Although electrical synapses have been widely investigated in invertebrates, their precise distribution and key physiological significance in the mammalian CNS are only now starting to be fully appreciated [7-11].
The apparent ‘passive’ nature of electrical synapses has led many to assume that they represent a means to hardwire discrete neuronal populations within the CNS. However, it is well known that in both invertebrates and non-mammalian vertebrates they can be subject to long-term modulation through a variety of pathways. For example, in the goldfish, high-frequency stimulation of eighth nerve fibers can lead to a long-term strengthening of the electrical synapse onto the Mauthner cell dendrite, which shares several features with LTP at chemical synapses [12]. Furthermore, if the stimulation protocol is appropriately altered, this electrical synapse can also be subject to an equivalent form of LTD [13]. A recent study by Landisman and Connors now reveals that long-term modulation of electrical synapses also occurs in the mammalian brain [14]. Writing in Science they describe how activation of metabotropic glutamate receptors (mGluRs) on neurons in the nucleus reticularis thalami (NRT) leads to a sustained reduction of the coupling strength between cells linked by electrical synapses (Figure 1). Although this is not the first indication that modulation of electrical synapses occurs in the mammalian CNS [see for example 15-20], it is significant for three reasons. First, it is based on direct electrical recordings of coupling strength. Second, it is induced by endogenous neurotransmitter release following stimulation of incoming fibres. Third, it is associated with measurable changes in neuronal population activity, i.e. modification of synchrony between rhythmically firing neurons. Thus, as with LTP and LTD at chemical synapses, this study shows that under certain conditions, afferent stimulation leads to persistent changes in the strength of mammalian electrical synapses, in this case a reduction of coupling strength.
Figure 1. The strength of electrical synapses between NRT neurons is persistently reduced following mGluR activation leading to a pronounced decrease in the size of coupling potentials.

(Note: although electrical synapses allow the bidirectional flow of ions, the effect of reduced coupling strength is only shown in one direction for simplicity).
Real vs apparent changes in electrical synapses
In order to argue that mGluR activation modulates electrical synapses, Landisman and Connors [14] make use of visualized whole-cell patch-clamp recordings from pairs of electrically coupled NRT neurons in rat brain slices. Using this technique a consistent voltage transient can be elicited at will in one of these neurons whilst scrutinizing the amplitude of the response in its partner neuron (Figure 1). A change in this amplitude is taken to signify a modulation of the electrical synapse between the two cells. This leads to an obvious question: how do we know that the change in the response amplitude is truly related to a modulation of the electrical synapse (i.e. the intercellular channels) rather than simply due to changes in the electrical properties of one or both of the individual neurons? The main concern is that mGluR activation might persistently alter the passive or active membrane properties of the neurons, and that it is this that affects the transmission of the electrical signal from one neuron to the other. The authors address this issue as follows. First, whilst the apparent input resistance (RN) of NRT neurons is certainly affected by transient mGluR activation in an immediate sense, this change is not sustained with any statistical significance, whereas the decrease in coupling strength is. This suggests that passive membrane properties are not persistently altered. Second, because the authors use an estimate of coupling conductance which takes into account any changes in RN, it is assumed that alterations in passive cell properties would only be meaningful if, i) they occur persistently in dendritic compartments, and ii) the neuron pairs under investigation are coupled at dendritic sites. If this were the case, then we would need to ask how voltage signals are attenuated during dendritic propagation in electrically non-compact NRT neurons, and also consider if mGluR-activation causes any long-lasting changes in the active properties of their dendrites. To avoid this complication the study is limited to pairs of neurons, the somata of which appear in close contact or within 5μm of each other. Of course, it is still possible that these NRT neurons are coupled at sites which are remote from the soma and so a clear issue for the future is determining their precise points of contact [see 21].
Future directions
Like all exciting discoveries, the persistent modulation of electrical synapses shown by Landisman and Connors [14] poses many additional questions. In the first instance, is it restricted to the NRT of immature (postnatal day 12 to 15) rats or is it also present in adults, in other brain areas and in other species. In situ hybridization and immunolabelling studies indicate that connexin 36 (Cx36), the connexin that is responsible for forming GJs in the rat NRT [22], peaks during the first two weeks of postnatal life and then sharply declines [21, 23, 24]. However, it is still present in mature animals and there is certainly evidence of electrical coupling between NRT neurons in the adult cat [25; unpublished observations]. Thus, it is likely that Cx36-based electrical synapses between NRT neurons are present in various species and into adulthood, but determining whether they can be subject to long-term modulation requires further detailed experimentation.
Since Cx36 is one of the major connexins expressed in other neurons of the mammalian CNS, it is likely that their electrical synapses could also be subject to persistent modification. Indeed, since many Cx36-expressing neuron types also possess postsynaptic mGluRs [e.g. 26], it might be surprising if this were not the case. More generally, it is interesting to speculate whether mammalian electrical synapses which putatively involve other members of the connexin and pannexin families, in particular Cx45 [10], Cx47 [27], Cx57 [10] and pannexin1 and 2 [28], might undergo long-term modulation following appropriate physiological stimuli. Most connexins possess multiple phosphorylation sites [29], and it has been shown for Cx43, a well-characterized connexin that is prevalent in glial cells, that several kinases can lead to phosphorylation and, consequently, changes in GJ communication [30]. This further hints at the possibility that other neurotransmitters that target G-protein coupled receptors can also elicit alterations in the coupling strength of electrical synapses. Indeed, perhaps this would not be particularly surprising given the known inhibitory effects of neurotransmitters such as dopamine, serotonin, noradrenaline and nitric oxide on tracer-coupling in the developing neocortex [15-18] and other brain regions [e.g. 19, 20].
In order to fully dissect the specific signal transduction mechanisms involved in electrical synapse modification in the NRT [14] it is imperative to know which subtypes of mGluRs are involved. The synaptically-induced modifications reported by Landisman and Connors [14] are mimicked by the non-specific Group I/II mGluR agonist, trans-ACPD, and prevented by the broad spectrum mGluR antagonist, MCPG. However, NRT neurons express both functional postsynaptic Group I and Group II mGluRs [31] (Figure 2). Work on various forms of LTD at chemical synapses in the CNS commonly supports a central role for Group I receptors [e.g. 32-35]. As such, we might assume that these are also responsible for initiating the modulation of electrical synapses. We can then speculate on which signalling molecules are central to this task (Figure 2). Group I mGluRs are preferentially coupled to phospholipase C (PLC) and so we might expect a key role for protein kinase C (PKC) [30, 36] and/or Ca2+ [37]. However, these receptors are also believed to couple positively to adenylyl cyclase (AC) [reviewed in 38], and have been linked to arachidonic acid (AA) formation, presumably through activation of phospholipase A2 activity [39]. The AC link might be important because hemi-channels formed in Xenopus oocytes by Cx35, the fish ortholog of Cx36, are inhibited by protein kinase A (PKA)-dependent phosphorylation [40], whereas AA is a well-known inhibitor of GJ function [reviewed in 41]. Alternatively, a reduction in electrical coupling may occur through additional pathways, as for LTD at certain types of chemical synapses [42, 43], or involve a postsynaptic activation of group II mGluRs [44, 45] and, possibly, a negative modulation of cyclic AMP (cAMP) and PKA. With the extensive armoury of pharmacological tools and knockout mice available, a detailed picture of the intracellular events that lead to electrical synapse modification should be easily obtainable and a priority for future work. It will also be important to determine exactly how coupling strength is reduced in terms of the properties of the electrical synapse itself. For example, does this occur through a decrease in the number of intercellular channels via internalization, a reduction in single channel conductance or through some other process [see 46].
Figure 2. Potential intracellular pathways that mediate the persistent, mGluR-induced reduction in electrical coupling strength.
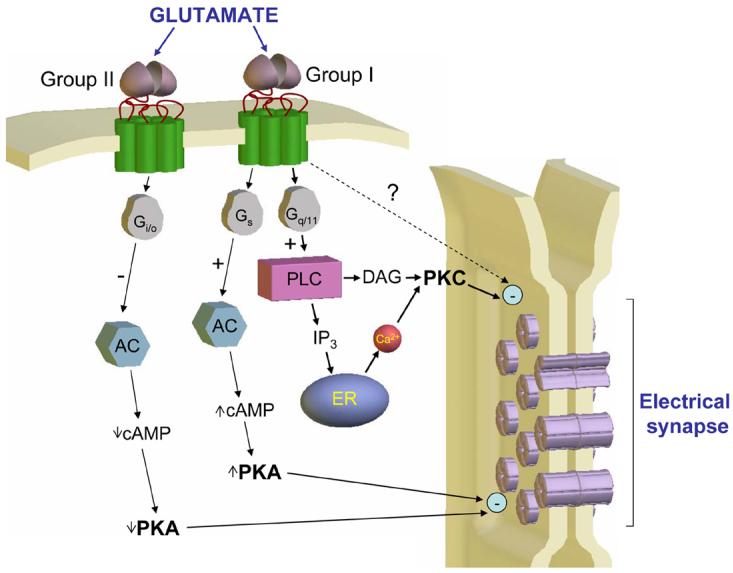
In addition to the mechanisms illustrated, activation of Group I receptors has also been linked to arachidonic acid formation [39]. For more information on Group I mGluR transduction pathways, see ref [38]. (DAG, diacylglycerol; ER, endoplasmic reticulum; other abbreviations in the text).
As noted above, the reduction in electrical synapse strength by mGluRs is related to a measurable decrease in the amplitude of electrical coupling potentials and neuronal synchrony [14]. Intriguingly, however, mGluR activation has been widely used as a tool for studying the functional relevance of electrical coupling between mammalian neurons [26, 47, 48], including those in the NRT [49]. This is because stimulation of these receptors commonly leads to depolarization of the neuronal population and, consequently, the instigation of spontaneous and continuous firing. The ability of electrical synapses to synchronize this firing within the population can then be easily examined. Could it therefore be that the degree of GJ-dependent synchrony observed in the presence of mGluR activation in other brain areas has been underestimated, as is the case in the NRT? With respect to this, it will be important in the future to determine the effect of different intensities and durations of tetanic stimulation or agonist concentration on electrical coupling strength as this will provide a clearer picture on the precise relationship between electrical synapse modification and mGluR activation. In particular, we would like to know in more detail exactly which types of stimulation lead to a reduction of coupling strength and which do not, so that these can be related to the different patterns of physiological and pathological activity that occur in afferent pathways.
Finally, how long does the reduction in coupling strength really last? In this respect, the present study [14] is unfortunately restricted by the inherent limitations of the recording technique, and thus modifications in coupling strength could only be assessed for 40 minutes at most. The authors show that the changes are clearly persistent within this time scale (though in one case a full reversal was achieved). Further studies are necessary to ascertain whether this time period is close to the limit of the time-course of the effect or whether these changes are truly ‘long-term’ in the way that early LTP researchers defined the term [1]. If the latter is true, it might be that we are dealing with a genuine mammalian ‘electrical LTD’ phenomenon. The obvious question then would be: how do we induce ‘electrical LTP’?
Possible significance of electrical synapse modulation in the NRT
At present, we can only speculate on the specific roles of electrical synapse modification in the NRT as these would strictly depend on its time-course, whether or not it is present in mature animals and the type of presynaptic activity most optimal for its instatement. If we are really dealing with ‘electrical LTD’ then we can conjecture that it might be a serious contender for underlying certain aspects of experience-dependent plasticity [50]. Another obvious function would be the increased effectiveness of the NRT as a pacemaker of some sleep rhythms [51-53], given that an enhanced electrical coupling between NRT neurons would result from the reduced mGluR activation during sleep compared to the wake state [48]. Indeed, in vivo evidence indicates that electrical coupling in the NRT has an important part to play in generating spindle waves during light sleep [25, 51]. Interestingly, it is thought that changes in GJ coupling can occur on a circadian basis in the suprachiasmatic nucleus [54].
In addition to an involvement in normal sleep rhythms, the NRT is also known to play a key role in the generation of spike and wave discharges of typical absence [55] and Lennox-Gastaut-like [53] seizures. In fact, an abnormally-elevated synchronized inhibition within the NRT and to adjacent thalamic relay nuclei has been widely proposed as one of the central components in bringing about the behavioural and EEG characteristics of these seizures [53, 56]. It is tempting to speculate that an aberrant modulation of electrical synapse strength might be involved in these scenarios. This is certainly consistent with recent evidence suggesting that electrical synapses in thalamic regions play an important role in both typical [57] and atypical absence paroxysms [58].
Concluding remarks
It is undeniable that research into the molecular, cellular and network aspects of LTP/LTD has delivered a wealth of invaluable information on the fundamental properties of chemical synapses. In this sense, electrical synapses have some serious catching up to do. However, the discovery of a long-term modulation of mammalian electrical synapses might now open up new research avenues, ultimately leading to a fuller understanding of the workings and significance of these neuronal devices.
Acknowledgements
We would like to thank Profs. David Spray and Kevin Fox for helpful comments.
References
- 1.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232(2):331–56. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collingridge GL, Bliss TV. Memories of NMDA receptors and LTP. Trends Neurosci. 1995;18(2):54–6. [PubMed] [Google Scholar]
- 3.Malenka RC. The long-term potential of LTP. Nat Rev Neurosci. 2003;4(11):923–6. doi: 10.1038/nrn1258. [DOI] [PubMed] [Google Scholar]
- 4.Barrionuevo G, Schottler F, Lynch G. The effects of repetitive low frequency stimulation on control and “potentiated” synaptic responses in the hippocampus. Life Sci. 1980;27(24):2385–91. doi: 10.1016/0024-3205(80)90509-3. [DOI] [PubMed] [Google Scholar]
- 5.Ito M. Long-term depression. Annu Rev Neurosci. 1989;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- 6.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Connors BW, Long MA. Electrical synapses in the mammalian brain. Annu Rev Neurosci. 2004;27:393–418. doi: 10.1146/annurev.neuro.26.041002.131128. [DOI] [PubMed] [Google Scholar]
- 8.Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the Mammalian brain. Neuron. 2004;41(4):495–511. doi: 10.1016/s0896-6273(04)00043-1. [DOI] [PubMed] [Google Scholar]
- 9.Rozental R, Giaume C, Spray DC. Gap junctions in the nervous system. Brain Res Brain Res Rev. 2000;32(1):11–5. doi: 10.1016/s0165-0173(99)00095-8. [DOI] [PubMed] [Google Scholar]
- 10.Sohl G, Maxeiner S, Willecke K. Expression and functions of neuronal gap junctions. Nat Rev Neurosci. 2005;6(3):191–200. doi: 10.1038/nrn1627. [DOI] [PubMed] [Google Scholar]
- 11.Hormuzdi SG, et al. Electrical synapses: a dynamic signaling system that shapes the activity of neuronal networks. Biochim Biophys Acta. 2004;1662(1-2):113–37. doi: 10.1016/j.bbamem.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 12.Yang XD, Korn H, Faber DS. Long-term potentiation of electrotonic coupling at mixed synapses. Nature. 1990;348(6301):542–5. doi: 10.1038/348542a0. [DOI] [PubMed] [Google Scholar]
- 13.Yang XD, Faber DS. Initial synaptic efficacy influences induction and expression of long-term changes in transmission. Proc Natl Acad Sci U S A. 1991;88(10):4299–303. doi: 10.1073/pnas.88.10.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landisman CE, Connors BW. Long-term modulation of electrical synapses in the mammalian thalamus. Science. 2005;310(5755):1809–13. doi: 10.1126/science.1114655. [DOI] [PubMed] [Google Scholar]
- 15.Rorig B, Klausa G, Sutor B. Dye coupling between pyramidal neurons in developing rat prefrontal and frontal cortex is reduced by protein kinase A activation and dopamine. J Neurosci. 1995;15(11):7386–400. doi: 10.1523/JNEUROSCI.15-11-07386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rorig B, Sutor B. Nitric oxide-stimulated increase in intracellular cGMP modulates gap junction coupling in rat neocortex. Neuroreport. 1996;7(2):569–72. doi: 10.1097/00001756-199601310-00046. [DOI] [PubMed] [Google Scholar]
- 17.Rorig B, Sutor B. Regulation of gap junction coupling in the developing neocortex. Mol Neurobiol. 1996;12(3):225–49. doi: 10.1007/BF02755590. [DOI] [PubMed] [Google Scholar]
- 18.Rorig B, Sutor B. Serotonin regulates gap junction coupling in the developing rat somatosensory cortex. Eur J Neurosci. 1996;8(8):1685–95. doi: 10.1111/j.1460-9568.1996.tb01312.x. [DOI] [PubMed] [Google Scholar]
- 19.O'Donnell P, Grace AA. Cortical afferents modulate striatal gap junction permeability via nitric oxide. Neuroscience. 1997;76(1):1–5. doi: 10.1016/s0306-4522(96)00433-2. [DOI] [PubMed] [Google Scholar]
- 20.O'Donnell P, Grace AA. Dopaminergic modulation of dye coupling between neurons in the core and shell regions of the nucleus accumbens. J Neurosci. 1993;13(8):3456–71. doi: 10.1523/JNEUROSCI.13-08-03456.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu XB, Jones EG. Fine structural localization of connexin-36 immunoreactivity in mouse cerebral cortex and thalamus. J Comp Neurol. 2003;466(4):457–67. doi: 10.1002/cne.10901. [DOI] [PubMed] [Google Scholar]
- 22.Landisman CE, et al. Electrical synapses in the thalamic reticular nucleus. J Neurosci. 2002;22(3):1002–9. doi: 10.1523/JNEUROSCI.22-03-01002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Condorelli DF, et al. Expression of Cx36 in mammalian neurons. Brain Res Brain Res Rev. 2000;32(1):72–85. doi: 10.1016/s0165-0173(99)00068-5. [DOI] [PubMed] [Google Scholar]
- 24.Belluardo N, et al. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865(1):121–38. doi: 10.1016/s0006-8993(00)02300-3. [DOI] [PubMed] [Google Scholar]
- 25.Fuentealba P, et al. Experimental evidence and modeling studies support a synchronizing role for electrical coupling in the cat thalamic reticular neurons in vivo. Eur J Neurosci. 2004;20(1):111–9. doi: 10.1111/j.1460-9568.2004.03462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beierlein M, Gibson JR, Connors BW. A network of electrically coupled interneurons drives synchronized inhibition in neocortex. Nat Neurosci. 2000;3(9):904–10. doi: 10.1038/78809. [DOI] [PubMed] [Google Scholar]
- 27.Teubner B, et al. Functional expression of the new gap junction gene connexin47 transcribed in mouse brain and spinal cord neurons. J Neurosci. 2001;21(4):1117–26. doi: 10.1523/JNEUROSCI.21-04-01117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruzzone R, et al. Pannexins, a family of gap junction proteins expressed in brain. Proc Natl Acad Sci U S A. 2003;100(23):13644–9. doi: 10.1073/pnas.2233464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper CD, et al. Analysis of connexin phosphorylation sites. Methods. 2000;20(2):196–204. doi: 10.1006/meth.1999.0937. [DOI] [PubMed] [Google Scholar]
- 30.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36(7):1171–86. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox CL, Sherman SM. Glutamate inhibits thalamic reticular neurons. J Neurosci. 1999;19(15):6694–9. doi: 10.1523/JNEUROSCI.19-15-06694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fitzjohn SM, et al. DHPG-induced LTD in area CA1 of juvenile rat hippocampus; characterisation and sensitivity to novel mGlu receptor antagonists. Neuropharmacology. 1999;38(10):1577–83. doi: 10.1016/s0028-3908(99)00123-9. [DOI] [PubMed] [Google Scholar]
- 33.Hansel C, Linden DJ. Long-term depression of the cerebellar climbing fiber--Purkinje neuron synapse. Neuron. 2000;26(2):473–82. doi: 10.1016/s0896-6273(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 34.Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically-induced hippocampal long-term depression. J Neurophysiol. 2006 doi: 10.1152/jn.00383.2005. [DOI] [PubMed] [Google Scholar]
- 35.Shigemoto R, et al. Antibodies inactivating mGluR1 metabotropic glutamate receptor block long-term depression in cultured Purkinje cells. Neuron. 1994;12(6):1245–55. doi: 10.1016/0896-6273(94)90441-3. [DOI] [PubMed] [Google Scholar]
- 36.Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18(6):969–82. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Heinke B, Sandkuhler J. Activation of group I metabotropic glutamate receptors induces long-term depression at sensory synapses in superficial spinal dorsal horn. Neuropharmacology. 2000;39(12):2231–43. doi: 10.1016/s0028-3908(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 38.Hermans E, Challiss RA. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. Biochem J. 2001;359(Pt 3):465–84. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aramori I, Nakanishi S. Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron. 1992;8(4):757–65. doi: 10.1016/0896-6273(92)90096-v. [DOI] [PubMed] [Google Scholar]
- 40.Mitropoulou G, Bruzzone R. Modulation of perch connexin35 hemi-channels by cyclic AMP requires a protein kinase A phosphorylation site. J Neurosci Res. 2003;72(2):147–57. doi: 10.1002/jnr.10572. [DOI] [PubMed] [Google Scholar]
- 41.Rozental R, Srinivas M, Spray DC. How to close a gap junction channel. Efficacies and potencies of uncoupling agents. Methods Mol Biol. 2001;154:447–76. doi: 10.1385/1-59259-043-8:447. [DOI] [PubMed] [Google Scholar]
- 42.Schnabel R, Kilpatrick IC, Collingridge GL. An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology. 1999;38(10):1585–96. doi: 10.1016/s0028-3908(99)00062-3. [DOI] [PubMed] [Google Scholar]
- 43.Schnabel R, Kilpatrick IC, Collingridge GL. Protein phosphatase inhibitors facilitate DHPG-induced LTD in the CA1 region of the hippocampus. Br J Pharmacol. 2001;132(5):1095–101. doi: 10.1038/sj.bjp.0703905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poschel B, et al. The metabotropic glutamate receptor mGluR3 is critically required for hippocampal long-term depression and modulates long-term potentiation in the dentate gyrus of freely moving rats. Cereb Cortex. 2005;15(9):1414–23. doi: 10.1093/cercor/bhi022. [DOI] [PubMed] [Google Scholar]
- 45.Cho K, et al. A new form of long-term depression in the perirhinal cortex. Nat Neurosci. 2000;3(2):150–6. doi: 10.1038/72093. [DOI] [PubMed] [Google Scholar]
- 46.Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Arch Biochem Biophys. 2000;384(2):205–15. doi: 10.1006/abbi.2000.2131. [DOI] [PubMed] [Google Scholar]
- 47.Hughes SW, et al. Properties and origin of spikelets in thalamocortical neurones in vitro. Neuroscience. 2002;110(3):395–401. doi: 10.1016/s0306-4522(01)00577-2. [DOI] [PubMed] [Google Scholar]
- 48.Hughes SW, et al. Synchronized oscillations at alpha and theta frequencies in the lateral geniculate nucleus. Neuron. 2004;42(2):253–68. doi: 10.1016/s0896-6273(04)00191-6. [DOI] [PubMed] [Google Scholar]
- 49.Long MA, Landisman CE, Connors BW. Small clusters of electrically coupled neurons generate synchronous rhythms in the thalamic reticular nucleus. J Neurosci. 2004;24(2):341–9. doi: 10.1523/JNEUROSCI.3358-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fox K, Wong RO. A comparison of experience-dependent plasticity in the visual and somatosensory systems. Neuron. 2005;48(3):465–77. doi: 10.1016/j.neuron.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 51.Steriade M, et al. The slow (< 1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. 1993;13(8):3284–99. doi: 10.1523/JNEUROSCI.13-08-03284.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuentealba P, Steriade M. The reticular nucleus revisited: intrinsic and network properties of a thalamic pacemaker. Prog Neurobiol. 2005;75(2):125–41. doi: 10.1016/j.pneurobio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 53.Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28(6):317–24. doi: 10.1016/j.tins.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 54.Colwell CS. Rhythmic coupling among cells in the suprachiasmatic nucleus. J Neurobiol. 2000;43(4):379–88. doi: 10.1002/1097-4695(20000615)43:4<379::aid-neu6>3.0.co;2-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slaght SJ, et al. Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges. J Neurosci. 2002;22(6):2323–34. doi: 10.1523/JNEUROSCI.22-06-02323.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nature Reviews Neuroscience. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- 57.Gareri P, et al. Antiabsence effects of carbenoxolone in two genetic animal models of absence epilepsy (WAG/Rij rats and lh/lh mice) Neuropharmacology. 2005;49(4):551–63. doi: 10.1016/j.neuropharm.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 58.Proulx E, et al. Functional contribution of specific brain areas to absence seizures: role of thalamic gap-junctional coupling. Eur J Neurosci. 2006;23(2):489–96. doi: 10.1111/j.1460-9568.2005.04558.x. [DOI] [PubMed] [Google Scholar]