

Abstract
Previous studies have shown that Prostaglandin E2 (PGE2) inhibits glucose-stimulated insulin secretion. However, the role of cyclooxygenase (COX)-1 vs. COX-2 derived PGE2 production in glucose-stimulated insulin secretion remains poorly understood. Here we investigated the expression of COX-1 and COX-2 in pancreatic islets and the effect of selective inhibition of COX-1 and COX-2 on glucose-stimulated insulin secretion using C57BL/6 (B6) mice. Although immunofluorescence histochemistry showed the constitutive expression of both COX-1 and COX-2 in B6 mouse pancreatic islets, insulin secretion and hyperglycemia after glucose loading were ameliorated in B6 mice treated with selective COX-2 inhibitor (SC58236) for 18 weeks. Interestingly, incubation with selective COX-2 inhibitor for 24 h led to a reduction in PGE2 production in pancreatic islets isolated from B6 mice. In addition, selective COX-2 inhibition enhanced insulin secretion from the isolated islets. These results collectively suggest that selective inhibition of COX-2 enhances glucose-stimulated insulin secretion through a reduction in PGE2 production in pancreatic islets.
Keywords: Cyclooxygenase-2, Prostaglandin E2, Insulin secretion, Pancreatic β-cell, C57BL/6 mouse
Cyclooxygenase (COX) is a enzyme which catalyzes the first step in the formation of prostaglandins (PGs), the conversion of arachidonic acid to PGH2, followed by the metabolism of PGH2 to biologically active end products, PGD2, PGE2, PGF2α, PGI2, or thromboxane A2 (TxA2) via specific synthases [1]. Two cyclooxygenase isoforms, COX-1 and COX-2, have been identified. COX-1 is considered to be a housekeeping enzyme responsible for basal physiological production of PGs, while COX-2 is a rapidly inducible enzyme contributing to the elevated production of PGs in the setting of disease and inflammation. In most tissues, COX-1 is dominantly expressed relative to COX-2. However, a previous study reported that COX-2 was the dominant isoform in Syrian hamster and human pancreatic islets under basal conditions through gene expression analysis [2]. In contrast, a recent study reported that COX-1 mRNA, rather than COX-2 mRNA, was abundantly expressed in freshly isolated mouse islets [3]. Thus, the expression and the localization of the two isoforms, COX-1 and COX-2, in the pancreatic islets remains controversial.
PGE2 is the major endogenous prostanoid derived from COX-1 and COX-2. Previous studies have shown that PGE2 inhibits glucose-induced insulin secretion in a pancreatic β-cell line, HIT cells [4,5] and also in humans [6]. Therefore, inhibition of COX-1 or COX-2 is expected to enhance insulin secretion by reducing PGE2 production. A recent in vitro study reported that treatment with a selective COX-2 inhibitor, celecoxib, increased glucose-stimulated insulin release in a pancreatic β-cell line, INS-1E cells [7]. However, in vivo effects of selective COX-1 or COX-2 inhibition on insulin secretion have not been examined. C57BL/6 mice represent an inbred mouse strain exhibiting poor glucose tolerance relative to other inbred mouse strains. C57BL/6 mice exhibit marked hyperglycemia and impaired insulin secretion after glucose loading relative to other inbred mouse strains [8,9]. Therefore, this mouse strain appears to be a suitable model for testing the in vivo effect of COX inhibition on glucose-stimulated insulin secretion.
The aim of the present study was to investigate the in vivo effect of selective inhibition of COX-1 and COX-2 on glucose tolerance and glucose-stimulated insulin secretion and to elucidate the mechanism by which selective inhibition of COX-1 or COX-2 may affect the insulin secretion.
Materials and methods
Experimental animals
Male C57BL/6J (B6) and DBA/2J (DBA/2) were purchased from Clea Japan (Tokyo, Japan) at 6 weeks of age. Genetically mixed background COX-2 knockout mouse was generated as described previously [10]. B6 background COX-2 knockout mouse was generated by backcrossing the mixed background COX-2 knockout mouse to a B6 strain for 10 generations. The mice were housed (n = 3–4 per cage) in a room with relative humidity of 50% and a 12/12-h light/dark cycle at 20–22 °C, and allowed unrestricted access to standard rodent chow and water. All animals were treated in accordance with the Animal Welfare Guidelines of Akita University and Vanderbilt University, and all procedures were approved by the Committee on Animal Experimentation of Akita University and Vanderbilt University.
Intraperitoneal glucose tolerance test (IPGTT) and blood parameter measurements
HbA1c measurement and IPGTT were performed at 10 weeks of age in both B6 and DBA/2 inbred mice. HbA1c levels were determined using a DCA 2000 Analyzer (Bayer, Elkhart, IN). Mice were fasted 6 h after daytime food withdrawal and then injected intraperitoneally with glucose in saline solution. Blood glucose was measured after glucose injection using Glucocard Diameter (Arkray, Tokyo, Japan). Plasma insulin levels after glucose injection were measured using a commercial insulin ELISA kit (Morinaga, Yokohama, Japan).
Immunofluorescence histochemistry
Following removal from 10-week-old B6 mice, the pancrease was perfusion-fixed with 4% paraformaldehyde. To assess the expression and localization of COX-2 in the pancreas, the sections were double labeled with guinea pig anti-insulin antibody (DakoCytomation, Carpinteria, CA) and rabbit anti-COX-2 antibody (Cayman Chemical, Ann Arbor, MI), and then incubated with Alexa Fluor 488-conjugated goat anti-guinea pig IgG (Molecular Probes, Eugene, OR) and Alexa Fluor 546-conjugated goat anti-rabbit IgG (Molecular Probes). In addition, the expression of COX-1 in the pancreas was evaluated by double labeling using guinea pig anti-insulin antibody (DakoCytomation) and goat anti-COX-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) as primary antibodies and Alexa Fluor 488-conjugated goat anti-guinea pig IgG (Molecular Probes) and Alexa Fluor 546-conjugated donkey anti-goat IgG (Molecular Probes) as secondary antibodies. The nuclei were stained using ToPro-3 (Molecular Probes). The pancreas sections were examined using confocal laser scanning microscopy (LSM510, Carl Zeiss, Oberkochen, Germany).
Protocol for treatment with selective COX-1 or COX-2 inhibitors in B6 mice
Selective COX-1 inhibitor (SC58560) and COX-2 inhibitor (SC58236) were kindly provided by Pfizer Inc. (Groton, CT). Stock solutions of selective COX-1 and COX-2 inhibitors were prepared by dissolving them in a solution of 95% polyethylene glycol 200 and 5% Tween 20. Selective COX-1 inhibitor (15 mg/ml in stock solution) and COX-2 inhibitor (3 mg/ml in stock solution) were then diluted 1:500 in distilled water and provided ad libitum in the drinking water. Control B6 mice were given the same solution without selective COX inhibitors diluted 1:500 in distilled water (vehicle). The treatment with either selective COX-1 inhibitor or selective COX-2 inhibitor was started at 12 weeks of age in B6 mice and continued for 18 weeks. At the end of the treatment for 18 weeks, HbA1c measurement and IPGTT (2 g/kg body weight) were performed to assess the effects of selective COX inhibitors on glucose tolerance and glucose-stimulated insulin secretion.
Measurement of gastric mucosal PGE2 levels
Gastric mucosal PGE2 synthesis was determined as a measure of endogenous COX-1 activity [11]. After the treatment with selective COX-1 or COX-2 inhibitors for 18 weeks, mucosal linings of stomachs from control and COX inhibitor-treated mice were harvested and homogenized in 0.1M phosphate buffer containing 1 mM EDTA and 10 µM indomethacin, followed by adding acetone to the samples. Precipitates were removed by centrifugation at 1500g for 10 min, and then supernatants were passed through C-18 SPE cartridges (Cayman Chemical). PGE2 was eluted with 5 ml ethyl acetate containing 1% methanol and determined using an enzyme immunosorbent assay kit (Cayman Chemical). Protein concentration of gastric mucosa was determined using a bicinochoninic acid protein assay (Sigma, St. Louis, MO). Gastric mucosal PGE2 levels were assessed using gastric mucosal PGE2 to protein ratio as described previously [10].
Islet isolation and culture
Pancreatic islets were isolated from male B6 mice aged 12 weeks as previously reported [12] with slight modification. Briefly, liberase RI (Roche Applied Science, Indianapolis, IN) was dissolved in Hepes–Krebs–Ringer bicarbonate buffer (HKRB; 10 mM Hepes, 129 mM NaCl, 5 mM NaHCO3, 4.7 mM KCl, 1.2 mM KH2PO4, 2.56 mM CaCl2, 1.2 mM MgSO4, 2.8 mM glucose, and 0.3% bovine serum albumin, pH 7.4). Mice were anesthetized with pentobarbital sodium (50 mg/kg body weight, intraperitoneal injection), and 2 ml liberase RI in HKRB (0.5 mg/ml) was directly infused into the pancreas through the common bile duct. The pancreas was removed and digested in 2 ml liberase RI (0.5 mg/ml) at 37 °C for 20 min. The islets were handpicked under microscopic visualization and washed three times in HKRB. Following the islet isolation procedure, the islets (20 per dish) were cultured in Eagle’s minimal essential medium (Nissui Pharmaceutical, Tokyo, Japan) containing 5.6 mM glucose supplemented with 2 mM l-glutamine, 16.7 mM NaHCO3, 10% fetal bovine serum, 100 µg/ml streptomycin, and 100 U/ml penicillin under an atmosphere of 95% air and 5% CO2 at 37 °C for 24 h. Following this 24-h period, the culture medium was replaced and supplemented with either 10 µM SC58560 or 10 µM SC58236 at 37 °C for 24 h, and medium PGE2 content was determined using an enzyme immunosorbent assay kit (Cayman Chemical). The islets were washed three times in HKRB and then incubated at 37 °C for 1 h in the same culture medium containing either 5.6 or 20 mM glucose with either 10 µM SC58560 or 10 µM SC58236. Medium insulin content was determined using an insulin ELISA kit (Morinaga).
Statistical analysis
Statistical analyses were performed with GraphPad Prism software system (GraphPad, San Diego, CA). All data are presented as means ± SE. Differences between groups were determined by paired t test, unpaired t test or ANOVA followed by Bonferroni’s multiple comparison test. A p value of less than 0.05 was considered statistically significant.
Results
Strain differences in glucose tolerance and glucose-stimulated insulin secretion
Although fasting blood glucose levels were similar between B6 and DBA/2 mice (153 ± 7 and 130 ± 7 mg/dl, respectively, NS), blood glucose levels at 20–120 min after IPGTT glucose injection were markedly elevated in B6 mice compared to DBA/2 mice (Fig. 1A). On the other hand, plasma insulin levels of B6 mice were not significantly elevated in response to glucose loading (Fig. 1B). In addition, insulin levels at 0–30 min after glucose injection were significantly lower in B6 mice than DBA/2 mice (Fig. 1B). HbA1c levels were significantly higher in B6 mice than DBA/2 mice, indicating that B6 mice were hyperglycemic relative to DBA/2 mice (Fig. 1C). Thus, B6 mice appear to represent an inbred mouse strain which has impaired glucose tolerance due to poor glucose-stimulated first-phase insulin secretion.
Fig 1.
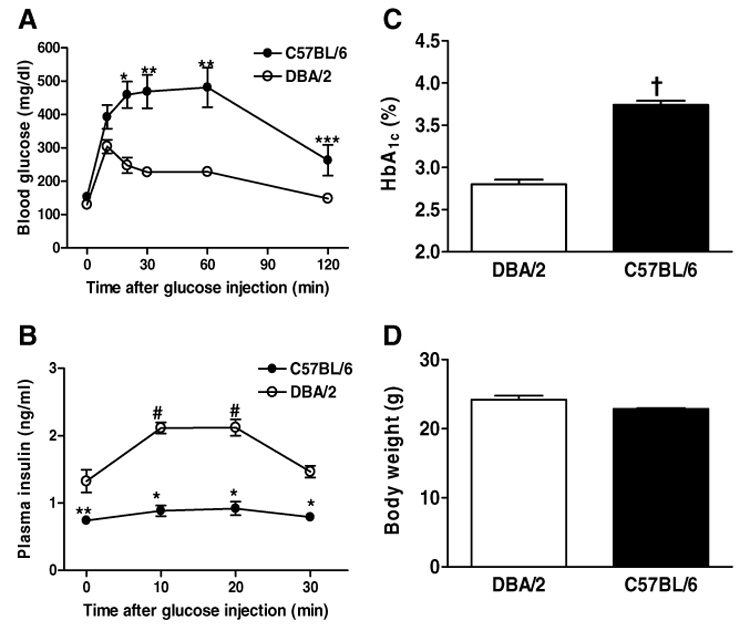
Deferences in glucose tolerance and glucose-stimulated insulin secretion between 10-week-old male C57BL/6 (n = 5) and DBA/2 mice (n = 5). (A,B) Changes in blood glucose and plasma insulin levels after glucose loading (2 g/kg body weight) are shown. *p < 0.001, **p < 0.01, ***p < 0.05 vs. DBA/2 at the same time. #p < 0.01 vs. value before glucose loading (0 min). (C,D) HbA1c and body weight are shown. †p < 0.001 vs. DBA/2.
Expression of COX-1 and COX-2 in B6 mouse pancreatic islets
Immunofluorescence histochemical examination revealed that both COX-1 and COX-2 were constitutively expressed in B6 mouse pancreatic islets under basal conditions (Fig. 2). Double labeling study with an insulin antibody that selectively binds to β cells of pancreatic islets indicated that both COX-1 and COX-2 were localized mainly in the insulin-producing β cells (Fig. 2).
Fig 2.
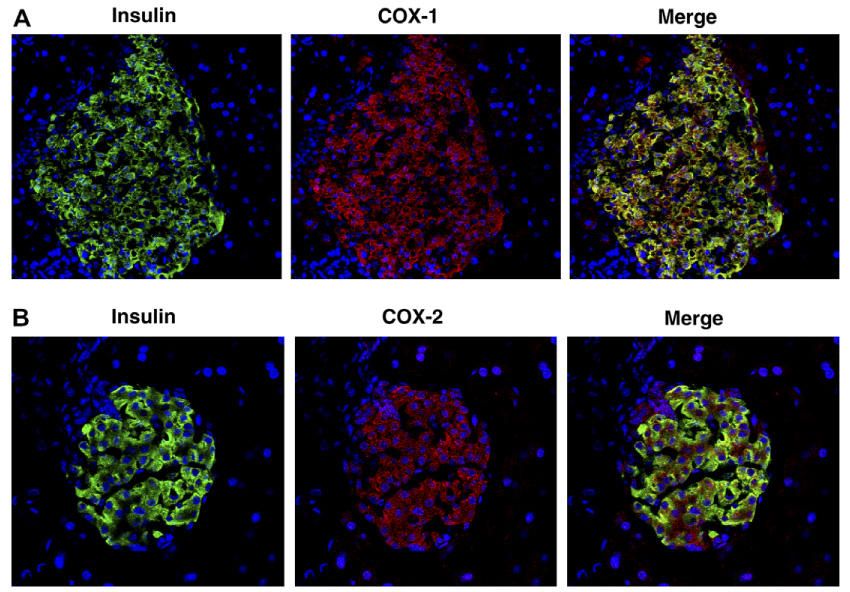
Immunofluorescence staining for insulin, COX-1 and COX-2 in 10-week-old male C57BL/6 mouse pancreatic islets. (A) Double labeling of insulin (green) and COX-1 (red). (B) Double labeling of insulin (green) and COX-2 (red). Magnification 400×. Nuclei are blue.
Effect of selective COX-1 or COX-2 inhibition on glucose tolerance and glucose-stimulated insulin secretion in B6 mice
To evaluate whether COX-1 and COX-2 expressed in pancreatic β cells affect insulin secretion in B6 mice, we treated the mice with either a selective COX-1 inhibitor or a selective COX-2 inhibitor. Following 18 weeks of COX-1 or COX-2 inhibitor treatment, IPGTT and HbA1c were determined (Fig. 3A, B and D). Blood glucose levels at 60 and 120 min after glucose injection were significantly lower in selective COX-2 inhibitor-treated mice than vehicle-treated mice. On the other hand, blood glucose levels during IPGTT were similar between selective COX-1 inhibitor-treated and vehicle-treated mice. In selective COX-2 inhibitor-treated mice, plasma insulin levels at 10 and 20 min after glucose injection were significantly elevated relative to baseline levels and significantly higher than those in vehicle-treated mice. In contrast, plasma insulin levels after glucose loading were not significantly elevated in selective COX-1 inhibitor-treated mice. Thus, we found that selective inhibition of COX-2, but not COX-1, reduces blood glucose levels by enhancing glucose-stimulated first-phase insulin secretion in B6 mice. Similar to B6 mice treated with selective COX-2 inhibitor, B6 background COX-2 knockout mice exhibited significantly lower blood glucose levels during IPGTT relative to the wild type mice (Fig. 3C). Following selective COX-2 inhibitor treatment for 18 weeks, HbA1c levels were also significantly lower in selective COX-2 inhibitor-treated mice than vehicle-treated mice (Fig. 3D). Body weight was not significantly different among three groups treated with vehicle, selective COX-1 inhibitor, and selective COX-2 inhibitor before and after the treatment (Fig. 3E).
Fig 3.

Effects of selective COX-1 vs. COX-2 inhibition on glucose tolerance and glucose-stimulated insulin secretion. (A,B) Intraperitoneal glucose tolerance test (IPGTT) in male C57BL/6 mice treated with vehicle (n = 8), COX-1 inhibitor (SC58560, n = 8), and COX-2 inhibitor (SC58236, n = 8). IPGTT was performed at the end of the treatment for 18 weeks. Changes in blood glucose levels (A) and plasma insulin levels (B) after glucose loading (2 g/kg body weight) are shown. *p < 0.001, **p < 0.01, ***p < 0.05 vs. vehicle at the same time. #p < 0.05 vs. value before glucose loading (0 min). (C) IPGTT in male C57BL/6 background COX-2 knockout mice (C57BL/6-COX-2−/−, n = 4) and male C57BL/6 wild type mice (C57BL/6-COX-2+/+, n = 4) at 12–16 weeks of age. Changes in blood glucose levels after glucose loading (1.5 g/kg body weight) are shown. †p < 0.05 vs. C57BL/6-COX-2+/+ at the same time. (D,E) Changes in HbA1c and body weight in male C57BL/6 mice treated with vehicle (n = 8), SC58560 (n = 8), and SC58236 (n = 8). ‡p < 0.001 vs. vehicle at 18 week of treatment. (F) Gastric mucosal PGE2 production in male C57BL/6 mice treated with vehicle (n = 8), SC58560 (n = 8), and SC58236 (n = 8). Gastric mucosal PGE2 levels were measured at the end of the treatment for 18 weeks. ##p < 0.05 vs. vehicle.
Biological activity of selective COX-1 inhibitor used
Since selective COX-1 inhibitor (SC58560) did not affect insulin secretion in B6 mice, we wished to confirm the in vivo activity of SC58560. COX-1 is dominantly expressed in gastric mucosa and mediates PGE2 production in this tissue [11]. Therefore, we determined the capacity of SC58560 to inhibit gastric mucosal PGE2 synthesis at the end of oral administration of selective COX inhibitors for 18 weeks. Mice treated with SC58560 exhibited a significantly reduced gastric mucosal PGE2 production compared to vehicle-treated mice, confirming that SC58560 successfully inhibits COX-1 (Fig. 3F). In contrast, the treatment with selective COX-2 inhibitor (SC58236) failed to reduce gastric mucosal PGE2 production, confirming the COX-2 selectivity of SC58236 (Fig. 3F).
Effect of selective COX-2 inhibition on PGE2 formation and glucose-stimulated insulin secretion by B6 mouse islets
To define the mechanism by which selective COX-2 inhibition ameliorates glucose-stimulated insulin secretion in B6 mice, we incubated the islets isolated from B6 mice with either selective COX-1 inhibitor (SC58560) or selective COX-2 inhibitor (SC58236) for 24 h. Treatment of islets with selective COX-2 inhibitor significantly reduced PGE2 production compared to controls (Fig. 4A). In contrast, selective COX-1 inhibitor failed to reduce PGE2 production by the islets (Fig. 4A). Following the incubation with selective COX inhibitors for 24 h, we examined insulin secretion by the islets in the presence of either 5.6 or 20 mM glucose. Similar to the present in vivo results, insulin secretion by the islets incubated with selective COX-2 inhibitor was significantly enhanced under both 5.6 and 20 mM glucose conditions relative to controls (Fig. 4B). Especially, incubation with selective COX-2 inhibitor induced a 4-fold increase in insulin secretion by the islets in the presence of 20 mM glucose (Fig. 4B). On the other hand, selective COX-1 inhibitor did not improve insulin secretion by the islets isolated from B6 mice.
Fig 4.
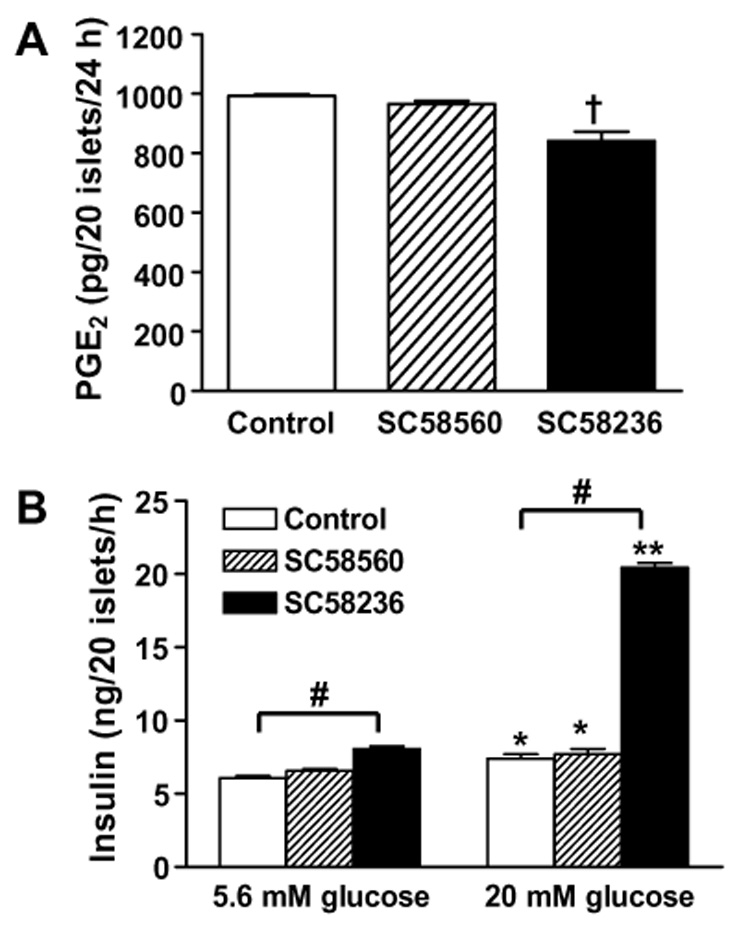
Effects of selective COX-1 vs. COX-2 inhibition on PGE2 production and insulin secretion in pancreatic islets isolated from male C57BL/6 mice. (A) The islets were incubated with either COX-1 inhibitor (SC58560) or COX-2 inhibitor (SC58236) for 24 h and medium PGE2 content was measured. †p < 0.001 vs. control. (B) The islets treated with either SC58560 or SC58236 for 24 h were incubated in culture medium containing either 5.6 or 20 mM glucose for 1 h and medium insulin content was measured. *p < 0.01, **p < 0.0001 vs. 5.6 mM glucose. #p < 0.001 vs. control.
Discussion
The present studies confirmed that B6 mice exhibit poor glucose-stimulated first-phase insulin secretion leading to hyperglycemia after glucose loading (Fig. 1A and B). Previous studies in B6 mice have reported the similar results [8,9]. Based on the physiological property regarding insulin secretion, we used B6 mouse as a suitable model for testing the in vivo effect of selective inhibition of COX-1 and COX-2 on glucose-stimulated insulin secretion in this study.
Before examining in vivo effect of selective COX-1 and COX-2 inhibition on insulin secretion, we wished to determine the expression of COX-1 and COX-2 in pancreatic islets of B6 mice, since the expression of the two COX iso-forms in pancreatic islets under basal conditions remains poorly defined. Immunofluorescence histochemical examination demonstrated that both COX-1 and COX-2 are expressed in pancreatic islets of B6 mice under basal conditions (Fig. 2). Interestingly, both COX-1 and COX-2 were localized mainly in the insulin-producing β cells as shown in merge images of Fig. 2. Therefore, it seems an important issue to be elucidated whether COX-1 and COX-2 are involved in the mechanism underlying glucose-stimulated insulin secretion.
Non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin, sodium salicylate, and indomethacin are used to treat inflammatory diseases including fever and rheumatoid arthritis. Importantly, low-dose of aspirin is widely used in type 1 and type 2 diabetic patients, reducing a risk for cardiovascular events by inhibiting production of thromboxane, a potent vasoconstrictor, and platelet aggregation in these patients [13]. The anti-inflammatory effects of NSAIDs have been attributed to inhibition of COX-1 and COX-2. Most of classical NSAIDs inhibit both COX-1 and COX-2 non-selectively [14]. Whether NSAIDs affect insulin secretion through inhibition of COX-1 or COX-2 remains less clearly defined.
In the present study, we investigated the in vivo effect of selective inhibition of COX-1 and COX-2 on insulin secretion and glucose tolerance in B6 mice using highly selective COX-1 inhibitor SC58560 [15] and COX-2 inhibitor SC58236 [16]. Through the results of IPGTT conducted after the treatment with SC58560 and SC58236, we found that selective inhibition of COX-2, but not COX-1, enhances glucose-stimulating insulin secretion in B6 mice, resulting in amelioration of hyperglycemia after glucose loading (Fig. 3A and B). In addition, we found that COX-2 gene disruption also ameliorates glucose tolerance in B6 mice (Fig. 3C). Thus, selective COX-2 inhibition with a compound or by gene disruption appears to provide a beneficial effect on the amelioration of glucose tolerance.
To further define the mechanism of present in vivo results, we investigated the response of pancreatic islets isolated from B6 mice to glucose following in vitro incubation with either selective COX-1 inhibitor SC58560 or COX-2 inhibitor SC58236 for 24 h. PGE2 production in B6 mouse islets was significantly reduced by incubation with a selective COX-2 inhibitor, whereas incubation with a selective COX-1 inhibitor did not affect PGE2 production in the islets (Fig. 4A). In support of the present results, a recent study also reported that selective COX-2 inhibitor SC58236 attenuates cytokine-induced PGE2 formation in rat islets [17]. Notably, B6 mouse islets incubated with selective COX-2 inhibitor exhibited a 4-fold increase in insulin secretion relative to control islets under high glucose condition (20 mM glucose) and a modest increase in insulin secretion under low-glucose condition (5.6 mM glucose), whereas incubation with selective COX-1 inhibitor did not affect insulin secretion from the islets (Fig. 4B). Thus, consistent with the present in vivo results, in vitro incubation of B6 mouse islets with a selective COX-2 inhibitor also augmented glucose-stimulated insulin secretion.
As demonstrated in previous in vivo and in vitro studies, PGE2 inhibits glucose-stimulated insulin secretion [4–6]. Therefore, reducing PGE2 production in pancreatic islets would be predicted to enhance glucose-stimulated insulin secretion. Although both COX-1 and COX-2 are expressed in normal murine islets, only COX-2 inhibition contributes to basal PGE2 production in isolated islets. This finding could explain why selective inhibition of COX-2, but not COX-1, enhanced glucose-stimulated insulin secretion in the present in vivo and in vitro examinations using B6 mice. We conclude that a reduction in PGE2 production in pancreatic islets by selective COX-2 inhibition may contribute to an amelioration of pancreatic β-cell dysfunction such as poor glucose-stimulated insulin secretion observed in B6 mice. Further studies will be required to elucidate the mechanisms by which COX-2-derived PGE2 modulates pancreatic islet function.
Acknowledgments
We thank Pfizer Inc. for kindly providing us with selective COX-1 inhibitor (SC58560) and selective COX-2 inhibitor (SC58236). The authors also acknowledge support of National Institutes of Health Grants DK37097 and DK74116 (to M.D.B.).
References
- 1.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 2.Sorli CH, Zhang HJ, Armstrong MB, Rajotte RV, Maclouf J, Robertson RP. Basal expression of cyclooxygenase-2 and nuclear factor-interleukin 6 are dominant and coordinately regulated by interleukin 1 in the pancreatic islet. Proc. Natl. Acad. Sci. USA. 1998;95:1788–1793. doi: 10.1073/pnas.95.4.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Persaud SJ, Burns CJ, Belin VD, Jones PM. Glucose-induced regulation of COX-2 expression in human islets of Langerhans. Diabetes. 2004;53 Suppl. 1:S190–S192. doi: 10.2337/diabetes.53.2007.s190. [DOI] [PubMed] [Google Scholar]
- 4.Robertson RP, Tsai P, Little SA, Zhang HJ, Walseth TF. Receptor-mediated adenylate cyclase-coupled mechanism for PGE2 inhibition of insulin secretion in HIT cells. Diabetes. 1987;36:1047–1053. doi: 10.2337/diab.36.9.1047. [DOI] [PubMed] [Google Scholar]
- 5.Seaquist ER, Walseth TF, Nelson DM, Robertson RP. Pertussis toxin-sensitive G protein mediation of PGE2 inhibition of cAMP metabolism and phasic glucose-induced insulin secretion in HIT cells. Diabetes. 1989;38:1439–1445. doi: 10.2337/diab.38.11.1439. [DOI] [PubMed] [Google Scholar]
- 6.Giugliano D, Di Pinto P, Torella R, Frascolla N, Saccomanno F, Passariello N, D’onofrio F. A role for endogenous prostaglandin E in biphasic pattern of insulin release in humans. Am. J. Physiol. 1983;245:E591–E597. doi: 10.1152/ajpendo.1983.245.6.E591. [DOI] [PubMed] [Google Scholar]
- 7.Luo C, Kallajoki M, Gross R, Mulari M, Teros T, Ylinen L, Mäkinen M, Laine J, Simell O. Cellular distribution and contribution of cyclooxygenase (COX)-2 to diabetogenesis in NOD mouse. Cell Tissue Res. 2002;310:169–175. doi: 10.1007/s00441-002-0628-6. [DOI] [PubMed] [Google Scholar]
- 8.Kaku K, Fiedorek FT, Province M, Permutt MA. Genetic analysis of glucose tolerance in inbred mouse strains. Diabetes. 1988;37:707–713. doi: 10.2337/diab.37.6.707. [DOI] [PubMed] [Google Scholar]
- 9.Kayo T, Fujita H, Nozaki J, E X, Koizumi A. Identification of two chromosomal loci determining glucose intolerance in a C57BL/6 mouse strain. Comp. Med. 2000;50:296–302. [PubMed] [Google Scholar]
- 10.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J. Clin. Invest. 2002;110:61–69. doi: 10.1172/JCI14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase 2 in vivo is anti-inflammatory and nonulcerogenic. Proc. Natl. Acad. Sci. USA. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dezaki K, Hosoda H, Kakei M, Hashiguchi S, Watanabe M, Kangawa K, Yada T. Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in β-cells. Diabetes. 2004;53:3142–3151. doi: 10.2337/diabetes.53.12.3142. [DOI] [PubMed] [Google Scholar]
- 13.American Diabetes Association. Aspirin therapy in diabetes. Diabetes Care. 2004;27 Suppl. 1:S72–S73. doi: 10.2337/diacare.27.2007.s72. [DOI] [PubMed] [Google Scholar]
- 14.Warner TD, Mitchell JA. Cyclooxygenase: new forms, new inhibitors, and lessons from the clinic. FASEB J. 2004;18:790–804. doi: 10.1096/fj.03-0645rev. [DOI] [PubMed] [Google Scholar]
- 15.Smith CJ, Zhang Y, Koboldt CM, Muhammad J, Zweifel BS, Shaffer A, Talley JJ, Masferrer JL, Seibert K, Isakson PC. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. USA. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, Anderson GD, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib) J. Med. Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 17.Heitmeier MR, Kelly CB, Ensor NJ, Gibson KA, Mullis KG, Corbett JA, Maziasz TJ. Role of cyclooxygenase-2 in cytokine-induced b-cell dysfunction and damage by isolated rat and human islets. J. Biol. Chem. 2004;279:53145–53151. doi: 10.1074/jbc.M410978200. [DOI] [PubMed] [Google Scholar]