

Abstract
Suppression of gene expression by small interfering RNA (siRNA) has proved to be a gene-specific and cost effective alternative to other gene suppression technologies. Short hairpin RNAs (shRNAs) generated from the vector-based expression are believed to be processed into functional siRNAs in vivo, leading to gene silencing. Since an shRNA library carries a large pool of potential siRNAs, such a library makes it possible to knock down gene expression at the genome wide scale. Although much of research has been focused on generating shRNA libraries from either individually made gene specific sequences or cDNA libraries, there is no report on constructing randomized shRNA libraries, which could provide a good alternative to these existing libraries. We have developed a method of constructing shRNAs from randomized oligonucleotides. Through this method, one can generate a partially or fully randomized shRNA library for various functional analyses. We validated this procedure by constructing a p53-specific shRNA. Western blot revealed that the p53-shRNA successfully suppressed expression of the endogenous p53 in MCF-7 cells. We then made a partially randomized shRNA library. Sequencing of 15 randomly picked cloned confirmed the randomness of the library. Therefore, the library can be used for various functional assays, such as target validation when a suitable screening or selection method is available.
Keywords: RNA Interference; RNA, Small Interfering; Oligonucleotides
Introduction
The recent development of short interfering RNA (siRNA) technology based on RNA interference (RNAi) enables the functional knock down of all genes in a systematic manner. RNAi is the mechanism of sequence-specific gene silencing by either the specific degradation of homologous mRNAs or transcription-mediated gene suppression (1-4). siRNAs are a new class of nucleic-acid-based gene-silencing molecule that could be more potent than ribozymes and antisense oligonucleotides in silencing gene expression because they use cellular machinery (5). Moreover, siRNA technology is more cost effective and efficient to create loss-of-function phenotypes, compared to antisense and knockout techniques. Because of its simplicity, effectiveness, specificity, and easy adaptation to high throughput, this technology holds enormous scientific, commercial, and therapeutic potential.
In research laboratories, two types of siRNAs have been widely used to induce specific gene silencing. While synthetic siRNAs are made in vitro chemically or enzymatically (e.g., in vitro transcription-based method) (6, 7), vector-based siRNAs are usually transcribed through members of the type III class of Pol III promoters such as the H1 promoter and U6 promoter (8, 9). Compared to synthetic siRNAs, vector-based siRNAs possess a unique feature that makes it possible to study gene silencing over a long period of time.
The majority of vector-based siRNAs are generated from short hairpin RNAs (shRNAs). In this system, a single Pol III promoter (e.g., H1 or U6) drives transcription of a single species of RNA molecule (8, 9) from a DNA fragment consisting of a 19-29 base pair stem with a tightly structured loop. Transcription of this highly structured fragment yields an shRNA, which is then processed into an siRNA through the conserved Dicer family of RNase III enzymes (10). Thus, the transcribed product mimics the synthetic siRNA duplex. Like the synthetic siRNA, the in vivo processed siRNA triggers mRNA degradation and gene silencing. In some cases, a double promoter system has also been used to drive vector-based siRNA expression, where two promoters are in tandem or in opposite directions (11-13). This system does not produce shRNAs; instead, siRNA is directly generated when two complementary RNA molecules come together and form a duplex, leading to gene silencing.
Mounting evidence has suggested base preference for the siRNA-mediated gene silencing. For instance, less than 50% of tested sequences of a gene are capable of generating functional siRNAs (14, 15). Therefore, selection of the right sequences is critical for gene silencing efficiency. Extensive studies have been done on selecting potential target sequences for siRNAs. Some guidelines have been suggested to increase the chance of success in silencing gene expression (16, 17). However, there are still no unified rules defining the functional siRNA sequences; different genes may have different sequence preferences. In many cases, functional siRNAs do not fall into these rules (18). Therefore, siRNAs derived from cDNA libraries would provide a good source to search for functional siRNAs because such libraries carry a variety of DNA sequences that would generate functional siRNAs.
Elucidation of gene function is a major challenge in the post genomic era. The concept of the siRNA library has emerged to meet such a demand because the siRNA library allows for evaluation of gene function at the genome wide scale. For instance, a retrovirus-based shRNA library has been constructed carrying 9,610 human and 5,563 mouse genes (19). However, although this approach is very attractive, generation of such a library is enormously time consuming and costly because potential siRNAs are designed and made individually against each gene. Therefore, an shRNA library made from cDNAs or cDNA libraries would offer a simple alternative to this approach (14, 15, 20). Although generation of these cDNA-based shRNA libraries is much more cost effective than an individually made gene-specific shRNA library, they still suffer from several limitations due to the inherited nature of cDNA libraries. For instance, not all genes are expressed in all tissues or cells; not all genes are expressed at the same level.
Ideally, a good shRNA library should carry all possible sequences with equal amount of copies against any gene, be easy to construct and be cost effective. A randomized shRNA library would meet these criteria. Toward this end, we have developed a method to construct shRNAs from randomized oligonucleotides, by which one can relatively easily generate a randomized shRNA library.
Materials and Methods
Cloning Strategy
As shown in Fig. 1, the entire cloning procedure involves the following 4 steps.
Fig. 1.
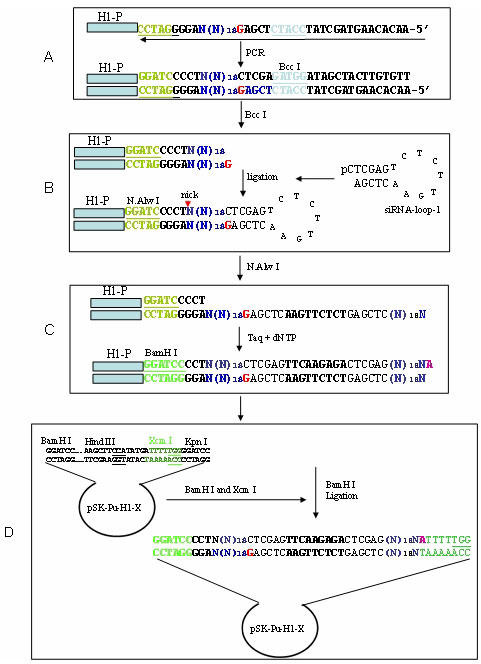
Illustration of cloning strategy. A: Incorporation of 19 randomized nucleotides into PCR products. PCR reactions were performed using the cloned H1 promoter as a template and primers H1-5.1 and H1-R-3.2. B: Ligation of siRNA-loop-1. After PCR, the PCR product was digested with Bcc I and then ligated to siRNA-loop-1. C: Generation of palindromic sequences. Digestion with the nicking enzyme N.Alw I generated a partially single-stranded and double-stranded structure. At 72°C Taq polymerase converted the single-stranded DNA to double stranded DNA. D: Cloning. The extended product was digested with BamH I and then ligated to pSK-H1-Pu-X that had been digested with BamH I and Xcm I.
A: Incorporation of 19 randomized nucleotides into PCR products: Since a sequence carrying 19-29 nucleotides has been shown to efficiently suppress its cognate genes (8, 21), we used a 19 base siRNA to illustrate how shRNAs can be generated from randomized oligonucleotides. First, we designed a primer that carries 19 randomized nucleotides, named R-H1-siRNA-3.2, 5’-AACACAAGTAGCTATCCATCTCGAGN19AGGGGATCCTGGTCTCATAC; part of its sequences is shown in Fig. 1 step A, as indicated by an arrow. This primer also carried other important features: 1) we introduced BamH I (GGATCC) at the end of the H1 promoter and before the potential transcription site for subsequent cloning and manipulations. 2) within the BamH I site was the N.Alw I recognized sequence (GGATCNNNN‸N), which makes a single cut on the double-stranded DNA and allows for opening of the loop structure at step C. We have previously used this nick enzyme for construction of an shRNA library from cDNA (22); 3) immediately after the randomized sequence was a 20 base sequence, which carries the Bcc I site (CCATC4/5) (Fig. 1, step A). The Bcc I-digested end carried a 5’ G (in red) overhang, which was compatible with the siRNA-loop-1 (step B). To incorporate this randomized oligonucleotide primer into the H1 promoter, we used it as an antisense PCR primer along with the sense primer H1-5.1, 5’-AGTGGCGCCCTGCAATATTTG. Thus, after PCR amplification, the randomized sequence was introduced into the PCR products; each molecule of the PCR products, in theory, would carry a unique sequence at the randomized sequences of the 3’ end, while the rest of sequences remain the same among all the molecules. Although we used the H1 promoter as an adaptor, any other DNA sequences should also work as long as they are compatible with the following steps.
B: Ligation of siRNA-loop-1: siRNA-loop-1 is a 21 base oligonucleotide that forms a 7 bp stem with a 7 base loop. The loop sequence was derived from a published one (8), but in a complementary form. When this loop was opened, the sequence of the filled strand would be exactly the same as the one previously described (8). The 7 bp stem of siRNA-loop-1 carried the Xho I site, which was designed to remove the loop in a later stage. The 5’ end of the oligonucleotide was phosphorylated to achieve efficient ligation. Thus, when it was self-annealed, siRNA-loop-1 formed a phosphorylated C overhang, which was complementary to the Bcc I-digested DNA fragment.
C: Generation of palindromic sequences: This step consisted of nicking and extension. To cut the top DNA strand between the end of the H1 promoter and the beginning of the target sequence, we digested the DNA fragment with the nicking enzyme N.Alw I, (Fig. 1, step B). We then incubated the nicked DNA at 72°C for 10 min. Because the upstream region from the nick was much larger than the downstream region (over 100 bp vs 25 bp) and thermodynamics favored the opening of the downstream region from the nick, the DNA fragment became partially single-stranded at the 3’ end (Fig. 1 step C). This partially double/single-stranded structure was essential to convert cloned sequences into a palindromic structure. Taq polymerase converted the single-stranded DNA into double-stranded DNA in the presence dNTP, using the bottom strand as a template and the top strand as a primer. The extended product carried an extra nucleotide A, which was compatible with the Xcm I site in pSK-H1-Pu-X (see below).
D: Cloning: To unidirectionally clone the potential DNA fragment for shRNAs, we digested the extended product with BamH I. The resulting 65 bp fragment was subsequently cloned into pSK-H1-Pu-X at BamH I and Xcm I sites. Thus, this unidirectional cloning procedure not only provided easy ligation but also allowed for transcription of the sequence with the desired length, because the Pol III enzyme recognizes 5Ts as a terminator. Finally, ligated DNA was introduced into competent E. coli cells.
PCR reactions and ligation
PCR was performed to introduce the target sequence into PCR products as the following conditions: 1 cycle of 94°C for 3 min, 30 cycles of 94°C for 0.5 min, 52°C for 1 min and 72°C for 0.5 min, followed by extension at 72°C for 10 min. The cloned H1 promoter DNA was used as a template. The PCR product was purified by PCR purification kit (Qiagen, Valencia, CA), then digested with Bcc I, and finally purified by PCR purification kit. To generate p53-specific shRNA, PCR was performed under the same conditions except that an antisense primer was p53-siRNA-3.1 (AACACAAGTAGCTATCCATCTCGAGUGTAGATTACCACTGGAGTCUAGGGG ATCCTGGTCTCATAC) where the p53-specific sequence was underlined.
A typical ligation reaction for siRNA-loop-1 was performed using Fast link ligation kit (Epicentre, Madison, WI) at 4°C overnight. The ligated DNA was separated in a 2% agarose gel and then purified using gel purification kit (Qiagen).
Construction of pSK-H1-Pu-X
To facilitate the final cloning of the DNA fragment carrying a 19-bp palindromic sequence, we constructed pSK-H1-Pu-X. First, we introduced Xcm I site between Hind III and Kpn I at pSK by two self-complementary oligonucleotides 5’-AGCTTUCCAUTATGATTTTUTGGUGGTAC and 5’-AUGGTUATACTAAAAUACCUC where Xcm I recognition site was underlined. The two oligonucleotides were first heated at 94°C for 5 min and then slowly cooled down to room temperature. The annealed oligonucleotides were subsequently ligated into pSK that was previously linearized by Hind III and Kpn I. The resulting plasmid pSK-X carried an Xcm I site, followed by 5Ts at 3’ region, serving a transcription terminator for the Pol III.
To introduce the H1 promoter and puromycin resistance gene into pSK-X, we amplified this segment using pSUPER-retro (Oligoengine, Seattle, WA) as a template. The primers were H1-3.3, 5’-GGATCCUTGGTCTCATACAGAACTTA, where a BamH I site (underlined) was introduced; and Pu-3.1 5’-TCTAGAUTTGCCAAACCTACAGGTGGG, where an Xba I site (underlined) was introduced. After this fragment was cloned into pSK-X at XbaI and Bam H1 sites, resulting plasmid pSK-H1-Pu-X would accommodate a DNA fragment that has a nucleotide A overhang at 3’ end and a BamH I site at the other end.
Cell culture and transfection
MCF-7 cells (American Type Cell Collection, Manassas, VA) were grown in RPMI medium (BioWhittaker, Walkersville, MD), supplemented with 10% FBS, 2 mM L-glutamine, 100 units of penicillin/ml and 100 ug streptomycin /ml (both from BioWhittaker). Cells were incubated at 37°C in a humidified chamber supplemented with 5% CO2.
Plasmid DNAs carrying an appropriate sequence were introduced into MCF-7 cells by Optifect (Invitrogen, Carlsbad, CA) per the manufacturer’s protocol in a 6-well plate. To determine the effect of p53-shRNA on expression of the endogenous p53, transfected MCF-7 cells were treated with doxorubincin at 0.4 ug/ml for 16 h. Cells were then harvested to extract total protein, which was quantified using a protein assay kit (Bio-Rad, Hercules, CA).
Western blot
Total protein was extracted and separated in 9% SDS-PAGE. Suppression of p53 protein was determined by anti-p53 antibody (Oncogene Science, Cambridge, MA). Signals were detected by ECL detection system (Pierce, Rockford, IL) and images were captured in Fluo-Chem imager (Alpha Innotech, San Leandro, CA).
Reagents and DNA sequencing
Oligonucleotides were synthesized through commercial sources (Sigma Genosys, Woodlands, TX or IDT, Coralville, IA). Restriction and nicking enzymes were obtained from New England Biolabs (Beverly, MA). Taq polymerase was purchased from ABI (Foster City, CA). DNA sequencing service was provided by Northwoods DNA (Solway, MN).
Results and Discussion
To determine whether functional shRNAs can be generated through this approach, we constructed a vector-based p53-shRNA following the strategy outlined in Fig. 1. To incorporate half of p53 shRNA palindromic sequence 5’-GACTCCAGTGGTAATCTAC (8) into PCR products, we used a sense primer, H1-5.1, and an antisense primer, p53-siRNA-3.1 (see Materials and Methods). Primer p53-siRNA-3.1 differed from R-H1-siRNA-3.2 in that the 19 base p53 sequence (8) replaced the 19 base randomized oligonucleotides of R-H1-siRNA-3.2. The amplified fragment was about 150 bp (Fig. 2A, lane 1). Following digestion with Bcc I, the digested fragment was 20 bp smaller than before digestion, as expected, resulting in a 130 bp fragment (Fig. 2A lane 2). After gel purification, the digested fragment was ligated to the 5’-phosphorylated siRNA-loop-1. Although ligation reactions are normally carried out at room temperature, or at 14-16°C, we carried the reaction out at 4°C overnight, due to the relatively small size of the loop. Under this condition, successful ligation was achieved because the 130 bp band was completely shifted to ~145 bp (Fig. 2B, lane 4). Since the Bcc I-digested fragment generated a G overhang at the digested end, this prevented end-end ligation of two molecules of the same species, thus, enhancing the ligation of siRNA-loop-1 and H1 promoter.
Fig. 2.
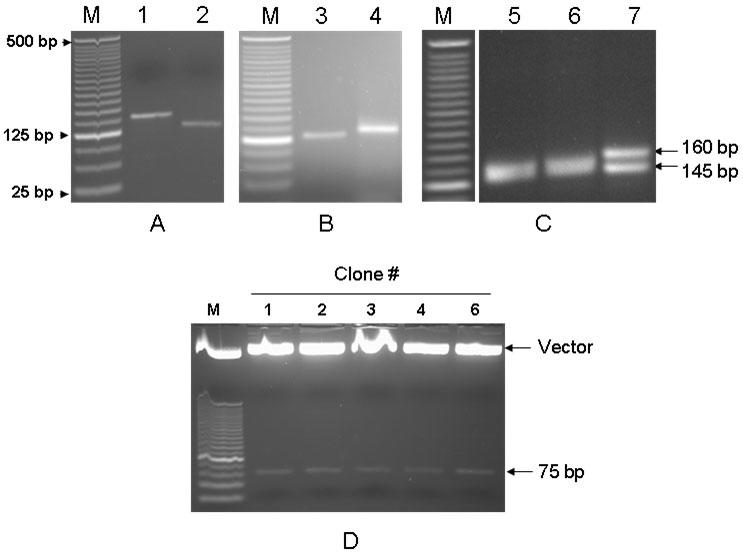
Generation of p53-shRNA. The same strategy described in Fig. 1 was used to construct p53-shRNA. A: PCR and Bcc I digestion. PCR amplification yielded a 150 bp band (lane 1) and digestion of the PCR product with Bcc I shifted the band to 130 bp (lane 2). B: Ligation. Bcc I-digested DNA fragment was ligated to siRNA-loop-1 at 4°C overnight. Lane 3, before ligation; lane 4, after ligation. C: Extension. Ligated DNA fragment was treated with the nicking enzyme N.Alw I and then extended by Taq polymerase in the presence of dNTPs. Lanes 5, before nicking; 6, after nicking; 7, after extension. The extended product was about 160 bp. D: Cloning. Plasmid DNA was isolated from five positive clones and then digested with BamH I and Kpn I. M, 25 bp DNA ladders. All gels are 2%.
The ligated products were gel-purified and were then treated with N.Alw I to make a nick at its recognition site. The nicking did not change migration of the DNA fragment; both the unnicked product and the nicked product migrated at ~145 bp (Fig. 2C, lanes 5&6). However, following extension in the presence of Taq polymerase and dNTP at 72°C, we detected two DNA fragments (Fig. 2C, lane 7): the lower band (~ 145 bp) co-migrated with the one without extension (Fig. 2C, lane 6) and the upper band (~160 bp) was about 15 bp larger than the lower band. PCR cloning and sequencing analysis indicated that the upper band was a truly extended product containing the ligated product of the Bcc I-digested fragment with siRNA-loop-1. The lower band was not clonable and was believed to be non-extended product presumably resulting from incomplete nicking. Alternatively, there is a possibility that a nick is made at the bottom strand, because the N.Alw I recognition sequence (GGATC) is also present at the bottom strand. Hence, the filling by Taq polymerase did not yield any extension. To clone the extended product, we gel-purified the upper band, subsequently digested it with BamH I, and finally ligated it to pSK-H1-Pu-X that had been digested with both BamH I and Xcm I. After the ligated DNA was introduced into E. coli cells, miniprep combined with restriction digestion showed that eight of ten randomly picked clones carried a 75 bp insert after digestion with BamH I and Kpn I. We used Kpn I here instead of Xcm I because the Xcm I site was eliminated after ligation (see Fig. 1). A result of restriction digestions for five of the eight positive clones is shown in Fig. 2D. Subsequent DNA sequencing confirmed that all five clones carried the right inserts, as expected.
To determine whether the p53-shRNA construct (p53-shRNA/pSK-H1-Pu-X) can generate functional siRNA to suppress p53 expression, we introduced p53-shRNA/pSK-H1-Pu-X into MCF-7 cells that express wild type p53 (23). We also made a p53 shRNA construct using the same p53 specific sequence based on pSUPER vector previously described (8), to serve as a positive control (p53-shRNA-p).We found that p53-shRNA/pSK-H1-Pu-X was able to inhibit the endogenous p53 expression, with over 80% suppression compared to the vector control (Fig. 3, lane 2). This was about comparable to that of the positive control p53-shRNA-p (Fig. 2, lane 3), suggesting that the p53 shRNA constructed through this presently described approach is functional.
Fig. 3.
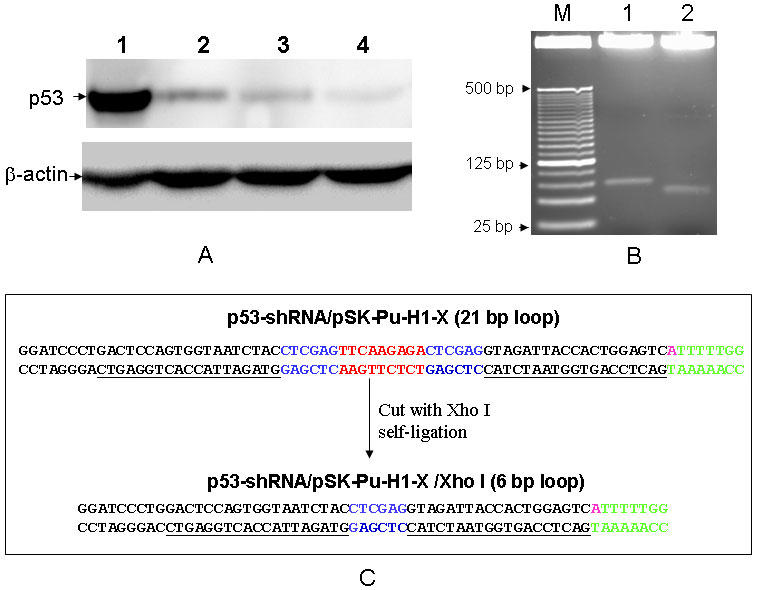
Suppression of p53 by p53-shRNA. A: Detection of p53 expression by Western blot. Plasmid carrying an appropriate DNA fragment was introduced into MCF-7 cells. Proteins samples were prepared as described in Materials and Methods. Lanes 1, vector control; 2, p53-shRNA/pSK-H1-Pu-X; 3, p53-shRNA-p (positive control) and 4, p53-shRNA/pSK-H1-Pu-X/Xho I. ?-actin serves as a loading control. B: Removal of siRNA-loop-1 in p53-shRNA. Both constructs were digested with BamH I and Kpn I. M, 25 bp DNA ladders; lanes 1, p53-shRNA/pSK-H1-Pu-X; 2, p53-shRNA/pSK-H1-Pu-X/Xho I. C: Sequence comparison of p53-shRNA/pSK-H1-Pu-X and p53-shRNA/pSK-H1-Pu-X/Xho I. The p53 specific sequence is underlined.
Next, we examined the effect of loop size in the p53-shRNA construct on the silencing efficiency. The loop in the original construct, i.e., p53-shRNA/pSK-H1-Pu-X (Fig. 2D), was 21 bp, which was larger than those commonly used loops. We first digested the clone with Xho I and then allowed it to be self-ligated. Since the Xho I site (CTCGAG) alone was successfully used as a loop to generate functional siRNAs (9), the construct generated by Xho I digestion and self-ligation is believed to be able to produce functional siRNAs. Compared to the original p53-shRNA/pSK-H1-Pu-X, the modified construct (p53-shRNA/pSK-H1-Pu-X/Xho I) caused a slightly higher suppression of p53 expression (Fig. 3, lane 4), suggesting that the larger loop (21 bp) may interfere with the gene silencing efficiency to some extent, but this interference seems to be marginal. Therefore, removal of siRNA-loop-1 during construction of an shRNA library can be eliminated, given that p53-shRNA with the 21-bp loop yielded over 80% of gene silencing efficiency.
After validation of this method, we decided to generate shRNAs from partially randomized oligonucleotides following the procedure in Fig. 1. To reduce the complexity, we designed an oligonucleotide primer carrying GNANNNNNNTNNHWWWWWA (sense strand), where H = A+C+T and W = A+T based on the general guidelines suggested by Reynolds et al. (16) and Ui-Tei et al. (17) combined with our own experience. We sequenced 15 of over 10,000 clones and found that all 15 clones have unique sequences in the randomized region (Fig. 4), demonstrating the random nature of this pooled shRNAs.
Fig. 4.

Sequences of 15 randomly picked clones from the library. N=A or G or C or T; H = A or C or T; W = A or T.
Until now, methods for construction of shRNA libraries are based on either known sequences for individual genes (19, 24) or sequences derived from mixed cDNAs or cDNA libraries (14, 15, 20). While cDNA-based shRNA libraries provide a good alternative to individually made gene-specific shRNA libraries in terms of costs, construction of such cDNA-based shRNA libraries can still be labor intensive. Furthermore, due to the nature of the cDNA library, these cDNA-based shRNA libraries could have several limitations including the unequal representation of genes in a cDNA library. In addition, when restriction enzymes are used to generate small cDNA fragments, some of the sequences might be missed because of their uneven distributions in the sequences.
Although it is generally believed that siRNA-mediated gene silencing is a posttranscriptional event, it has been recently shown that siRNAs can also induce transcriptional gene silencing (1, 2). While the mechanism of siRNA-induced transcriptional gene silencing is not fully understood, it is clear that the promoter regions are also targets for siRNAs. Since the shRNA libraries derived from randomized oligonucleotides could carry promoter-specific siRNA sequences, such libraries would offer another advantage over the cDNA-based shRNA libraries that target only cDNA sequences.
It has been reported that randomized siRNAs can be made from randomized oligonucleotides through the double promoter system (11, 13). However, gene silencing efficiency for siRNAs derived from the two promoter system is not as high as the shRNAs from the single promoter system, especially at the lower concentrations (25-27). This could be a problem for stable transfectants where usually a single copy of the cloned sequence is present in each cell and thus, the gene silencing efficiency could be compromised. A possible explanation is that the single promoter system generates a single RNA molecule, which is easy to form a self-complementary double-stranded structure. In contrast, the double promoter system generates two RNA molecules, one sense and another antisense. In order for these two RNA molecules to efficiently form a duplex, the amount of RNA molecules would be much higher than a single molecule.
To determine the effect of shRNA loop size on gene silencing, we compared the construct carrying the original size of loop (21 bp) with one carrying a smaller loop (6 bp). Since a commonly used loop is about 9 bases, a large loop in shRNAs is believed to interfere with the shRNA-induced gene silencing efficiency. Hence, such a large loop is removed by digestion with a restriction enzyme and then self-ligation (14, 15, 20). To determine whether this step is necessary to achieve efficient gene silencing in the present approach, we also eliminated the initial loop sequence by digesting the plasmid with Xho I, followed by self-ligation. Our results indicate only a slight increase in suppressing p53 expression by the p53 shRNA construct carrying a small loop. In fact, this result is also consistent with a report that an shRNA with a 23 base loop is not different from one with a 9 base loop in suppressing the luciferase gene (25). This could be explained by the fact that Dicer processes the dsRNA stepwise from the terminus (28). Therefore, we suggest that it is not necessary to remove the original loop during construction of randomized shRNA libraries.
In summary, we have described a simple approach to generate shRNAs, which can be used to construct a randomized shRNA library. This approach should be easily applied to viral vectors so that shRNAs can be used to infect virtually any type of cells with high efficiency. Given the wide use of siRNA technology to evaluate gene function, the randomized shRNA library will facilitate studies of gene function in human and mice, as well as other organisms. The method described in this study will also stimulate improvement of siRNA technology and development of its new applications.
Acknowledgments
This work was supported in part by NIH grant R01-CA102630 and the Excellence in Academic Medicine Program of SIU School of Medicine, and in part by SIU Cancer Institute.
Abbreviations
- bp
base pair
- RNAi
RNA interference
- shRNA
short hairpin RNA
- siRNA
short interfering RNA
References
- Induction of DNA methylation and gene silencing by short interfering RNAs in human cells. Kawasaki H, Taira K. Nature. 2004;431:211–217. doi: 10.1038/nature02889. [DOI] [PubMed] [Google Scholar]
- Small interfering RNA-induced transcriptional gene silencing in human cells. Morris KV, Chan SW, Jacobsen SE, Looney DJ. Science. 2004;305:1289–1292. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]
- RNA interference. Hannon GJ. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- The genetics of RNA silencing. Tijsterman M, Ketting RF, Plasterk RH. Annu Rev Genet. 2002;36:489–519. doi: 10.1146/annurev.genet.36.043002.091619. [DOI] [PubMed] [Google Scholar]
- siRNAs: applications in functional genomics and potential as therapeutics. Dorsett Y, Tuschl T. Nat Rev Drug Discov. 2004;3:318–329. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- A system for stable expression of short interfering RNAs in mammalian cells. Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Sui G, Soohoo C, Affar el B, Gay F, Shi Y, Forrester WC. Proc Natl Acad Sci USA. 2002;99:5515–5520. doi: 10.1073/pnas.082117599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene silencing in mammals by small interfering RNAs. McManus MT, Sharp PA. Nat Rev Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- An approach to genomewide screens of expressed small interfering RNAs in mammalian cells. Zheng L, Liu J, Batalov S, Zhou D, Orth A, Ding S, Schultz PG. Proc Natl Acad Sci USA. 2004;101:135–140. doi: 10.1073/pnas.2136685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Generation of an shRNAi expression library against the whole human transcripts. Miyagishi M, Matsumoto S, Taira K. Virus Res. 2004;102:117–124. doi: 10.1016/j.virusres.2004.01.022. [DOI] [PubMed] [Google Scholar]
- A plasmid-based system for expressing small interfering RNA libraries in mammalian cells. Kaykas A, Moon RT. BMC Cell Biol. 2004;5:16. doi: 10.1186/1471-2121-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restriction enzyme-generated siRNA (REGS) vectors and libraries. Sen G, Wehrman TS, Myers JW, Blau HM. Nat Genet. 2004;36:183–189. doi: 10.1038/ng1288. [DOI] [PubMed] [Google Scholar]
- Enzymatic production of RNAi libraries from cDNAs. Shirane D, Sugao K, Namiki S, Tanabe M, Iino M, Hirose K. Nat Genet. 2004;36:190–196. doi: 10.1038/ng1290. [DOI] [PubMed] [Google Scholar]
- Rational siRNA design for RNA interference. Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Ui-Tei K, Naito Y, Takahashi F, Haraguchi T, Ohki-Hamazaki H, Juni A, Ueda R, Saigo K. Nucleic Acids Res. 2004;32:936–948. doi: 10.1093/nar/gkh247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase. Donze O, Picard D. Nucleic Acids Res. 2002;30:e46. doi: 10.1093/nar/30.10.e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A resource for large-scale RNA-interference-based screens in mammals. Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O'Shaughnessy A, Gnoj L, Scobie K et al. Nature. 2004;428:427–431. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- Small interfering RNA production by enzymatic engineering of DNA (SPEED). Luo B, Heard AD, Lodish HF. Proc Natl Acad Sci USA. 2004;101:5494–5499. doi: 10.1073/pnas.0400551101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alternative approach to generate shRNA from cDNA. Dinh A, Mo YY. Biotechniques. 2005;38:629–632. doi: 10.2144/05384RN02. [DOI] [PubMed] [Google Scholar]
- Relationship between radiation-induced G1 phase arrest and p53 function in human tumor cells. Nagasawa H, Li CY, Maki CG, Imrich AC, Little JB. Cancer Res. 1995;55:1842–1846. [PubMed] [Google Scholar]
- A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B et al. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- Optimization of an siRNA-expression system with an improved hairpin and its significant suppressive effects in mammalian cells. Miyagishi M, Sumimoto H, Miyoshi H, Kawakami Y, Taira K. J Gene Med. 2004;6:715–723. doi: 10.1002/jgm.556. [DOI] [PubMed] [Google Scholar]
- Mammalian RNAi for the masses. Shi Y. Trends Genet. 2003;19:9–12. doi: 10.1016/S0168-9525(02)00005-7. [DOI] [PubMed] [Google Scholar]
- Identification of a network involved in thapsigargin-induced apoptosis using a library of small interfering RNA expression vectors. Futami T, Miyagishi M, Taira K. J Biol Chem. 2005;280:826–831. doi: 10.1074/jbc.M409948200. [DOI] [PubMed] [Google Scholar]
- Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W. Embo J. 2002;21:5875–5885. doi: 10.1093/emboj/cdf582. [DOI] [PMC free article] [PubMed] [Google Scholar]