

Abstract
The purpose of this study was to determine the effect of thymosin beta 4 (Tβ4) on NFκB protein levels, activation, phosphorylation, and nuclear translocation in a model of tumor necrosis factor (TNF)-α-mediated corneal inflammation. Transformed and primary (HCET and HCEC) human corneal epithelial cells were stimulated with the pro-inflammatory cytokine TNF-α and treated or not with Tβ4. Nuclear NFκB p65 subunit protein levels were assayed using ELISA, and activity was measured by determining NFκB binding to consensus oligonucleotides. NFκB p65 protein phosphorylation was also measured by ELISA. Nuclear translocation of NFκB p65 subunit was assayed by immunofluorescence microscopy. Compared to non-treated controls, Tβ4 treatment significantly decreased nuclear NFκB protein levels, NFκB activity and p65 subunit phosphorylation in corneal epithelial cells after TNF-α stimulation. In TNF-α-stimulated corneal epithelial cells, NFκB p65 subunit translocation to the nucleus was observed using immunofluorescence microscopy. In contrast, Tβ4 blocked nuclear translocation of the NFκB p65 subunit in TNF-α-stimulated corneal epithelial cells. TNF-α initiates cell signaling pathways that converge on the activation of NFκB, thus both are known mediators of the inflammatory process. Tβ4, a protein with diverse cellular functions including wound healing and suppression of inflammation, inhibits the activation of NFκB in TNF-α-stimulated cells. These results have important clinical implications for the potential role of Tβ4 as a corneal anti-inflammatory agent.
Keywords: corneal epithelium, inflammation, nuclear factor kappa B, thymosin beta 4, tumor necrosis factor alpha
1. Introduction
The corneal wound repair process is intricately linked to a complex inflammatory response that must be precisely regulated to ensure proper healing and optimal visual outcome. Many studies have contributed information regarding the roles of cytokine and chemokine expression and activity in the post-wound corneal inflammatory response in a wide array of clinical pathologies (Gillitzer and Goebeler, 2001; Wilson et al., 2003; Agrawal and Tsai, 2003; Stramer et al., 2004;). For example, in chemically injured corneal epithelial cells, the levels of pro-inflammatory cytokines and chemokines are upregulated (Planck et al., 1997; Sotozono et al., 1997; Sosne et al., 2002). Dry eye induced experimentally in mice, also stimulates production and expression of TNF-α (Luo et al., 2004). Additionally corneal epithelial monolayers infected with Pseudomonas aeruginosa demonstrated increased expression and secretion of IL-6, IL-8, and TNF-α (Zhang et al., 2005).
Many studies suggest that TNF-α is a potent pro-inflammatory cytokine considered to be a central mediator of the inflammatory response. It regulates antimicrobial defenses, wound healing, defense against malignancies, and apoptotic cell death (Zhang et al., 2004). One consequence of the activation of signal transduction pathways subsequent to TNF-α stimulation is the activation of transcription factors necessary for the induction of chemokine gene expression (Baud and Karin 2001; Ritchie et al., 2004). One major transcription factor is NFκB, formed by the heterodimerization or homodimerization of proteins of the Rel family, the two most important of which are p50 and p65 (Hanada and Yoshimura, 2002). NFκB mediates diverse biological processes, from inflammation to apoptosis (Hayden and Ghosh, 2004). In unstimulated cells, NFκB dimers are located in the cytoplasm. The family of inhibitory proteins, IκBs, binds to NFκB, masks its nuclear localization signal, and keeps it in the cytoplasm. Various extracellular stimuli, including TNF-α act through different signaling pathways to converge on the activation of IκB kinase (IKK). Phosphorylated IκB is degraded and released from the NFκB dimer, permitting the translocation of NFκB to the nucleus. Nuclear NFκB subsequently binds to κB enhancer elements of target genes (Karin and Ben-Neriah, 2000). Because of its ability to regulate the expression of inflammatory proteins, NFκB is believed to play a major role in the inflammatory process.
Thymosin beta-4 (Tβ4) is a water-soluble, 43-amino acid acidic polypeptide (pI 5.1) with a molecular weight of 4.9 kDa (Low et al., 1981; Low and Goldstein, 1982; Goodall et al., 1983; Yu et al., 1994). Tβ4 is a ubiquitous polypeptide, highly conserved across species, and is found at concentrations of 1 × 10−5 to 5.6 × 10−1 M in a variety of tissues and cell types (Hannappel et al., 1982; Hannappel and Leibold, 1985; Hannappel and van Kampen, 1987). Previously, we reported that Tβ4 promotes corneal wound healing, decreases inflammation, and modulates the MMP/TIMP balance in a mouse model of corneal alkali injury (Sosne et al., 2002, 2005). Although the mechanism(s) of action of exogenous Tβ4 on corneal wound repair and suppression of inflammation remain unclear, we hypothesize that Tβ4 may interfere with NFκB signaling pathways that are central to the inflammatory response. In this report, we extend our analysis of the interrelationship between Tβ4 and corneal inflammation by providing evidence that Tβ4 suppresses NFκB phosphorylation, activity, and nuclear translocation in cultured human corneal epithelial cells stimulated with TNF-α. Our results suggest that Tβ4 may exert its anti-inflammatory effects by regulating the activity of NFκB, a key modulator of inflammation.
2. Methods
2.1. Human Corneal Epithelial Cell Culture
Non-transformed human corneal epithelial cells (HCEC) at passage 3 were purchased from Cascade Biologics (Portland, OR). HCEC were rapidly thawed, seeded onto the appropriate tissue culture plastic substrate, and cultured in serum-free EpiLife medium containing human corneal growth supplement as suggested by Cascade. HCEC were used for experiments at passage 4. The transformed human corneal epithelial cell line 10.014 pRSV-T (HCET) was additionally used in this study (Kurpakus et al., 1999). HCET were maintained in serum-free KGM on tissue culture plastic coated with a fibronectin-collagen type I matrix. Cells were cultured in a standard tissue culture incubator at 37°C and in an atmosphere of 5% CO2 and 95% room air with medium changes every two days.
2.2. NFκB Activity Assay
HCET or HCEC at approximately 80% confluence were pre-treated with synthetic Tβ4 (1 µg/ml, a gift from RegeneRx, Inc., Bethesda, MD) for 1 h. Tβ4 was used at a concentration of 1 µg/ml in these studies because we have demonstrated this to be an optimum dosage for epithelial cells (Sosne et al., 2004, 2006). Dose-response assays showed that in corneal epithelium Tβ4 at a concentration of 1 µg/ml provides maximum inhibitory protection against the pro-inflammatory effects of TNF-α compared to lower or higher concentrations (results not shown). Cells were then treated with TNF-α (10 ng/ml, R&D Systems, Minneapolis, MN) in the presence or absence of Tβ4 for 5, 15, 30, 45, or 60 min. The concentration of TNF-α used in these studies has been shown to be an effective pro-inflammatory agent in other cell systems (Zhang et al., 2005). Dose response studies with corneal epithelium (results not shown) showed that similarly to other studies (Zhang et al., 2006), the 10 ng/ml concentration produced a significant inflammatory response in vitro but did not result in cytotoxicity. Cells maintained in culture medium only, or Tβ4 only, for 30 min were used as controls. At each time point the culture medium was removed and the adherent cells were scraped from the dish into 1 ml of cold PBS. The solution was centrifuged at 3000 rpm for 5 min. The cell pellet was lysed in 1 ml of cell extraction buffer provided with the activity assay kit (TransAm NFκB p65 Transcription Factor Assay Kit, Active Motif, Carlsbad, CA) on ice for 30 min, with vortexing every 10 min. The extract was centrifuged at 13,000 rpm for 10 min at 4°C and the supernatant was used for activity assays following the kit protocol. In addition to the activity assay, the ELISA for total NFκB p65 subunit protein was completed simultaneously using aliquots of the same supernatants. ELISA values were used to standardize the activity values to correct for differences in protein concentration in each sample. The assays were repeated three times (n = 3) and the results of a representative assay are reported as NFκB activity in arbitrary units ± SEM. Statistical analysis was performed using the unpaired Student’s t-test with significance set at P < 0.05.
2.3. NFκB Phosphorylation Assay
HCET were cultured in wells of a 96-well plate until approximately 80% confluent. Cells were pre-treated with Tβ4 (1 µg/ml, produced synthetically by RegeneRx, Inc., Bethesda, MD) for 1 h. Cells were then treated with TNF-α (10 ng/ml) in the presence or absence of Tβ4 for 5, 15, 30, 45, or 60 min. The concentration of TNF-α used in these studies produces a significant inflammatory response but does not result in cytotoxicity. Cells maintained in culture medium only, or Tβ4 only, for 30 min were used as controls. At each time point adherent cells were fixed in cell fixing buffer containing 4% glutaraldehyde according to the phosphorylation assay kit protocol (CASE, cellular activation of signaling ELISA for phosphoserine 276 of NFκB p65 subunit, SuperArray, Frederick, MD). Fixed cells were then processed using phospho-specific (phosphoserine 276 p65) or pan-protein specific (nonphosphorylated p65) primary antibodies and HRP-conjugated secondary antibodies. Following incubation with developing and stop solutions provided with the kit, individual well absorbance was read at 450 nm. Variation in cell number in individual wells was corrected by assaying the OD 540 nm of stained cells following completion of the phosphorylation assay and performing numerical correction of the OD 450 nm values. The assay was repeated twice (n = 8) and a representative result is presented as relative amounts of phosphorylated and nonphosphorylated NFκB in arbitrary units ± SEM. Statistical analysis was performed using the unpaired Student’s t-test with significance set at P < 0.05.
2.4. NFκB Protein Levels
HCET were cultured until approximately 80% confluent. Cells were pre-treated with Tβ4 (1 µg/ml) for 1 h. Cells were then treated with TNF-α (10 ng/ml) in the presence or absence of Tβ4 for various times. Cells maintained in culture medium only, or Tβ4 only were used as controls. The culture medium was removed and the adherent cells were scraped from the dish into 1 ml of cold PBS. The solution was centrifuged at 3000 rpm for 5 min. The cell pellet was resuspended in 0.5 ml hypotonic buffer (20 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2) and incubated on ice for 15 min. 25 µl of 10% NP-40 was added with vortexing. The homogenate was centrifuged for 10 min at 3000 rpm at 4°C. The pellet was resuspended in 100 µl of the cell extraction buffer and incubated for 30 min on ice with vortexing every 10 min. The homogenate was centrifuged at 14,000 rpm for 30 min at 4°C. The resulting supernatant was used for the NFκB total p65 subunit ELISA. A representative analysis is presented as NFκB protein (pg/ml) ± SEM. Statistical analysis was performed using the unpaired Student’s t-test with significance set at P < 0.05.
2.5. Nuclear Translocation of NFκB
HCET were cultured on collagen-coated 4 chambered glass microscope slides to approximately 80% confluence. Cells were pre-treated with Tβ4 (1 µg/ml) for 1 h. Cells were then treated with TNF-α (10 ng/ml) in the presence or absence of Tβ4 for 30 min. Cells maintained in culture medium only, or Tβ4 only were used as controls. Cells were fixed in 3.7% glutaraldehyde/PBS for 10 min then washed in PBS. Cells were permeabilized in 1% Triton X-100 for 5 min and washed again. The cells were then incubated with rabbit polyclonal antibodies specific for the N-terminus of human p65 NFκB (Santa Cruz Biotechnologies, Santa Cruz, CA) followed by FITC-conjugated goat anti-rabbit secondary antibodies. Digitized images were captured and stored (Axiophot fluorescence microscopy and imaging system, Carl Zeiss Medicine, Inc., Thornwood, NY).
3. Results
3.1. NFκB Activity in Human Corneal Epithelial Cells
We hypothesized that Tβ4 functions as a transcription factor mediator in addition to its other known cellular functions. To provide support for this hypothesis, we examined the effect of Tβ4 on the activity of NFκB in human corneal epithelial cells stimulated with TNF-α in vitro. Cultured cells were employed for these studies because sufficient material for detailed biochemical analyses could be easily generated. Transcription factor activity was analyzed colorimetrically using an assay that measured the ability of NFκB to bind to oligonucleotides containing consensus kB binding motifs immobilized to a plastic substrate. Whole-cell lysates were used for these analyses per protocol since activated NFκB is presumed to be located only within the nucleus.
In the transformed human corneal epithelial cell line HCET, treatment of cells with TNF-α resulted in a progressively increased level of NFκB activity commensurate with time of stimulation (Fig. 1). The treatment of TNF-α stimulated cells with Tβ4 resulted in increased NFκB activity at 5, 10 (results not shown), 15 (Fig. 1), and 20 (results not shown) min of stimulation compared to cells stimulated with TNF-α. However, at 30 and 60 min, Tβ4 treatment significantly suppressed TNF-α induced activation of NFκB, and activity approached that of cells treated with Tβ4 alone.
Fig. 1.
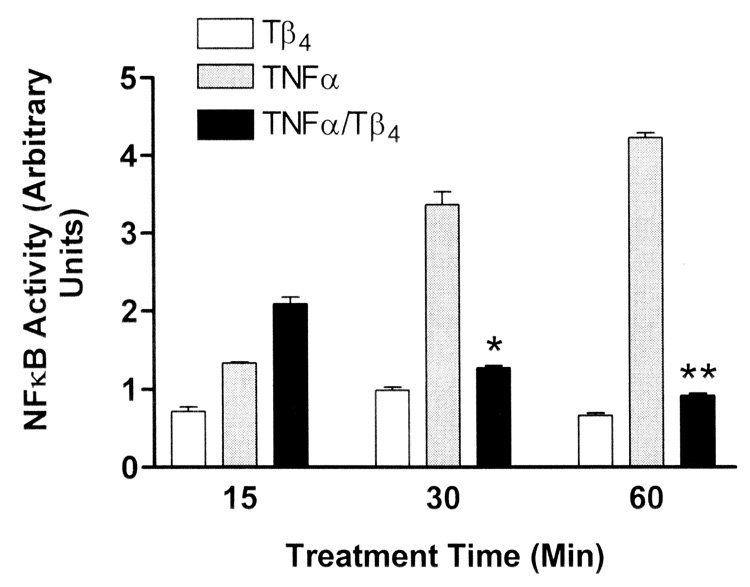
Tβ4 suppresses NFκB activity in human corneal epithelial cells in vitro. Transformed human corneal epithelial cells (HCET) were treated with 1 µg/ml Tβ4 only (open bars) or stimulated with 10 ng/ml TNF-α (gray bars) only. HCET were also stimulated with TNF-α and simultaneously treated with Tβ4 (black bars) for the times indicated. At assay times less than 20 min, Tβ4 treatment resulted in increased NFκB activity in TNF-α stimulated cells (only the 15 min data is shown). In contrast by 30 min of Tβ4 treatment, NFκB activity was significantly suppressed in TNF-α stimulated cells (* P = 0.0068, ** P < 0.0001). Note that NFκB activity in HCET treated with Tβ4 alone is consistently lower than in cells treated with TNF-α. NFκB activity in media control cells was negligible and is not shown.
Although transformed cell lines provide ample material for analyses, experimental results can be of concern since it is not clearly known how the cells may differ from in vivo counterparts. To complement NFκB activity assays in transformed human corneal epithelial cells, we also performed analyses in non-transformed human corneal epithelial culture and found similar results of decreased NFκB activity with Tβ4 treatment after TNF-α stimulation (results not shown). Based on these results, we were confident that the transformed line, HCET, could be used for further analysis of NFκB dynamics in Tβ4-treated, and TNF-α-stimulated, conditions.
3.2. Effect of Tβ4 on NFκB Phosphorylation
In addition to nuclear translocation, the p65 subunit of NFκB must undergo phosphorylation on several serine residues in order to be activated. It has been reported that serine 276 is the major phosphorylation site of p65 and its phosphorylation is essential for p65-dependent cellular responses (Okazaki et al., 2003). Since our studies suggested that Tβ4 could suppress inflammatory mediator induced NFκB activation, we next determined the ability of Tβ4 to regulate the phosphorylation of the serine 276 residue of the p65 subunit of NFκB. We used an ELISA approach that allowed for the measurement of phosphorylated and nonphosphorylated NFκB levels on groups of identically treated cells. Commensurate with increased NFκB activity in corneal epithelial cells treated with TNF-α, we observed an increase in the relative percentage of cellular NFκB that was phosphorylated (Fig. 2). At 30 and 60 min of TNF-α stimulation, essentially all of the NFκB was phosphorylated. At these time points, treatment of TNF-α-stimulated corneal epithelial cells with Tβ4 significantly reduced NFκB phosphorylation levels (statistical analysis shown in Figure 2 legend). In contrast to the 30 and 60 min time points, at 5 and 45 min time points, treatment of TNF-α-stimulated corneal epithelial cells with Tβ4 resulted in a slight increase in NFκB phosphorylation (statistical analysis shown in Figure 2 legend). Evidence for oscillatory dynamics of NFκB nuclear translocation and phosphorylation levels has been published (Nelson et al., 2005; Chickarmane et al., 2006). Our NFκB phosphorylation patterns observed at the 5 and 45 min time points suggest that similar oscillatory dynamics are evident in our model.
Fig. 2.
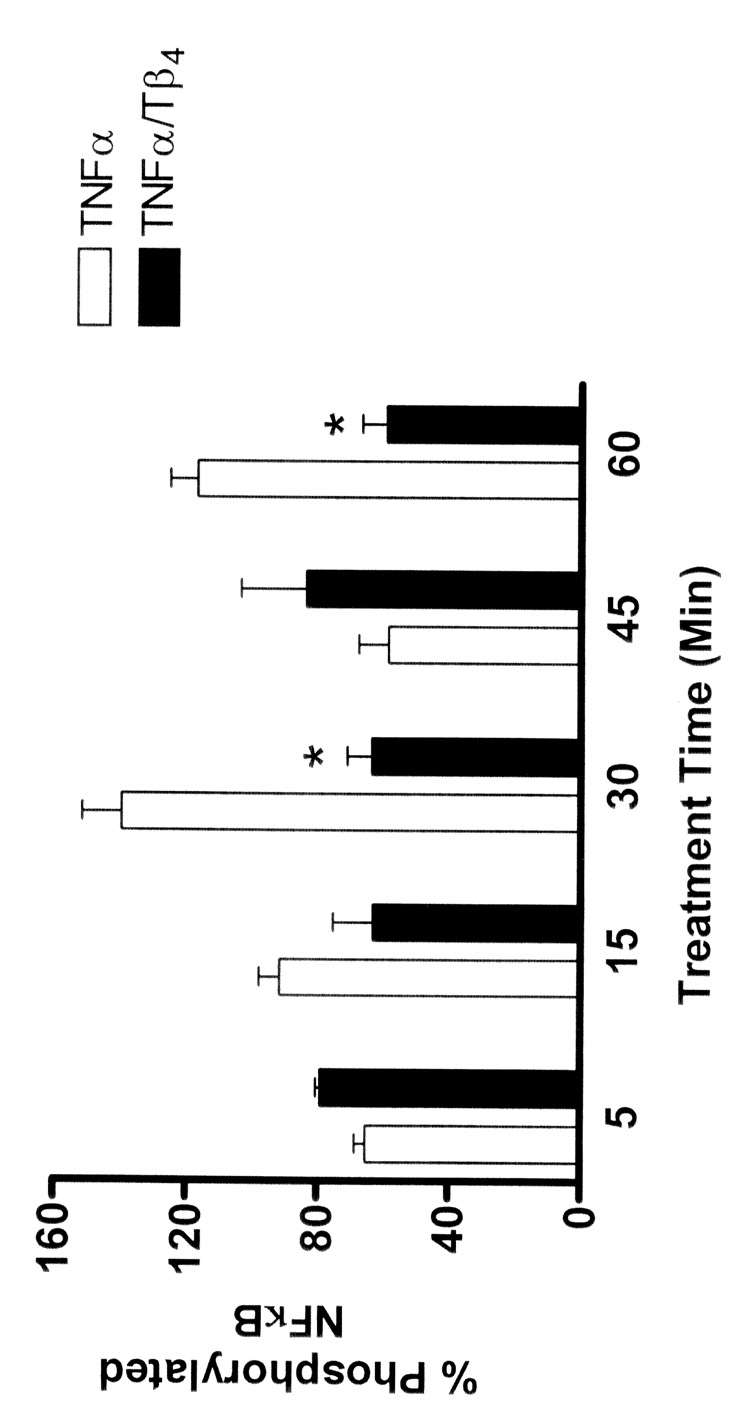
Tβ4 suppresses NFκB phosphorylation in human corneal epithelial cells in vitro. HCET were stimulated with TNF-α in the presence or absence of Tβ4 for the times indicated. Using an ELISA approach, relative levels of phosphorylated (serine 279 of p65) and nonphosphorylated NFκB were measured. In cells stimulated with TNF-α essentially all of the NFκB p65 is phosphorylated by 30 min. Treatment of TNF-α stimulated cells with Tβ4 significantly inhibits the phosphorylation of NFκB (* at 30 min P = 0.0056, * at 60 min P = 0.0066). In contrast, at 5 and 45 min, treatment of TNF-α stimulated cells with Tβ4 increased the phosphorylation of NFκB (P = 0.0181 and P = 0.3133, respectively).
We focused on an analysis of phosphorylation levels at 30 minutes of TNF-α stimulation because our studies as shown in Figure 1 revealed that Tβ4 could significantly suppress NFκB activity at this time point. In media control cells and in cells treated with Tβ4 only, levels of nonphosphorylated NFκB were significantly higher than levels of phosphorylated NFκB (Fig. 3). Conversely, in cells stimulated with TNF-α for 30 min essentially all of the NFκB was in the phosphorylated form. In contrast, in cells that were treated with Tβ4 during TNF-α stimulation, the levels of phosphorylated NFκB were significantly decreased.
Fig. 3.
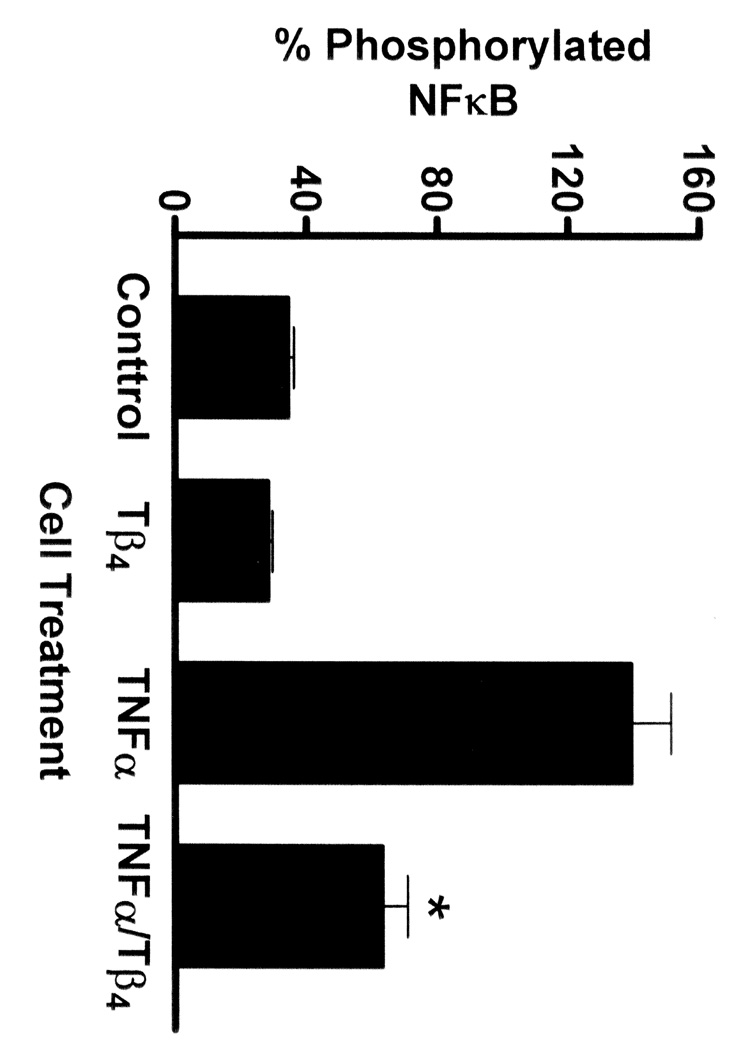
Tβ4 suppresses NFκB phosphorylation in human corneal epithelial cells in vitro. HCET were treated for 30 min as indicated. Using an ELISA approach, relative levels of phosphorylated (serine 279 of p65) and nonphosphorylated NFκB were measured. In media control cells and in cells treated with Tβ4 only, most of the NFκB exists in the nonphosphorylated form. In contrast, in cells stimulated with TNF-α essentially all of the NFκB p65 is phosphorylated. Treatment of TNF-α stimulated cells with Tβ4 significantly inhibits the phosphorylation of NFκB (* P = 0.0056).
3.3. Effect of Tβ4 on NFκB Nuclear Translocation
Nuclear translocation is an essential downstream event in NFκB mediated gene transcription. Inhibition of nuclear translocation by Tβ4 would provide additional support to demonstrate that this molecule plays a role in the regulation of NFκB inflammatory processes. HCET were treated for 30 min and the amount of NFκB protein in nuclear fractions was measured by ELISA. Only activated, phosphorylated NFκB is capable of translocating to the nucleus. Compared to media control cells or cells treated with Tβ4 only, the amount of NFκB protein measured in the nuclear fraction of cells treated with TNF-α was significantly increased (Fig. 4). However, treatment of cells with Tβ4 during TNF-α activation resulted in significant decrease of measurable NFκB protein in nuclear fractions. This suggests that Tβ4 suppressed nuclear translocation of NFκB under conditions of TNF-α stimulation.
Fig. 4.
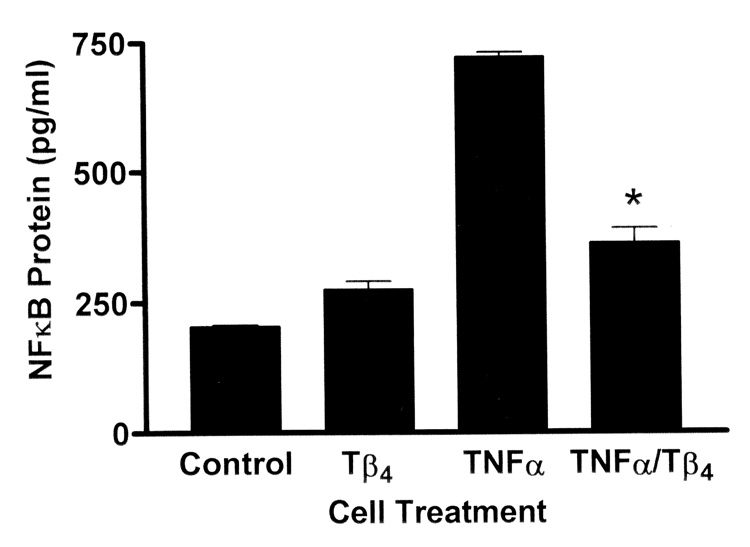
Tβ4 suppresses NFκB nuclear translocation in human corneal epithelial cells in vitro. HCET were treated for 30 min as indicated. Using an ELISA approach, the relative levels of NFκB in the nuclear fractions of cell lysates were measured. Note the significant increase in nuclear NFκB protein in cells treated with TNF-α only compared to media control cells or cells treated with Tβ4 only. Treatment of TNF-α stimulated cells with Tβ4 significantly reduces the level of nuclear NFκB protein (* P = 0.0066).
The nuclear translocation of NFkB was also examined morphologically in HCET by immunofluoresence microscopy using an antibody specific for the p65 subunit (Fig. 5). In both media control cells (Fig. 5A) and cells treated with Tβ4 only (Fig. 5B), NFκB p65 protein was localized to the cytoplasm. In cells treated with TNF-α only NFκB showed a predominantly nuclear localization, indicating its translocation in response to TNF-α signaling (Fig. 5C). In cells treated with Tβ4 during the period of TNF-α stimulation the fluorescence pattern appeared to be mainly perinuclear (Fig. 5D), although evidence of both cytoplasmic and nuclear NFκB localization could be observed.
Fig. 5.
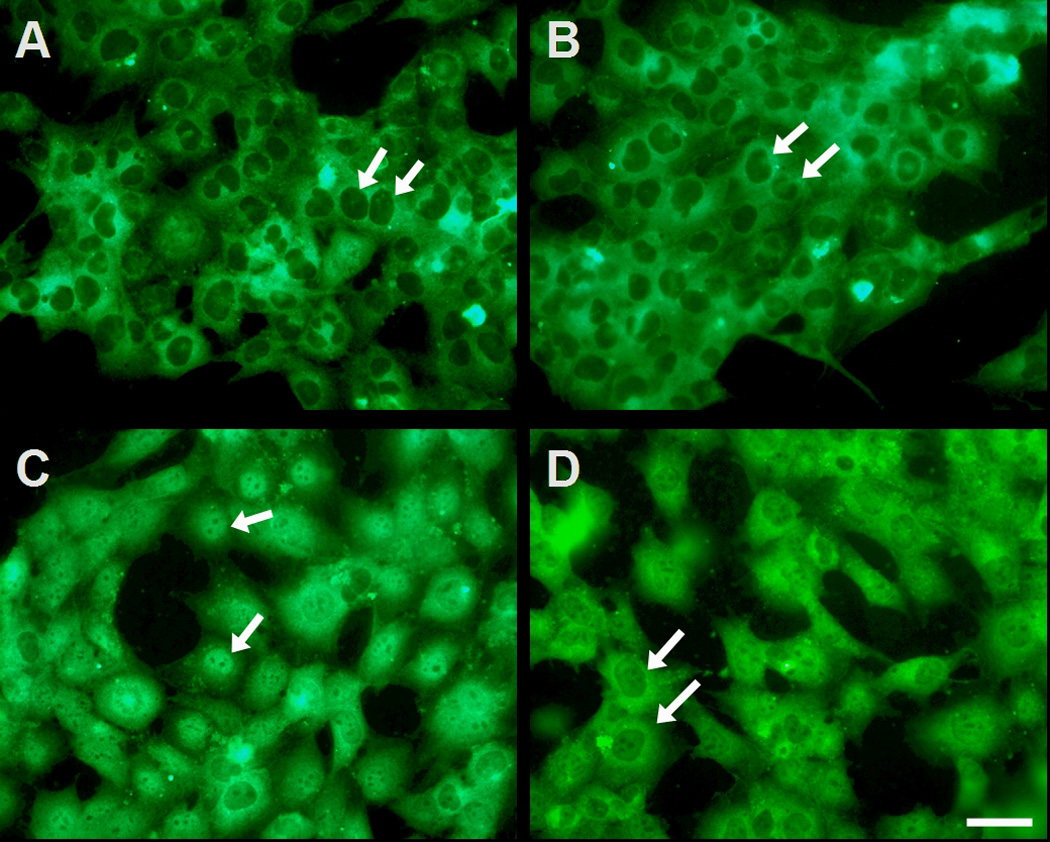
Tβ4 suppresses NFκB nuclear translocation in human corneal epithelial cells in vitro. HCET were cultured on glass microscope slides and maintained in culture medium (A), treated with Tβ4 only (B), with TNF-α only (C), or with TNF-α and Tβ4 (D). Cells were processed for immunofluorescence microscopy using an antibody specific for p65 subunit of NFκB. Note the lack of nuclear labeling in media control cells (arrows in A) or cells treated with Tβ4 only (arrows in B). In contrast note the almost exclusive nuclear localization of NFκB in cells treated with TNF-α (arrows in C). The presence of Tβ4 during TNF-α stimulation appears to prohibit the nuclear translocation of NFκB, as there is a predominant perinuclear localization pattern in these cells (arrows in D). Bar, 50 µm.
4. Discussion
When epithelium is injured, the cornea responds by synthesizing several cytokines, growth factors and tissue remodeling molecules. Pro-inflammatory cytokines have been implicated in the inflammation that follows corneal epithelial injury, and cytokine-mediated processes play a significant role in corneal epithelial wound healing (Brazzell et al., 1991; Sotozono et al., 1995; Lambiase et al., 1998; Chandrasekher et al., 2002). In addition, the transcription factor, NFκB, regulates various genes involved in inflammatory responses and its activation is important for host defense and repair after insult (Hayden and Ghosh, 2004).
As reviewed recently, (Goldstein et al., 2005), Tβ4 has clear anti-inflammatory and wound healing activities. We previously reported that topical application of Tβ4 accelerates corneal re-epithelialization and modulates corneal cytokine production such as interleukin (IL)-1β and IL-18) in rat corneas following scrape wound and it also stimulates in vitro human corneal epithelial cell migration (Sosne et al., 2001). We further demonstrated in vivo using an inflammatory corneal alkali injury model that Tβ4 promotes corneal wound healing and down-regulates a number of key inflammatory cytokines, e.g., IL-1β, and chemokines, e.g., macrophage inflammatory (MIP)-1α, MIP-1β, and MIP-2 (Sosne et al., 2002, 2005).
Despite our advances in establishing the anti-inflammatory and pro-wound healing properties of Tβ4, the mechanism of how Tβ4 exerts these effects remains unclear. It is well established by other investigators that NFκB activation plays a central role in inflammatory aspects of the wound healing process (Baldwin, 1996). A majority of inflammatory cytokines and chemokines use NFκB related pathways for signaling following ligand binding to cell surface receptors (Schmidt et al., 2003). To extend our work regarding the role of Tβ4 as an anti-inflammatory and wound healing agent, the present study explores the effects of Tβ4 on suppressing NFκB levels and activity following TNF-α stimulation in corneal epithelial cells. This is the first report, to our knowledge, that documents that Tβ4 can suppress NFκB activation after a pro-inflammatory stimulation with TNF-α.
The use of TNF-α as a pro-inflammatory stimulus in our cell culture model has important physiological relevance to corneal inflammation and epithelial wound healing. This is because TNF-α is increased in tears during infections and inflammations of the cornea, suggesting its importance as a mediator of inflammation in vitro as well as in vivo (Bitko et al., 2004; Kumar et al., 2004; Saika et al., 2005). In addition, recent studies suggest a connection between TNF-α and NFκB activity in corneal wound healing after alkali injury (Saika et al., 2005). Several other studies document that TNF-α activates NFκB in many different cell types and additionally corneal epithelial wound healing may be modulated by a mechanism involving TNF-α/NFκB regulation of cell-cycle, and therefore proliferation and migration (Wang et al., 2005). In agreement, here we report that in human corneal epithelial cells (both primary and transformed), TNF-α-induced NFκB activation is rapid and sustained. NFκB-induced gene expression requires that NFκB not only becomes activated, but it must also be phosphorylated and must physically translocate from cytoplasm to the nucleus (Karin and Ben-Neriah, 2000). Our studies demonstrate that Tβ4 treatment inhibits all three of these central events, suggesting that Tβ4 may exert its anti-inflammatory effects via NFκB-related signaling pathways.
We also found that Tβ4 suppression of TNF-α-induced NFκB activation is not immediate as maximal effects of Tβ4 were time related. Although Tβ4 is intracellular, following microinjection into intact cells, fluorescently labeled Tβ4 has both a diffuse cytoplasmic and a pronounced nuclear staining (Huff et al., 2004). Since Tβ4 can also assume a nuclear localization it is interesting to speculate that the time-related inhibition of NFκB activation may represent Tβ4 blocking the cyclic reentry of activated NFκB into the nucleus. We cannot discount the possibility however that Tβ4 inhibits events upstream of NFκB nuclear translocation, including the phosphorylation and activation of IκB kinase and we are investigating these two possibilities.
In conclusion, we present the first evidence that Tβ4 inhibits NFκB activity and cellular function, suggesting a possible mechanistic pathway by which Tβ4 exerts its anti-inflammatory properties. In addition to our previous studies showing the anti-inflammatory and pro-wound healing effects of Tβ4 after alkali burn (Sosne et al., 2002, 2005), it has been demonstrated by others (Saika et al., 2005) that blocking the NFκB pathway might be beneficial in treating corneal alkali burns. To our knowledge, our studies are novel because they are the first to propose NFκB-mediated signaling events downstream of Tβ4 treatment in the cornea. Tβ4, the major actin sequestering protein in eukaryotic cells, has several potential clinical applications in the repair and remodeling of ulcerated tissues and solid organs following hypoxic injuries, such as myocardial infarction and stroke (Goldstein et al., 2005). A therapeutic agent such as Tβ4, that promotes corneal re-epithelialization, wound healing and cell survival while regulating inflammation after trauma or surgery without adverse side effects would be a major clinical advance. This hitherto unrecognized anti-inflammatory capability of Tβ4 in cornea is particularly provocative and clinically relevant because of its potential usage as a potent anti-inflammatory therapy in a wide array of inflammatory corneal conditions.
Acknowledgments
Grant Information: Supported by National Eye Institute Grant KO8 EY13412 (GS), Core Vision Grant P30EY04068, and a Research to Prevent Blindness Career Development Award (GS). GS is a Career Development Award Recipient from Research to Prevent Blindness
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrawal VB, Tsai RJ. Corneal epithelial wound healing. Indian J. Ophthalmol. 2003;51:5–15. [PubMed] [Google Scholar]
- Baldwin AS. The NF-kB and I kappaB proteins: new discoveries and insights. Ann. Rev. Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- Bitko V, Garmon NE, Cao T, Estrada B, Oakes JE, Lausch RN, Barik S. Activation of cytokines and NF-kappa B in corneal epithelial cells infected by respiratory syncytial virus: potential relevance in ocular inflammation and respiratory infection. BMC Microbiol. 2004;15:4–28. doi: 10.1186/1471-2180-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazzell RK, Stern ME, Aquavella JV, Beuerman RW, Baird L. Human recombinant epidermal growth factor in experimental corneal wound healing. Invest. Ophthalmol. Vis. Sci. 1991;32:336–340. [PubMed] [Google Scholar]
- Chandrasekher G, Ma X, Lallier TE, Bazan HE. Delay of corneal epithelial wound healing and induction of keratocyte apoptosis by platelet-activating factor. Invest. Ophthalmol. Vis. Sci. 2002;43:1422–1428. [PubMed] [Google Scholar]
- Chickarmane V, Kholodenko BN, Sauro HM. Oscillatory dynamics arising from competitive inhibition and multisite phosphorylation. J. Theor. Biol. 2006 doi: 10.1016/j.jtbi.2006.05.013. May 23 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J. Leukoc. Biol. 2001;69:513–521. [PubMed] [Google Scholar]
- Goldstein AL, Hannappel E, Kleinman HK. Thymosin beta4: actin-sequestering protein moonlights to repair injured tissues. Trends Mol. Med. 2005;11:421–429. doi: 10.1016/j.molmed.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Goodall GJ, Morgan JI, Horecker BL. Thymosin beta 4 in cultured mammalian cell lines. Arch. Biochem. Biophys. 1983;221:598–601. doi: 10.1016/0003-9861(83)90182-0. [DOI] [PubMed] [Google Scholar]
- Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine & Growth Fact. Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- Hannappel E, Xu GJ, Morgan J, Hempstead J, Horecker BL. Thymosin beta 4: a ubiquitous peptide in rat and mouse tissues. Proc. Natl. Acad. Sci. U. S. A. 1982;79:2172–2175. doi: 10.1073/pnas.79.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannappel E, Leibold W. Biosynthesis rates and content of thymosin beta 4 in cell lines. Arch. Biochem. Biophys. 1985;240:236–241. doi: 10.1016/0003-9861(85)90028-1. [DOI] [PubMed] [Google Scholar]
- Hannappel E, van Kampen M. Determination of thymosin beta 4 in human blood cells and serum. J. Chromatogr. 1987;397:279–285. doi: 10.1016/s0021-9673(01)85010-x. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Huff T, Rosorius O, Otto AM, Muller CS, Ballweber E, Hannappel E, Mannherz HG. Nuclear localisation of the G-actin sequestering peptide thymosin beta4. J. Cell Sci. 2004;117:5333–5341. doi: 10.1242/jcs.01404. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Ann. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Kumar A, Zhang J, Yu FS. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: role of peptidoglycan in stimulating proinflammatory cytokine secretion. Invest. Ophthalmol. Vis. Sci. 2004;45:3513–3522. doi: 10.1167/iovs.04-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurpakus MA, Daneshvar C, Davenport J, Kim A. Human corneal epithelial cell adhesion to laminins. Curr. Eye Res. 1999;19:106–114. doi: 10.1076/ceyr.19.2.106.5330. [DOI] [PubMed] [Google Scholar]
- Lambiase A, Bonini S, Micera A, Rama P, Bonini S, Aloe L. Expression of nerve growth factor receptors on the ocular surface in healthy subjects and during manifestation of inflammatory diseases. Invest. Ophthalmol. Vis. Sci. 1998;39:1272–1275. [PubMed] [Google Scholar]
- Low TL, Hu SK, Goldstein AL. Complete amino acid sequence of bovine thymosin beta 4: a thymic hormone that induces terminal deoxynucleotidyl transferase activity in thymocyte populations. Proc. Natl. Acad. Sci. U. S. A. 1981;78:1162–1166. doi: 10.1073/pnas.78.2.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low TL, Goldstein AL. Chemical characterization of thymosin beta 4. J. Biol. Chem. 1982;257:1000–1006. [PubMed] [Google Scholar]
- Luo L, Li DQ, Doshi A, Farley W, Corrales RM, Pflugfelder SC. Experimental dry eye stimulates production of inflammatory cytokines and MMP-9 and activates MAPK signaling pathways on the ocular surface. Invest. Ophthalmol. Vis. Sci. 2004;45:4293–4301. doi: 10.1167/iovs.03-1145. [DOI] [PubMed] [Google Scholar]
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Sakon S, Sasazuki T, Sakurai H, Doi T, Yagita H, Okumura K, Nakano H. Phosphorylation of serine 276 is essential for p65 NF-kappaB subunit-dependent cellular responses. Biochem. Biophys. Res. Commun. 2003;300:807–812. doi: 10.1016/s0006-291x(02)02932-7. [DOI] [PubMed] [Google Scholar]
- Planck SR, Rich LF, Ansel JC, Huang XN, Rosenbaum JT. Trauma and alkali burns induce distinct patterns of cytokine gene expression in the rat cornea. Ocul. Immunol. Inflamm. 1997;5:95–100. doi: 10.3109/09273949709085057. [DOI] [PubMed] [Google Scholar]
- Ritchie MH, Fillmore RA, Lausch RN, Oakes JE. A role for NF-κB binding motifs in the differential induction of chemokine gene expression in human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 2004;45:2299–2305. doi: 10.1167/iovs.03-0367. [DOI] [PubMed] [Google Scholar]
- Saika S, Miyamoto T, Yamanaka O, Kato T, Ohnishi Y, Flanders KC, Ikeda K, Nakajima Y, Kao WW, Sato M, Muragaki Y, Ooshima A. Therapeutic effect of topical administration of SN50, an inhibitor of nuclear factor-kappaB, in treatment of corneal alkali burns in mice. Am. J. Pathol. 2005;166:1393–1403. doi: 10.1016/s0002-9440(10)62357-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Peng B, Li Z, Sclabas GM, Fujioka S, Niu J, Schmidt-Supprian M, Evans DB, Abbruzzese JL, Chiao PJ. Mechanisms of proinflammatory cytokine-induced biphasic NF-kappaB activation. Mol. Cell. 2003;12:1287–1300. doi: 10.1016/s1097-2765(03)00390-3. [DOI] [PubMed] [Google Scholar]
- Sosne G, Chan CC, Thai K, Kennedy M, Szliter EA, Hazlett LD, Kleinman HK. Thymosin beta 4 promotes corneal wound healing and modulates inflammatory mediators in vivo. Exp. Eye Res. 2001;72:605–608. doi: 10.1006/exer.2000.0985. [DOI] [PubMed] [Google Scholar]
- Sosne G, Szliter EA, Barrett R, Kernacki KA, Kleinman H, Hazlett LD. Thymosin beta 4 promotes corneal wound healing and decreases inflammation in vivo following alkali injury. Exp. Eye Res. 2002;74:293–299. doi: 10.1006/exer.2001.1125. [DOI] [PubMed] [Google Scholar]
- Sosne G, Xu L, Prach L, Mrock LK, Kleinman HK, Letterio JJ, Hazlett LD, Kurpakus-Wheater M. Thymosin beta 4 stimulates laminin-5 production independent of TGF-beta. Exp. Cell Res. 2004;293:175–183. doi: 10.1016/j.yexcr.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Sosne G, Christopherson L, Barrett RP, Fridman R. Thy mosin-β4 modulates corneal matrix metalloproteinase levels and polymorphonuclear cell infiltration after alkali injury. Invest. Ophthalmol. Vis. Sci. 2005;46:2388–2395. doi: 10.1167/iovs.04-1368. [DOI] [PubMed] [Google Scholar]
- Sosne G, Albeiruti AR, Hollis B, Siddiqi A, Ellenberg D, Kurpakus-Wheater M. Thymosin beta4 inhibits benzalkonium chloride-mediated apoptosis in corneal and conjunctival epithelial cells in vitro. Exp. Eye Res. 2006;83:502–507. doi: 10.1016/j.exer.2006.02.001. Epub 2006 Apr 21. [DOI] [PubMed] [Google Scholar]
- Sotozono C, Inatomi T, Nakamura M, Kinoshita S. Keratinocyte growth factor accelerates corneal epithelial wound healing in vivo. Invest. Ophthalmol. Vis. Sci. 1995;36:1524–1529. [PubMed] [Google Scholar]
- Sotozono C, He J, Matsumoto Y, Kita M, Imanishi J, Kinoshita S. Cytokine expression in the alkali-burned cornea. Curr. Eye Res. 1997;16:670–676. doi: 10.1076/ceyr.16.7.670.5057. [DOI] [PubMed] [Google Scholar]
- Stramer BM, Fini ME. Uncoupling keratocyte loss of corneal crystallin from markers of fibrotic repair. Invest. Ophthalmol. Vis. Sci. 2004;45:4010–4015. doi: 10.1167/iovs.03-1057. [DOI] [PubMed] [Google Scholar]
- Wang L, Reinach P, Lu L. TNF-alpha promotes cell survival through stimulation of K+ channel and NFkappaB activity in corneal epithelial cells. Exp. Cell Res. 2005;311:39–48. doi: 10.1016/j.yexcr.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SE, Netto M, Ambrosio R., Jr Corneal cells: chatty in development, homeostasis, wound healing, and disease. Am. J. Ophthalmol. 2003;136:530–536. doi: 10.1016/s0002-9394(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Yu FX, Lin SC, Morrison-Bogorad M, Yin HL. Effects of thymosin beta 4 and thymosin beta 10 on actin structures in living cells. Cell Motil. Cytoskeleton. 1994;27:13–25. doi: 10.1002/cm.970270103. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kohli M, Zhou Q, Graves DT, Amar S. Short- and long-term effects of IL-1 and TNF antagonists on periodontal wound healing. J. Immunol. 2004;173:3514–3523. doi: 10.4049/jimmunol.173.5.3514. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wu XY, Yu FS. Inflammatory responses of corneal epithelial cells to Pseudomonas aeruginosa infection. Curr. Eye Res. 2005;30:527–534. doi: 10.1080/02713680590968150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liu Z, Meier KE. Lysophosphatidic Acid as a Mediator for Proinflammatory Agonists in a Human Corneal Epithelial Cell Line. Am. J. Physiol. Cell Physiol. 2006 doi: 10.1152/ajpcell.00523.2005. Jun 7; Epub ahead of print. [DOI] [PubMed] [Google Scholar]