

Abstract
In the development of anti-cancer drugs, it is important to yield selective cytotoxicity primarily against tumor tissues. To achieve this goal, the use of a polymer-drug conjugate appears to be appealing, simply because it can take the advantage of the so-called enhanced permeability and retention (EPR) effect due to vascular leak in tumors. Among various types of polymers, polyrotaxane (PR) is an interesting candidate and warrants further consideration. It is a self-assembled polymer made entirely of biocompatible components, by threading α-cyclodextrin (α-CD) molecules with the poly(ethylene glycol) (PEG) chain. The abundance in functional –OH groups on the CD residues render PR the capability of carrying a large dose of small anti-tumor agents for delivery.
Herein, we presented a novel PR-based delivery system using doxorubicin (DOX) as the model anti-cancer drug. Daunorubicin (DNR) was conjugated to the PR polymer via hydrolysable linkages, and upon hydrolysis, doxorubicin was released as the cytotoxic drug. To facilitate an intracellular uptake by the tumor cells of the PR-DOX conjugates, a cell-penetrating low molecular weight protamine (LMWP) peptide was further attached to the two termini of the PR chain. Using an innovative principle established in our laboratory, such as via the inhibition of the cell-penetrating activity by binding with heparin and reversal of this inhibition by subsequent addition of protamine, cellular uptake of the polymer-drug conjugates could be readily regulated.
In this paper, we performed in vitro studies to demonstrate the feasibility of this delivery system. The LMWP-PR-DOX conjugates, which yielded a sustained release of DOX over a period of greater than 4 days, were successfully synthesized. Intracellular uptake of these conjugates by A2780 human ovarian cancer cells and regulation of such uptake by heparin and protamine were confirmed by using the MTT assay and also the confocal microscopy method.
Keywords: Daunorubicin, Doxorubicin, Polyrotaxane, Low molecular weight protamine, Heparin Inhibition
1. INTRODUCTION
To succeed in chemotherapy, two requirements are needed: 1) the anticancer drug must be effective in the in vivo microenvironment of tumors, and 2) it must reach the target cells in vivo in sufficient quantities [1]. In addition, the drug must be able to minimize non-specific toxicity toward normal cells. Unfortunately, the half-life of the most low molecular weight anticancer drugs (< 500 Da) is short in the blood stream. In addition, they are not able to selectively invade tumor tissue because they can penetrate other healthy cells by diffusion. Therefore, it is essential to target tumor cells and enhance the permeability of drugs in order to achieve effective cancer therapy.
Macromolecular drug carriers have been known to be effective in overcoming some of these obstacles posed in the conventional chemotherapy. A macromolecular drug carrier can easily permeate into the tumor and induce its passive accumulation due to the vascular leakiness and impaired lymphatic drainage in solid tumors [2, 3]. This phenomenon is now known as the enhanced permeability and retention (EPR) effect [4]. Based on this rationale, macromolecular drug carrier systems for cancer treatment have been actively pursued [5–8]. The synthetic polymers such as poly(styrene-co-maleic acid) [5], 2-hydroxypropyl methacrylate (HPMA) [6], polyethylene glycol(PEG) [7], and poly(glutamic acid) [8] have already been widely attempted to formulate drug-polymer conjugates. As a part of this type of drug-polymer conjugate research, studies on the pharmaceutical applications of supramolecular assemblies have recently been exploited. A typical example is the assembly of polyrotaxane (PR), which is characterized by a unique structure in which cyclic cyclodextrin molecules are threaded onto a linear macromolecular PEG chain and then capped with bulky end groups [9].
However, the delivery of hydrophilic or macromolecular drugs into cells is limited by their poor cell membrane permeability. Cell entry of such drugs or drug carriers is conventionally accomplished via adsorptive or receptor-mediated endocytosis, which may not necessarily be effective because those entities are subject to endolysosomal trafficking upon entry. An alternative strategy, which has recently been drawing significant attentions, is to overcome this problem by chemically conjugating the large proteins or hydrophilic drugs to a class of so-called cell-penetrating peptides (CPPs). CPPs are small protein transduction domains (PTDs), generally around 10–16 amino acids in length, that can efficiently deliver the attached macromolecules (e.g. proteins, DNAs) and even nanoparticle carriers (e.g. polymers, liposomes) into cells by crossing the biological membrane barriers [10–12]. These peptides are generally characterized by the presence of basic amino acids, and include TAT (YGRKKRRQRRR) from human immunodeficiency virus (HIV-1) [13], VP22 from herpes simplex virus [14], Drosophila Antennapedia (ANTP) homeotic transcription factor [15], synthetic arginine-rich peptides [16], and the low molecular weight protamine (LMWP) peptide developed in our laboratory by enzymatic digestion from native protamine [17]. Although the exact mechanism in cell translocation remains unclear, the presence of basic amino acids appears essential for lipid interactions and penetration of the cell membrane [11, 12, 16]. Because of this unique property, the use of CPP-drug conjugates for tumor penetration has been examined. However, despite their unmatched ability to efficiently translocate cargo molecules into cells, these CPPs do not possess target selectivity in cell entry, which all but significantly hinders their potential clinical applications [18].
Proposed herein is a novel polyrotaxane-based carrier system for effective delivery of anti-tumor agents. As shown in Figure 1 and using doxorubicin (DOX) as a model drug, daunorubicin (DNR) is derivatized and chemically conjugated to PR which, upon release via hydrolysis, yields the parent drug DOX. To facilitate cellular uptake of the PR-drug conjugates, the cell-penetrating LMWP peptide is linked to the PR-DOX conjugates for intracellular delivery of DOX. Prior to administration, the cell-penetrating function of the cationic LMWP peptide will be masked by binding with a negatively-charged heparin molecule. Overall, this system is deliberately designed in such a manner that after accumulation of the conjugates at the tumor site by EPR-mediated passive targeting, protamine sulfate, a clinical heparin antagonist that binds heparin stronger than LMWP, will be administered subsequently to release heparin inhibition on LMWP, allowing an effective intracellular uptake of the LMWP-PR-DOX conjugates. Once inside tumor cells, DOX molecules will be released naturally from the PR polymer chain by hydrolysis, yielding a sustained level of DOX within the tumor cells to exert cytotoxic effects.
Figure 1.

Schematic illustration of the polyrotaxane-doxorubicin conjugates.
In the present study, in vitro cell culture results were presented to demonstrate the feasibility of this PR-based DOX delivery system. The LMWP-PR-DOX conjugates, which yielded a linear and sustained release of anti-cancer drug over a period greater than 4 days, were successfully synthesized and characterized. Furthermore, the controlled regulation of intracellular uptake of these conjugates using heparin and protamine was also demonstrated.
2. MATERIALS AND METHODS
Materials
Daunorubicin HCl (DNR-HCl) and doxorubicin HCl (DOX-HCl) were purchased from the Hospital Pharmacy at the University of Michigan. Boc-L-tyrosine hydroxysuccinimide ester (BOC-Tyr-OSu) was purchased from Fluka (Allentown, PA). Unless otherwise stated, all chemicals including (polyoxyethylene glycol)bisamine (PEG-BA; Average Mn: 3,350 Da), and alpha-cyclodextrin (α-CD) were purchased from Sigma Chemical Co. (St. Louis, MO), and all solvents were obtained from Aldrich Chemical Co. (Milwaukee, WI). Water was distilled and deionized (ddH2O)
Synthesis of Polyrotanxane
A schematic illustration of the synthesis of polyrotaxane is shown in Figure 2. Pseudopolyrotaxane was first prepared according to the procedures of Eguchi et. al. [9]. In brief, 5 ml PEG-BA(1 g, 3.0 × 10−4mol) aqueous solution was added to a saturated aqueous solution of α-CD (12 g, 1.2 × 10−2 mol) at room temperature and ultrasonically agitated for 1 h, followed by stirring for 24 h. Precipitated pseudopolyrotaxane was collected by centrifugation and dried in vacuo at room temperature. (see Step 1 in Figure 2).
Figure 2.

Schematic illustration of the synthesis of polyrotaxane-doxorubicin conjugates.
Boc-Tyr-OSu (5 g, 1.3 × 10−2 mol) dissolved in DMSO was added to saturated solution of pseudopolyrotaxane (12 g, 4 × 10−4 mol) in DMSO and the mixture was stirred at room temperature for 96 h. The reaction mixture was then poured into excess acetone, and the precipitate was collected by centrifugation and washed with ethanol and water. The washed precipitate was collected by centrifugation and dried in vacuo at room temperature. (Step 2).
Boc-Tyr-terminated polyrotaxane (1 g, 3.0 × 10−5 mol) and succinic anhydride (5 g, 5.0 × 10−2 mol) were dissolved in pyridine and stirred at room temperature for 72 h, and the reaction mixture was poured into excess ether. The precipitate was collected by centrifugation, and all products were analyzed using 1H nuclear magnetic resonance spectroscopy, Bruker Avance DRX-500 NMR spectrometer performed at the Biomedical Research Core Facilities, University of Michigan. using DMSO-d6 as the solvent for structural analysis (Step 3).
Synthesis of Doxorubicin-Polyrotaxane Conjugate
Tert-butoxycarbonyl (Boc) group protected PR was subject to removal of the Boc group according to the method of Han et al. [19]. Boc-PR was poured into 4 N HCl in dioxane solution and stirred at room temperature for 40 min. Solvent was removed in vacuo, and the product was precipitated by the addition of ether and dried under vacuum (Step 4). PR-DOX conjugate was then prepared by following the method of Arcamone et al. [20] and Tanaka et. al. [21]. Briefly, lyophilized powder obtained from the solution of DNR-HCl (20mg, 3.5 × 10−5 mol) was dissolved in 25% methanol in dioxane (3 mL). The dissolved powder was filtered and filtrate was removed. One hundred milligrams (6.3 × 10−4 mol) of bromine dissolved in chloroform was added to DNR solution and stirred at 4ºC for 72 h, and the solvent was removed in vacuo. DNR-Br thus prepared was then dissolved in DMF, following by the addition of PR carboxyethylester (100 mg, 2.5 × 10−6 mol). The mixture was stirred at room temperature for 24 h, and solvent was removed in vacuo and subjected to DRX-500 1H NMR using DMSO-d6 as the solvent for structural analysis (Step 5).
Preparation of LMWP Peptide
LMWP was prepared according to previously described procedures [17]. In brief, thermolysin and protamine were mixed in 50mM Tris buffer (pH 7.5) and incubated at room temperature for 1 h, followed by quenching of thermolysin activity with 50 mM EDTA. The reaction mixture was purified by heparin affinity chromatography and a total of 5 peptides, denoted thermolysin-digested segment of protamine (TDSP) 1–5 according to the order of elution from heparin column, were obtained. Arginine-rich TDSP 5 (VSRRRRRRGGRRRR) was used for all subsequent studies.
Preparation of LMWP-Polyrotaxane-Doxorubicin Conjugate
To prepare the LMWP-PR-DOX conjugate, LMWP was first modified at its N-terminus by SPDP. Briefly, LMWP (5 mg/ml in phosphate buffer, pH 8.0) was mixed with tenfold molar excess of N-succinimidyl-3-(2-pyridyldithio)propionate (SPDP), dissolved in dimethylsulfoxide (DMSO), and reacted at room temperature for 2 h. SPDP-modified LMWP was then treated with dithiothreitol (DTT) at room temperature for 2 h to link the free sulfhydryl group at the N-terminus of LMWP. The product was then concentrated and lyophilized. To prepare PR-DOX, deprotected PR (20 mg/ml in phosphate buffer, pH 8.0) was mixed with a twenty-fold molar excess of SPDP, dissolved in DMSO and reacted at room temperature for 2 h. The hydrolytic product of unreacted SPDP was removed from the solution by size exclusion chromatography, and fractions containing conjugates were pooled. DNR was then conjugated to purify SPDP-PR by using the same method described above in PR-DOX conjugation. To synthesize the final LMWP-PR-DOX conjugates, activated PR-DOX conjugate solution was mixed with sulfhydryl(SH)-LMWP (twenty-fold molar excess of SH-SPDP:PR-DOX conjugate) and phosphate buffer (pH 7.4), and reacted at 4ºC for 8 h. LMWP-PR-DOX was purified by using a heparin-affinity column and excessive LMWP was further removed by utilizing ultrafiltration method (molecular weight cut off: 3 kDa). Pooled LMWP-PR-DOX conjugates were concentrated and lyophilized.
Fluorescence Labeling
Daunorubicin and doxorubicin possess their own fluorescence (excitation 470 nm/emission 580 nm). Polyrotaxane was labeled with Lissamine rhodamine B ethylenediamine (RHO, excitation 570 nm/emission 590nm) at its carboxylic acid groups. To perform this labeling, RHO was first dissolved in DMSO at a concentration of 10 mg/ml. Twenty microliter of the dye solution was then mixed with the PR-drug conjugates (with or without the attachment of LMWP) in labeling buffer (0.24 M methylimidazole, pH 9.0, 0.32 M 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC)) at a final DNR concentration of 2 μM. The mixture was reacted at room temperature for 4 h. Fluorescence-labeled drug conjugates were separated by size exclusion chromatography.
Content determination
Approximately 2 mg polyrotaxane-DOX was placed in a 100 ml volumetric flask, and 20 ml of 1:1 mixture of 50 mM borate buffer (pH 9.5)/methanol solution was added. The flask was shaken vigorously at 37 ºC for 24 h and the reaction was stopped by adding concentrated H3PO4. One milliliter of this solution was collected and diluted with water prior to fluorometrical analysis (excitation 470 nm/emission 580 nm) [22].
In vitro drug release studies
The release of doxorubicin from PR-DOX by hydrolysis was evaluated at 37°C in phosphate buffer (pH 7.4). Approximately 1 mg of PR-DOX was weighed and suspended in 1 mL of the buffer. The mixture was placed in a dialysis bag (MWCO 3000) and dialyzed against 30 ml of the same medium at an agitation rate of 100 rpm. One milliliter of the buffer solution was collected at designated time intervals and same volume of fresh buffer was added to the release medium. All samples were analyzed by fluorometry (excitation 470 nm/emission 580 nm).
In Vitro Cytotoxicity
DOX-induced cytotoxicity was assessed by a calorimetric assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). In brief, 1 × 105 A2780 human ovarian cancer cells (ECACC, Salisbury, UK) were seeded into 96 well plates. Cells were maintained in RPMI 1640 (Gibco, Carlsbad, CA) supplemented with 10 % heat-inactivated fetal calf serum, penicillin and streptomycin, and were then grown at 37 ºC in a humidified atmosphere of 5% CO2. Test samples were sterilized by using 0.22 μm sterilized syringe filter. After 24 h incubation at 37ºC, the medium was aspirated, and 8 different concentrations of sterile test substances (free DOX, PR-DOX and LMWP-PR-DOX) in fresh media were added. Control cells were treated with an equivalent volume of Dulbecco’s phosphate buffered saline (DPBS). Cells were subsequently cultured for 2 h, the medium was aspirated, and 100 μL of fresh medium and 25 μL of 5 mg/ml MTT solution were added to each well. The plates were incubated for 4 hours at 37ºC and formazan crystals were dissolved by the addition of DMSO. Finally, absorbance was measured at 570 nm.
Cell uptake assays
A2780 cells were plated on 4-well chamber slides at a density of 1 × 104 cells/slide and incubated at 37 ºC in a humidified atmosphere of 5% CO2. Cells were maintained in RPMI 1640 (Gibco, Carlsbad, CA) supplemented with 10 % heat-inactivated fetal calf serum, penicillin and streptomycin, and were then grown at 37 ºC in a humidified atmosphere of 5% CO2. Test samples were sterilized by using 0.22 μm sterilized syringe filter. After complete adhesion, the cell medium was removed. Five test samples, containing RHO-labeled PR, including: (1) free DOX; (2) PR-DOX (3) LMWP-PR-DOX; (4) LMWP-PR-DOX + heparin; and (5) LMWP-PR-DOX + heparin + protamine, were added to the cells at final DOX, heparin, and protamine concentration of 2 μM, 2 μM, and 4 μM, respectively. Following incubation for 2 h at 37ºC, cells were washed with PBS, fixed with 1% paraformaldehyde, and visualized using confocal laser scanning microscopy (Carl Zeiss, Gottingen, Germany).
Statistical Analysis
Student’s t-test was used to test for statistical difference between samples. A p value of ≤0.05 was considered to be statistically significant.
RESULTS AND DISCUSSION
Synthesis and characterization of polyrotaxane-DOX and LMWP-polyroxane-DOX Conjugates
Doxorubicin is a common anti-tumor agent which is effective to most cancers including breast, lung, ovary, and so on. However, it could cause significant toxic effects such as myelosuppression and cardiac toxicity, due to lack of selectivity on their therapeutic actions. Conjugation of polymer to doxorubicin is expected to improve the pharmacokinetic profile or therapeutic index, based on the effect of the so-called EPR passive tumor targeting.
Figure 3 shows the NMR spectrum of the synthesized PR-DOX conjugates, with peaks representing: 1H NMR (DMSO): δ (ppm) = 1.1 (s, C(5′)H1 of daunosamine), 1.3(d, C(2′)H2 of daunosamine), 2.2 (d, C(8)H2 of anthracycline), 2.3 (s, CH2 of CEE), 3.5 (s, CH2 of PEG), 4.0(d, C(5) H1 of CD), 7.0 (dd, aromatics of Tyr), 7.9 (s, C(3)H1 of anthracycline). The overall yield of the PR-DOX conjugate was 68%, whereas the average weight content of DNR in the conjugate was 12 % (w/w). The final yield of LMWP-PR-DOX was approximately 34%. The purity for both PR-DOX and LMWP-PR-DOX was > 95 %, as determined by gel permeation chromatography (Sephadex G-25 and Bio-Rad SEC 300). The molecular weight of the PR-DOX conjugate, as determined by the 1H-NMR data, was approximately 50 kDa and confirmed by gel permeation chromatography by utilizing poly (2-vinylpyridine) standard. Based on these results, each PR-DOX conjugate was calculated to contain 1 PEG chain, 30 α-CD residues, and 12 DNR drug molecules. The molecular weight of PR-DOX was also estimated by measurement of the amine groups of tyrosine capping residues exposed after deprotection by using the 2,4,6-trinitrobenzenesulfonic acid (TNBS) method [23, 24], or by utilizing the pyridine-2-thione assay of the SPDP-activated PR [25]. The molecular weight of PR-DOX determined by these two methods was 53 kDa and 48 kDa, respectively, which was consistent with the molecular weight estimated from NMR spectra. The average molar ratio of ethylene oxide/CD was 2.5, which was in reasonable agreement with the theoretical value of 2.0 ~2.2 suggested by other investigators [26, 27]. Deprotection process for PR-DOX was different from that for LMWP-PR-DOX, because the amine groups could react with the N-hydroxysuccinimidyl (NHS) group of SPDP.
Figure 3.
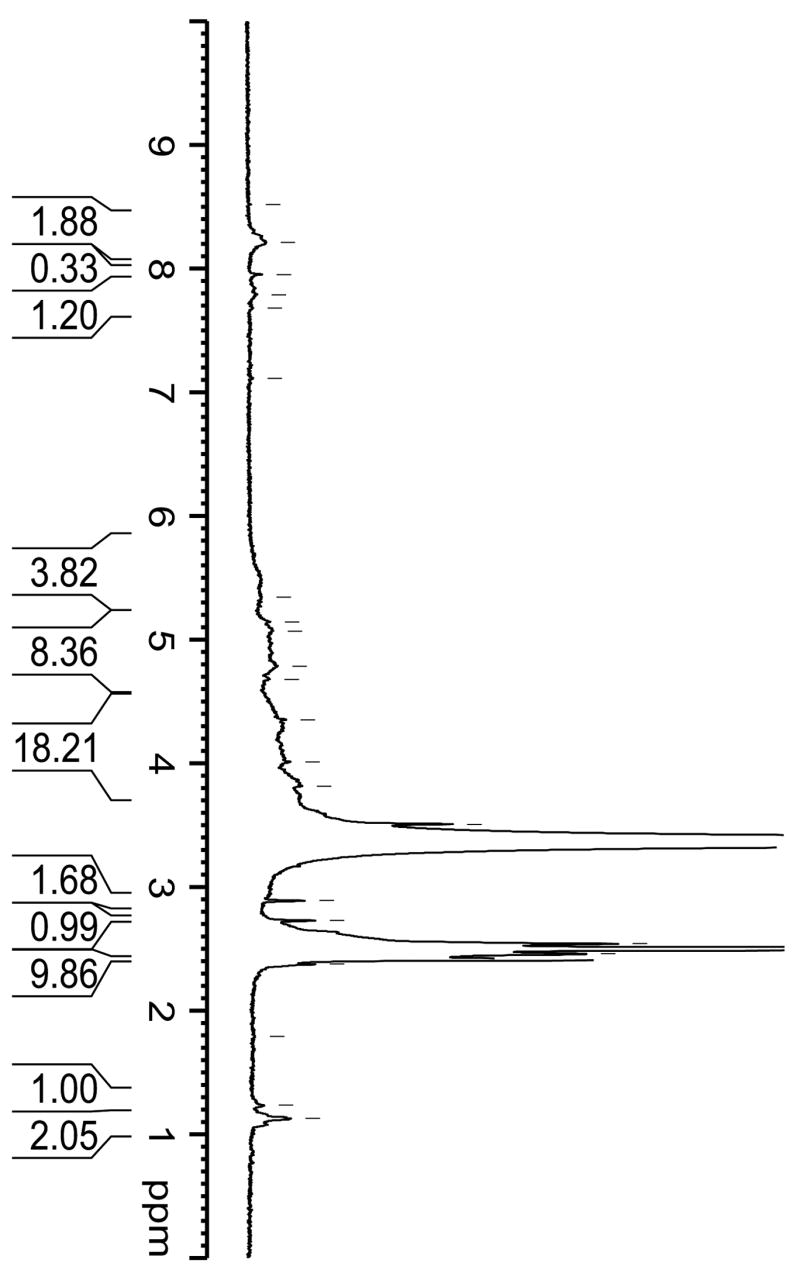
NMR spectrum of the synthesized polyrotaxane-doxorubicin conjugates.
As described previously, the PR-DOX conjugates were synthesized by threading the α-CD residues onto a PEG chain, blocking the two ends of the PEG chain with bulky L-Tyr groups, activating the resulting PR polymer with succinic anhydride (SA), and then linking DNR on the SA-activated groups via hydrolysable ester linkages. Doxorubicin was released by hydrolysis from PR-DOX. The two primary building blocks of PR, α-CD and PEG, are both used widely in pharmaceutical industry and thus considered to be safe and relatively nontoxic. PEG, a well-known biocompatible polymer, only has two functional groups at the terminal ends, whereas PR has abundant functional groups (e.g. –OH), rendering it suitable for multi-component conjugation. Furthermore, both the size and molecular weight of the PR carrier can be easily controlled, either by altering the molecular weight of PEG or by changing the molar ratio between α-CD and PEG during the threading process, to produce the most desirable platform in avoiding renal filtration and reticuloendothelial system clearance.
The release of DOX by hydrolysis from the PR-DOX conjugates was examined in vitro under physiological conditions. As seen in Figure 4, for the first 48 h, a constant release profile was observed without significant burst release of the drug molecules. Approximately, 50% of DOX was released within a period of 30 hours. After 4 days, more than 80% of DOX on the conjugate had been released by hydrolysis. It is fitted with the other drug release kinetics from drug-polymer conjugate linked with ester bond by hydrolysis [28, 29]. This release rate, which was governed primarily by the rate of hydrolysis of the ester bond between DOX and PR, could be readily manipulated by adopting different chemical linkages. An ester bond is weaker than an amide bond and, as a consequence, the release rate of doxorubicin from the PR-drug conjugate connected via amide bond was very slow [29]. To this regard, conjugation method between the PR polymer and doxorubicin via amide bond was generally used. However, in this case the drug content was normally less than 5%. When conjugation was made by using the bromination of the 14th-position in the anthracycline structure of DNR, higher drug content was obtained. Although each PR has about 100 functional groups, 10% of the carboxyethylester (CEE) groups were actually used for conjugation with DNR. This low utilization rate might be attributed to the steric hindrance caused by the attached DNR molecules.
Figure 4.

Doxorubicin release from polyrotaxane-doxorubicin conjugate in PBS (pH 7.4). (n=3)
Selection of the linkage among each component of the polymer-drug conjugate is crucial. It was reported that when DOX was conjugated to polyglutamic acid (PGA) via the amide bond, the PGA-DOX conjugate was not necessarily cytotoxic, since not all the amide bonds were degraded so that a sufficient amount of DOX still stayed on the conjugates and was not released [30]. On the other hand, DNR had been conjugated to PGA via hydrolyzable ester linkage by the reaction of 14-bromodaunorubicin with the carboxylic group of PGA; the same conjugation method employed herein in PR and DNR conjugation. Although the cytotoxic effects were not as potent as that of free DOX against leukemia cells, the PGA-DOX conjugates nevertheless showed antitumor activity along with a low systemic toxicity. Therefore, in designing the polymer-drug conjugates, it is important to select the most desirable linkage with the consideration of several important factors.
In Vitro Cytotoxicity
Cytotoxicity of the PR-drug conjugates were examined in vitro against the A2780 human ovarian cancer cell line using the MTT method. The ID50 values for free DOX, PR-DOX and LMWP-PR-DOX, as determined from the results in Figure 5, were 9.03 (± 5.07), 282.0 (± 78.5), and 60.5 (± 12.2) μM, respectively. While the LMWP-PR-DNR conjugate was about 7-fold less potent in killing cancer cells than free DOX, it nevertheless yielded an approximately 5-fold increase in cytotoxic effect over that of the conjugate without the attachment of the cell-penetrating LMWP peptide. The differences in the in vitro cytotoxicity of these three DOX-containing compounds can be accounted for in terms of the following reasons. Free DOX, a small hydrophobic molecule, can be taken up by cells easily by passive diffusion, whereas macromolecular PR-DOX conjugates are hard to diffuse into the cells. Even though LMWP-PR–DOX conjugate is a water soluble macromolecule and therefore cell impermeable, it was nevertheless taken up by cells to a much greater extent compared to PR-DOX conjugates, apparently due to the demonstrated trans-membrane activity of LMWP [17].
Figure 5.

Cytotoxicity of doxorubicin-polymer conjugates. Free DOX (◆), polyrotaxane-DOX (■), and LMWP-polyrotaxane-DOX (▲) conjugates against A2780 human ovarian cancer cells in log-phase culture. Values represented as mean ± SD. Each experiment was performed in triplicate.
LMWP-Mediated Intracellular Delivery of DOX and Regulation by Heparin and Protamine
DOX and DNR exhibited its own fluorescence with an excitation and emission wavelengths of 470 and 580 nm, respectively. To monitor LMWP-mediated intracellular delivery of the conjugates, the PR chain was labeled with Lissamine Rhodamine B ethylenediamine (Rho; excited at 570 nm and emitted at 590 nm) at the carboxylic acid groups. As shown by the confocal results in Figure 6, DOX molecules were able to diffuse into the cells with ease following 2 hour of incubation with these carcinoma cells, as reflected by the high intensity of green fluorescence within the exposed cells (Figure 6.1). On the other hand, the PR-DOX conjugates were not taken up by the carcinoma cells (Figure 6.2), primarily due to the macromolecular structure of the PR polymer. Conversely, the LMWP-PR-DOX conjugates were able to internalize into the cells, as the green DNR-derived fluorescence and red fluorescence from Rho-labeled polyrotaxane were seen to be overlaid inside the cells (Figure 6.3); implicating that the PR-DOX conjugates successfully internalized the cells due to the cell-penetrating activity of LMWP. When heparin was added, this LMWP-induced cell internalization was markedly abolished (Figure 6.4), apparently due to the electrostatic interactions between the positively-charged LWMP and negatively-charged heparin molecules. Some translocation seemed to occur with the heparin-masked LMWP-PR-DOX conjugates, as a low intensity of both the red and green fluorescence was observed inside the cells. One hypothesis was that an insufficient amount of heparin was added to the LMWP-PR-DOX conjugates, rendering its inhibition on LMWP incomplete. Another possible explanation may be competition of cell surface heparin sulfate moieties with the administered heparin inhibitor. When protamine was added, the heparin-induced inhibition was nearly completely reversed (Figure 6.5), as an overlaid image of strong intensities of both the green fluorescence from DOX and red fluorescence from the Rho marker on the PR chain re-appeared.
Figure 6.
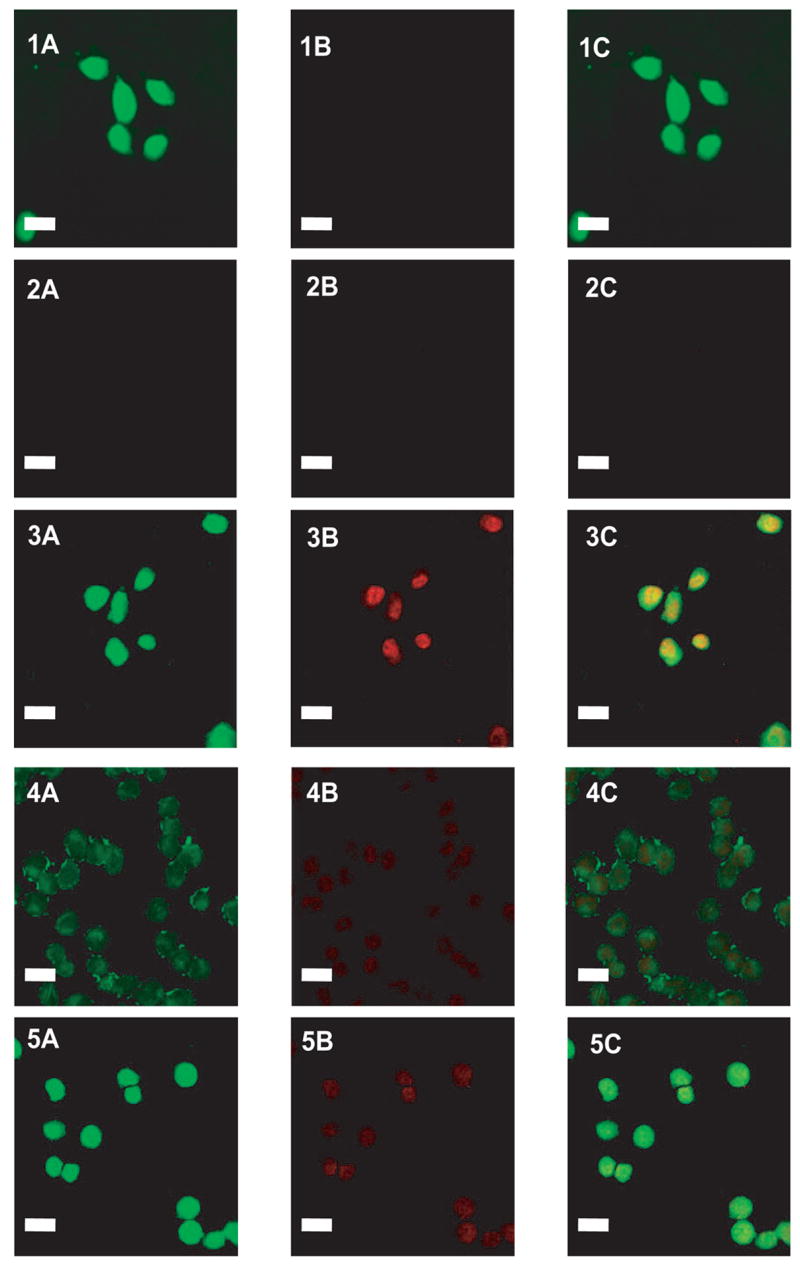
Cellular localization of Lissamine Rhodamine B ethylenediamine-labeled LMWP-drug conjugates in A2780 human ovarian carcinoma cells. Free DOX, Lissamine Rhodamine B ethylenediamine(RHO)-labeled polyrotaxane-DOX, Rho-labeled LMWP-polyrotaxane-DOX, Rho-labeled LMWP-polyrotaxane-DOX and heparin mixture, and Rho-labeled LMWP-polyrotaxane-DOX, heparin, and protamine mixture were overlaid onto cultured A2780 cells in the presence of 10% FBS. Cellular localization was monitored by confocal microscopy. (1) Free DOX; (2) Rho-labeled polyrotaxane-DOX; (3) Rho-labeled LMWP-polyrotaxane-DOX; (4) Rho-labeled LMWP-polyrotaxane-DOX and heparin mixture; (5) Rho-labeled LMWP-polyrotaxane-DOX, heparin, and protamine mixture. (A) 488 nm (green) detection; (B) 560 nm (red) detection; (C) overlaid (A+B) Bar is 20 μm.
Some DOX molecules would indeed be released from LMWP-PR-DOX by hydrolysis. Nevertheless, the fluorescence seen inside the cells after 2 hr incubation was primarily attributed to the conjugates rather than free drug, simply because less than 10% of DOX was hydrolyzed the conjugates after 2 h. The Z-section method of confocal microscopy was performed and it gave the same pattern as those seen in Figure 6, indicating that the images were from internalized conjugates rather than those adsorbed in cell surfaces. Consistent with these findings, flow cytometric analysis also confirmed that LMWP was able to ferry polyrotaxane into the carcinoma cells (data was not shown). Further validation of this heparin- and protamine-induced regulation on the uptake of the PR-drug conjugates by tumor cells will be carried out in animal studies.
Whereas the well-known cell-penetrating peptides such as TAT, and VP-22 were derived from viral resources, LMWP was obtained directly from protamine; a widely employedclinical drug that is relatively non-toxic [17]. In addition, heparin and protamine are extensively used clinically following certain major surgical procedures (e.g. cardiopulmonary bypass). Therefore, all the components of this new drug delivery are deemed to be both nontoxic and safe.
Taken together, the LMWP-PR-DOX conjugate appears to offer a great potential for intracellular delivery of therapeutic agents into tumor cells in achieving highly effective yet safe drug therapy. Extensive animal studies are currently in progress to further validate this novel drug delivery system.
Acknowledgments
This work was supported in part by NIH Grants R01 HL55461 and R01 CA114612. Dr. Victor C. Yang is currently a Cheung Kong Scholar offered by the Chinese Ministry of Education at the School of Chemical Engineering, Tianjin University.
The authors would like to appreciate Mr. Bruce Donohue (Microscopy and Image Analysis Laboratory, Department of Cell and Development Biology, University of Michigan) for technical assistances in conducting confocal microscopy observation, and Dr. Scott Woehler (Biomedical Research Core Facilities, University of Michigan) for conducting NMR works.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jain RK. Delivery of molecular medicine to solid tumors: lessons from in vivo imaging of gene expression and function. J Control Release. 2001;74(1–3):7–25. doi: 10.1016/s0168-3659(01)00306-6. [DOI] [PubMed] [Google Scholar]
- 2.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 3.Dvorak +HF, Nagy JA, Dvorak JT, Dvorak AM. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am J Pathol. 1988;133(1):95–109. [PMC free article] [PubMed] [Google Scholar]
- 4.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. [PubMed] [Google Scholar]
- 5.Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74(1–3):47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- 6.Greco F, Vicent MJ, Gee S, Jones AT, Gee J, Nicholson RI, Duncan R. Investigating the mechanism of enhanced cytotoxicity of HPMA copolymer-Dox-AGM in breast cancer cells. J Control Release. 2007;117(1):28–39. doi: 10.1016/j.jconrel.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Greenwald RB. PEG drugs: an overview. J Control Release. 2001;74(1–3):159–171. doi: 10.1016/s0168-3659(01)00331-5. [DOI] [PubMed] [Google Scholar]
- 8.Singer JW. Paclitaxel poliglumex (XYOTAX, CT-2103): a macromolecular taxane. J Control Release. 2005;109(1–3):120–126. doi: 10.1016/j.jconrel.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 9.Eguchi M, Ooya T, Yui N. Controlling the mechanism of trypsin inhibition by the numbers of alpha-cyclodextrins and carboxyl groups in carboxyethylester-polyrotaxanes. J Control Release. 2004;96(2):301–307. doi: 10.1016/j.jconrel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27(2):85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285(5433):1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 12.Chauhan A, Tikoo A, Kapur AK, Singh M. The taming of the cell penetrating domain of the HIV Tat: myths and realities. J Control Release. 2007;117(2):148–162. doi: 10.1016/j.jconrel.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272(25):16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 14.Elliott G, O’Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88(2):223–233. doi: 10.1016/s0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 15.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269(14):10444–10450. [PubMed] [Google Scholar]
- 16.Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem. 2002;277(4):2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 17.Park YJ, Chang LC, Liang JF, Moon C, Chung CP, Yang VC. Nontoxic membrane translocation peptide from protamine, low molecular weight protamine (LMWP), for enhanced intracellular protein delivery: in vitro and in vivo study. Faseb J. 2005;19(11):1555–1557. doi: 10.1096/fj.04-2322fje. [DOI] [PubMed] [Google Scholar]
- 18.Vives E. Present and future of cell-penetrating peptide mediated delivery systems: “is the Trojan horse too wild to go only to Troy?”. J Control Release. 2005;109(1–3):77–85. doi: 10.1016/j.jconrel.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 19.Han G, Tamaki M, Hruby VJ. Fast, efficient and selective deprotection of the tert-butoxycarbonyl (Boc) group using HCl/dioxane (4 m) J Pept Res. 2001;58(4):338–341. doi: 10.1034/j.1399-3011.2001.00935.x. [DOI] [PubMed] [Google Scholar]
- 20.Arcamone F, Franceschi G, Penco S. Process for the preparation of adriamycin and adriamycinone and adriamycin derivatives. 3803124. US Patent. 1974 April 9;
- 21.Tanaka H, Kominato K, Yamamoto R, Yoshioka T, Nishida H, Tone H, Okamoto R. Synthesis of doxorubicin-cyclodextrin conjugates. J Antibiotics. 1994;47(9):1025–1029. doi: 10.7164/antibiotics.47.1025. [DOI] [PubMed] [Google Scholar]
- 22.Hruby M, Konak C, Ulbrich K. Polymeric micellar pH-sensitive drug delivery system for doxorubicin. J Control Release. 2005;103(1):137–148. doi: 10.1016/j.jconrel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Snyder SL, Sobocinski PZ. An improved 2,4,6-trinitrobenzenesulfonic acid method for the determination of amines. Anal Biochem. 1975;64(1):284–288. doi: 10.1016/0003-2697(75)90431-5. [DOI] [PubMed] [Google Scholar]
- 24.Kang HC, Kim S, Lee M, Bae YH. Polymeric gene carrier for insulin secreting cells: poly(L-lysine)-g-sulfonylurea for receptor mediated transfection. J Control Release. 2005;105(1–2):164–176. doi: 10.1016/j.jconrel.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 25.Ramanathan S, Qiu B, Pooyan S, Zhang G, Stein S, Leibowitz MJ, Sinko PJ. Targeted PEG-based bioconjugates enhance the cellular uptake and transport of a HIV-1 TAT nonapeptide. J Control Release. 2001;77(3):199–212. doi: 10.1016/s0168-3659(01)00474-6. [DOI] [PubMed] [Google Scholar]
- 26.Harada A, Kamachi M. Complex formation between poly(ethylene glycol) and -cyclodextrin. Macromolecules. 1990;23(10):2821–2823. [Google Scholar]
- 27.Ooya T, Yui N. Multivalent interactions between biotin-polyrotaxane conjugates and streptavidin as a model of new targeting for transporters. J Control Release. 2002;80(1–3):219–228. doi: 10.1016/s0168-3659(02)00030-5. [DOI] [PubMed] [Google Scholar]
- 28.Sugahara S, Kajiki M, Kuriyama H, Kobayashi TR. Paclitaxel delivery systems: the use of amino acid linkers in the conjugation of paclitaxel with carboxymethyldextran to create prodrugs. Biol Pharm Bull. 2002;25(5):632–641. doi: 10.1248/bpb.25.632. [DOI] [PubMed] [Google Scholar]
- 29.Schoenmakers RG, van de Wetering P, Elbert DL, Hubbell JA. The effect of the linker on the hydrolysis rate of drug-linked ester bonds. J Control Release. 2004;95(2):291–300. doi: 10.1016/j.jconrel.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Hoes CJT, Potman W, van Heeswijk WAR, Mud J, de Grooth BG, Greve J, Feijen J. Optimization of macromolecular prodrugs of the antitumor antibiotic Adriamycin. J Control Release. 1985;2:205–213. [Google Scholar]