

Abstract
Serpins inhibit serine proteases by mechanically disrupting the protease active site. The protease first reacts with the serpin's reactive center loop (RCL) to form an acylenzyme. Then the RCL inserts into a β-sheet in the body of the serpin, translocating the attached protease ∼70 Å and deforming the protease active site, thereby trapping the acylenzyme. Loop insertion (∼1 s−1) is an order of magnitude slower than hydrolysis of a typical substrate acylenzyme (∼50 s−1), indicating that the protease is inhibited during translocation. We have previously trapped a partially translocated covalent complex of rat trypsin and α1-proteinase inhibitor (EpartI*) resulting from attractive interactions between cationic dyes and anionic rat trypsin. Here, using single pair Förster resonance energy transfer, we demonstrate that EpartI* is a metastable complex that can dissociate to free protease and cleaved serpin (I*) as well as convert to the canonical fully translocated complex EfullI*. The partitioning between these two pathways is pH dependent, with conversion favored at low pH and dissociation favored at high pH. The short lifetime of EpartI* (∼3 h at pH 7.4) and the pH dependence of EpartI* dissociation suggest that, unlike in EfullI*, the catalytic triad is intact in EpartI*. These results also demonstrate that interactions between target proteases and the body of the serpin can hinder protease translocation leading to short-lived covalent complexes.
Keywords: conformational changes, fluorescence, FRET, serpin, protease, single molecule fluorescence
Inhibitory serpins are ubiquitous regulators of serine and cysteine proteases involved in development, blood coagulation, fibrinolysis, inflammation, and many other physiological processes (Carrell and Travis 1985; Ray et al. 1992; Silverman et al. 2001; Hashimoto et al. 2003). Serpins use a unique, kinetically controlled mechanism in which remodeling of the serpin structure leads to mechanical disruption of the protease active site. As in the normal substrate reaction, the protease docks with the serpin's RCL to form the encounter complex, E•I (Scheme 1; Fig. 1; Ye et al. 2001; Dementiev et al. 2003). The RCL is then cleaved resulting in the acylenzyme intermediate, E–I, where the C terminus of the cleaved RCL is attached to Ser195 of the protease. The catalytic His57 can then act as a general base to activate water, leading to hydrolysis of the acylenzyme and release of the protease and I*. Alternatively, the RCL can insert into a β-sheet in the body of the serpin translocating the attached protease ∼70 Å. Translocation disrupts the protease active site generating the covalent complex, EfullI*, and trapping the acylenzyme intermediate (Huntington et al. 2000; Calugaru et al. 2001; Gettins 2002a; Dementiev et al. 2006).
Figure 1.

Protease inactivation by serpins. The protease, E, and serpin, I, dock to form the encounter complex, E•I, shown on the left (Dementiev et al. 2003) (loph.pdb). The RCL is then cleaved, resulting in the acylenzyme intermediate, E–I, which partitions between a substrate-like pathway yielding E and I*, and the inhibitory pathway where the RCL also inserts into β-sheet A and the attached protease is translocated, forming EfullI*, shown on the right (Huntington et al. 2000) (1ezx.pdb). The protease, BTryp, is green with the catalytic triad in red. The Ile16–Asp194 salt bridge is purple, and the location of Alexa Fluor 555 is in green space fill. The serpin, α1PI, is shown in blue with the RCL in yellow, β-sheet A in red, and Cys232, to which Atto 610 is attached, is in crimson space fill. Protein figures were made using PyMOL (DeLano Scientific). The generally accepted scheme for the protease–serpin reaction is shown at the bottom.
Structures of covalent complexes between bovine trypsin (BTryp) and the serpin α1-proteinase inhibitor (α1PI, also known as α1-antitrypsin) as well as those between porcine pancreatic elastase and α1PI show fairly extensive distortions of the protease structure relative to a substrate acylenzyme that can easily account for the stability of the acylenzyme in the serpin complex (Huntington et al. 2000; Dementiev et al. 2006). The oxyanion hole and the S1 site are disrupted, and Ser195 is located ∼6 Å from His57. The salt bridge between the N-terminal Ile16 and Asp194 is also missing, as is the 142–149 loop, and the 218–225 loop is displaced. These distortions appear to result from steric clashes between the protease and the distal portion of the serpin. These clashes would only occur upon full insertion of the RCL into β-sheet A, and therefore require full translocation of the protease (Huntington et al. 2000; Olson et al. 2001; Zhou et al. 2001; Gettins 2002b; Dementiev et al. 2006). The BTryp–α1PI complex shows more extensive loss of secondary structure (Huntington et al. 2000), although the physiological relevance of this observation is disputed (Dementiev et al. 2006). The energy driving this structural remodeling arises from structural defects in I, which are resolved in I* (Im et al. 1999; Seo et al. 2000, 2002; Lee et al. 2001) as well as from favorable interactions that are unique to I* (Fulton et al. 2005). Thus, cleavage of the RCL allows the serpin to convert to the more stable I* conformation, inhibiting the protease like a molecular mousetrap (Huntington and Carrell 2001).
The partitioning between the substrate and inhibitor pathways (Scheme 1; Fig. 1) is determined by the rate of structural remodeling. However, while full disruption of the protease active site likely occurs late in translocation (Tew and Bottomley 2001; Zhou et al. 2001; Gettins 2002b; Cabrita et al. 2004), the rate constants for insertion of the RCL into β-sheet A are at least an order of magnitude slower than the rate constants of substrate deacylation (∼1 s−1 vs. ∼50 s−1) (Hedstrom 2002; Mellet et al. 2002; Shin and Yu 2006). Therefore, the active site must be perturbed early in the translocation process. Using single pair Förster resonance energy transfer (spFRET), we have previously observed a partially translocated complex EpartI* of rat anionic trypsin (RTryp) and α1PI (Liu et al. 2006). The peak spFRET efficiency for EpartI* suggest that no more than the first two residues of the cleaved RCL may be inserted into β-sheet A. Partial translocation is not observed with BTryp, and is only found when α1PI is labeled with cationic dyes at Cys232. Therefore, we proposed that an interaction between the cationic dye and anionic RTryp provides a restraining force that prevents full translocation. Since the protease is only partially translocated, the active site should not be completely disrupted; in fact, EpartI* has a lifetime of hundreds of minutes at pH 7.4 rather than days (Liu et al. 2006).
While our previous work shows that EpartI* can dissociate to E and I*, if EpartI* is trapped in a local energy minimum, it may also convert to EfullI*. To investigate this possibility and to further characterize this intriguing, short-lived covalent complex, we have monitored the pH dependence of EpartI* decomposition. Here we report spFRET experiments which demonstrate that EpartI* decays to both I* and EfullI*, and that the deacylation of EpartI* is mediated by His57, indicating that the protease is only partially disrupted. To the best of our knowledge, this work is the first characterization of a partially translocated, partially disrupted protease–serpin covalent complex.
Results
Dissociation of bovine trypsin (BTryp) covalent complexes
Covalent complexes were formed by preincubating BTryp and α1PI at pH 7.4. After a 15-min incubation, these samples were diluted to 50–100 pM with the appropriate buffer. The apparent rate constants for complex dissociation, k app, were determined by measuring the spFRET at times ranging from 15 min to 70 h after complex formation. Initially, the spFRET histograms show two distinct peaks (Fig. 2). The peak centers reflect the average distance between the donor and acceptor fluorophores while the half-widths are determined by the relative mobility of the two fluorophores as well as by photon shot noise and detector noise. The peak at 0% spFRET efficiency arises from complexes that only contain donor fluorophores and complexes in which the acceptor fluorophore has photobleached. At longer times, this zero peak also contains contributions from the BTryp released when BTryp-α1PI covalent complexes dissociate. The wide peak centered at 27% spFRET efficiency arises from EfullI* (Liu et al. 2006), and corresponds to the fully translocated conformation observed in X-ray crystal structures of α1PI complexes (Huntington et al. 2000; Dementiev et al. 2006). The locations and half-widths of the spFRET peaks are independent of pH (Supplemental Table S1). At pH 7.4, the EfullI* peak disappears over four days and the zero peak grows as the complex dissociates. The plot of the area under the EfullI* peak versus time yields a reaction progress curve that is well described by a single-exponential equation (k app = [6.1 ± 0.3] × 10−6 s−1) (Fig. 2). The value of k app is pH dependent, increasing at pH 8.5 and decreasing at pH 5.5 (Fig. 2). These values are ∼10-fold greater than those determined by monitoring the initial rates of BTryp–α1PI dissociation in bulk experiments (Calugaru et al. 2001), which is reasonable given differences in experimental conditions and design.
Figure 2.

Monitoring dissociation of BTryp–α1PI covalent complexes using spFRET. (A) Changes in BTryp spFRET histograms over time in 50 mM HEPES, pH 7.4, 100 mM NaCl with the Gaussian fits used to determine peak areas shown as lines. (B) Changes in BTryp spFRET histograms over time in 50 mM Bis-Tris, pH 5.5, 100 mM NaCl with the Gaussian fits used to determine peak areas shown as lines. (C) Area of the BTryp EfullI* spFRET peak over time at pH 7.4. (D) Area of the BTryp EfullI* spFRET peak over time at pH 5.5. For C and D, the line shows the fit to a single exponential used to determine kapp at pH 7.4 and pH 5.5, respectively.
Disappearance of rat trypsin (RTryp) complexes
RTryp–α1PI complexes were formed as described above. Initially, RTryp–α1PI spFRET histograms show three distinct peaks (Fig. 3A): the zero peak, a peak centered at 14% corresponding to EfullI*, and a third peak centered at 58%. We have established previously that the 58% peak arises from covalent complexes in which RTryp is only partially translocated relative to α1PI, EpartI* (Liu et al. 2006). EpartI* arises from interactions between anionic RTryp and cationic dyes (including Atto610, Texas Red, and tetramethylrhodamine) located at residue 232, and these interactions counteract the force exerted by the inserting RCL (Mellet et al. 2002; Liu et al. 2006).
Figure 3.

Monitoring dissociation of RTryp–α1PI covalent complexes using spFRET. (A) Changes in RTryp spFRET histograms over time in 50 mM HEPES, pH 7.4, 100 mM NaCl with Gaussian fits used to determine peak areas shown as lines. (B) Changes in RTryp spFRET histograms over time in 50 mM Bis-Tris, pH 5.5, 100 mM NaCl with Gaussian fits shown as lines. Note the pronounced growth in the EfullI* peak, 14% spFRET efficiency, at short times. (C) Area of the RTryp EfullI* spFRET peak (filled squares) and the EpartI* peak (open circles) over time at pH 7.4. (D) Area of the RTryp EfullI* spFRET peak (filled squares) and the EpartI* peak (open circles) over time at pH 5.5. For EfullI* in C and D, the line shows the fit to a double-exponential that accounts for both the conversion of EpartI* to EfullI* at short times and the dissociation of EfullI* at long times. For EpartI* in C and D, the line is a single-exponential fit to determine kapp,part. The insets shows the EpartI* data at higher resolution. Differences in the Y-axis scale of C and D simply reflect the larger number of photons collected at pH 7.4.
As seen for the BTryp complexes, the locations and half-widths of the spFRET peaks are independent of pH (Supplemental Table S1). Fully translocated RTryp complexes consistently show narrower half-widths than BTryp complexes, independent of the location of the acceptor in α1PI, reflecting the differences in mobility around trypsin residue 113 in the covalent complexes (Liu et al. 2006).
The distribution of RTryp complexes changes over time (Fig. 3). EpartI* disappears most rapidly under all conditions, and the data are well described by a single-exponential equation with rate constant k app,part (Figs. 3, 4A). The value of k app,part displays a bowl-shaped dependence on pH. At pH > 8, only the dissociation of EpartI* to free E and I* is observed. However, at pH < 8, the decrease in the 58% peak is accompanied by increases in both the 0% and 14% peaks, indicating that EpartI* both dissociates and converts to EfullI* (Fig. 3). The conversion of EpartI* to EfullI* is a single-exponential process described by the rate constant kc. The value of kc decreases with increasing pH, reaching an apparent minimum near pH 8 (Figs. 3, 4B). To determine the pKa and identify the residue responsible for the decrease in kc with increasing pH, the pH dependence of kc was fit to Equation 4 (Fig. 4B) resulting in k 1c ≥ 0.02 s−1, k 2c = (1.4 ± 1.2) × 10−5 s−1, and pKa ≤ 4, where k 1c and k 2c are the conversion rate constants for protonated and deprotonated EpartI* species, respectively (Scheme 3).
Figure 4.

(A) The pH dependence of k app, derived from the dissociation of EfullI*, with filled squares for RTryp and open squares for BTryp. The line shows the fit to Equation 1 for the RTryp data. Also shown is the pH dependence of k app,part derived from the disappearance of the EpartI* for RTryp covalent complexes (open circles). As shown in Scheme 2, EpartI* can disappear by converting to EfullI* with rate constant kc or by dissociating to E and I* with rate constant k dp. (B) The pH dependence of conversion, kc. The line is the fit to Equation 4. (C) The pH dependence of dissociation, k dp (k app,part -kc). The line is the fit to Equation 6. Compared to dissociation of EfullI* (k app), the pH-dependent dissociation of EpartI* is less steep indicating that the RTryp active site is not fully deformed.
Scheme 3.

The rate constant for the dissociation of EpartI* (k dp) can be obtained from the difference between the rate constants for the disappearance of EpartI* (k app,part) and that for the conversion to EfullI* (kc) (Equation 5), (Fig. 4C). The value of k dp is relatively pH independent in the neutral pH region, but increases at higher pH. Equation 6 was used to model the pH dependence of k dp resulting in: k 1 = 0 s−1, k 2 = (1.16 ± 0.06) × 10−4 s−1, k OH = 46 ± 3 M−1s−1, and pKa = 6.6 ± 0.1, where the uncertainties are based on the 95% confidence intervals. The reported pKas for RTryp's His57 range from 6.8 for free RTryp (Hedstrom et al. 1996) to ∼8 for RTryp-α1PI EfullI* (Kaslik et al. 1997), suggesting that the active site in EpartI* is not disrupted enough to alter the pKa of His57.
The dissociation of RTryp EfullI* occurs on a much slower timescale than the disappearance of EpartI*, so the rate constant for this process could be obtained from single-exponential fits to data collected after the EpartI* reaction was complete. Remarkably, the kinetics of RTryp EfullI* dissociation are indistinguishable from the dissociation of BTryp complexes (Figs. 2–4). The value of k app is pH independent when pH <5, and increases linearly with pH from pH 6–9 (Fig. 4A), unlike previous experiments involving BTryp complexes where an inflection is observed (Calugaru et al. 2001). Above pH 9, the reaction becomes too fast to measure with diffusion-based spFRET, and the lack of high pH data may obscure this modest inflection point. The pH dependence of k app is well described by the following equation (Calugaru et al. 2001; Plotnick et al. 2002):
with the water-mediated dissociation rate constant kH2O = (3.8±0.5) × 10−7s−1, and the hydroxide ion-mediated rate constant k OH = 17 ± 1 M−1s−1 (Fig. 4A). The value of kH2O is very similar to that determined for the dissociation of BTryp–α1PI complexes in bulk experiments kH2O = (3.2±0.7) × 10−7s−1 (Calugaru et al. 2001). However, the value of k OH determined from bulk experiments is slower (kOH = 0.4 ± 0.6 M−1s−1) (Calugaru et al. 2001). This discrepancy may arise from differences in buffer conditions as well as differences in the techniques used to monitor complex dissociation. There are slight differences in the k OH values for EpartI* (k OH = 46 ± 3 M−1s−1) and EfullI* suggesting that the acyl linkage may be more solvent accessible in EpartI*.
Discussion
Comparison of BTryp and RTryp EfullI* complexes
As observed previously, the spFRET histogram EfullI* peaks for BTryp complexes are much wider than those for RTryp complexes, indicating that the region around residue 113 is more mobile in BTryp complexes. These observations suggest that the structure of BTryp is more disrupted than that of RTryp, which might predict that RTryp EfullI* complexes should hydrolyze faster than BTryp complexes. Instead, we observe that the values of kH2O and k OH are very similar for RTryp and BTryp complexes. These values are also are very similar to those for the hydrolysis of a simple alkylester (Calugaru et al. 2001; Plotnick et al. 2002), and therefore, most probably reflect the intrinsic reactivity of the acylester rather than a protease mediated process. Indeed, the rate of hydrolysis of porcine pancreatic elastase and BTryp covalent complexes with α1PI are very similar despite the less extensive disruption seen in the elastase complexes (Huntington et al. 2000; Dementiev et al. 2006).
The coupling of translocation and disruption of the protease active site
The EpartI* complex of RTryp–α1PI is the first example of a partially translocated complex with sufficient stability to permit detailed characterization. As shown in Scheme 2 (Fig. 4), EpartI* can either dissociate to E and I* or convert to EfullI*. The rapid dissociation of EpartI* at neutral pH is readily explained by the presence of a relatively intact active site, and the pKa, ∼6.6, is most likely due to ionization of the catalytic His57. But, what accounts for the facile conversion of EpartI* to EfullI*? One possible explanation is that low pH decreases the attractive force between the fluorophore and RTryp thus increasing kc. Alternatively, lowering the pH may further destabilize the RTryp active site. In common with other serine proteases, RTryp has a stabilizing salt bridge between the N-terminal Ile16 and Asp194 (Hedstrom et al. 1996). We demonstrated previously that mutations which destabilize this salt bridge increase the initial population of EfullI* at the expense of EpartI* (Liu et al. 2006). The pKa ≤ 4 determined from the pH dependence of conversion suggests that protonation of an Asp or Glu residue is involved in conversion. Perhaps the protonation of Asp194 facilitates conversion by increasing the compliance of the acylenzyme linkage between RTryp's Ser195 and α1PI's RCL.
The specificity of the interactions required to trap EpartI* are reminiscent of exosite interactions between positively charged residues in variable region 1 of tissue type plasminogen activator (tPA) and negatively charged residues in the RCL of plasminogen activator inhibitor-1 (PAI-1) C-terminal to the cleavage site (Madison et al. 1989, 1990a,b; Tachias and Madison 1997; Ibarra et al. 2004). In both cases, electrostatic interactions target specific proteases (RTryp but not BTryp for EpartI*) (Liu et al. 2006) (and tPA but not urokinase-type plasminogen activator or trypsin for exosite interactions) (Madison et al. 1990a; Olson et al. 2001; Ibarra et al. 2004) and make loop insertion rate limiting (Kvassman et al. 1998; Olson et al. 2001). Exosite interactions lead to a rapid equilibrium between the tPA–PAI-1 encounter complex and the initial acylenzyme, indicating that the protease active site is intact (Kvassman et al. 1998; Olson et al. 2001). In contrast, in EpartI* the acylenzyme is likely displaced from its location in the encounter complex and the protease active site is partially disrupted.
Shorter-lived α1PI covalent complexes also occur when one or two Ala residues are added to the RCL N-terminal to the cleavage site (Zhou et al. 2001). Lengthening the RCL increases the distance between the body of α1PI and the protease in EfullI*, altering the strain on the acyl linkage and/or its solvent accessibility. Changing the interactions between the serpin and the protease in EfullI* reduces the half-life of covalent complexes containing the α1PI Pittsburgh variant of (Met358Arg, P1) from weeks to hours for thrombin and from weeks to minutes for factor Xa (Zhou et al. 2001). The short half-lives observed for RCL extensions and EpartI* illustrate the importance of specific protease–serpin interactions in determining the extent of protease active-site disruption, and thus the lifetime of covalent complexes. These and the exosite interactions provide a way for a single serpin to modulate its interactions with different proteases.
Partial disruption of the RTryp active site in EpartI* supports models of serpin inhibition in which full inhibition requires full translocation (Gettins 2002b; Shin and Yu 2006), and reflects the fact that partial translocation does not allow α1PI to fully exploit the free energy difference between I and I*. Trypsin experiences two opposing forces during translocation: the force exerted by the serpin pulling on the protease and the opposing force of viscous drag from the solvent. Decreasing the energy stored in the initial, metastable serpin structure or increasing the drag decreases the rate of translocation (Shin and Yu 2006). The cationic dye label augments the opposing drag force (Liu et al. 2006). Unlike viscous drag, which affects the entire protein surface, the force from the dye arises from a stationary position. In effect, RTryp serves as the rope in a tug of war, with the RCL pulling in one direction and the fluorophore in another, trapping EpartI* before the RCL fully inserts and while the protease active site retains some structure and catalytic power. However, due to unrelieved strain in the α1PI structure and the lower energy of the fully remodeled α1PI structure, EpartI* is only metastable and it must either dissociate or convert to EfullI*.
This work adds to the growing chain of evidence demonstrating that the stability of protease–serpin complexes is modulated by specific interactions that determine the extent of loop insertion, which in turn, determines the extent of deformation of the active site. While long inhibition requires full insertion of the RCL, short-lived covalent complexes can be generated by adding one or two residues to the RCL (Zhou et al. 2001) or by attractive interactions between the body of the serpin and the protease that counteract full insertion. These interactions can be quite specific, as demonstrated by our ability to trap EpartI* for RTryp but not BTryp complexes, and the fate of the short-lived complexes can be manipulated by changing the solution conditions. This ability to trap partially translocated complexes may allow us to follow serpin conformational changes along the insertion pathway and to engineer short-lived serpin complexes for different proteases.
Materials and Methods
Trypsin preparation
Chymotrypsinogen amino acid numbering is used for all trypsin variants. The Ser113Cys (S113C) bovine trypsinogen variant was expressed as inclusion bodies in Escherichia coli strain SG13009, refolded, purified, and activated to bovine trypsin (BTryp) as described previously (Peterson et al. 2001). The Lys113Cys (K113C) Rat anionic trypsinogen II variant was expressed in Saccharomyces cerevisiae strain DM101α, purified, and activated to RTryp as described previously (Hedstrom et al. 1992; Pasternak et al. 1998).
α1PI preparation
The wild-type α1PI gene in the plasmid pEAT8–137 (Kwon et al. 1994; Laska 2001) contains two natural sequence variations, a His101Arg mutation and a Glu376Asp mutation, which do not affect function (Huber and Carrell 1989). All α1PI variants were expressed as inclusion bodies in E. coli BL21(DE3) cells, refolded, and purified using a combination of methods previously published (Kwon et al. 1994, 1995; Griffiths and Cooney 2002) as described previously (Liu et al. 2006).
Stoichiometry of inhibition (SI)
Trypsin variants were titrated with p-nitrophenyl-p′-guanidinobenzoate HCl to determine the concentration of the active enzyme (Chase and Shaw 1967). Active enzymes comprised >90% of all trypsin samples both before and after fluorescent labeling. The SI, the ratio of moles of serpin needed to inactivate 1 mol of active trypsin, was determined by titrating trypsin with α1PI as described previously (Rubin et al. 1990). The values of SI are 1 for unlabeled and labeled trypsins and α1PIs.
Protein labeling
Trypsin variants were labeled at residue 113 with Alexa Fluor 555 maleimide (Invitrogen) according to the manufacturer's instructions. Free dye was separated from trypsin using a soybean trypsin inhibitor column (Sigma Chemical) that binds trypsin. Wild-type α1PI was labeled with Atto 610 maleimide (Atto-tec) according to the manufacturer's instructions. A PD-10 gel filtration column (GE Healthcare) was used to separate α1PI from the free dye. The labeling efficiencies were 0.1–0.3 for trypsin and 0.5–0.8 for α1PI. The value of Ro, the dye separation at which there is 50% energy transfer, is 59 Å for the Alexa Fluor 555-Atto 610 dye pair (Liu et al. 2006).
spFRET data collection and analysis
Donor-labeled trypsin variants (0.5–2 μM) were mixed at a ratio of 1:2 with acceptor-labeled α1PI in 50 mM HEPES, 100 mM NaCl, pH 7.4 at room temperature. After a 15-min incubation, the samples were diluted to 50–100 pM in 50 mM buffer containing 100 mM NaCl and 10 mM of the trypsin inhibitor benzamidine. The following buffers were used: sodium acetate, pH 4.5 and 5.5; Bis-Tris, pH 6.3; HEPES, pH 7.4 and 8.0; and TAPS, pH 8.5 and 9.1. Samples (300 μL) were applied to coverglass-mounted wells (Nalgene) coated with 1 mg/mL bovine serum albumin (Sigma Chemical) to prevent protein adsorption unto the sides of the wells. The wells were sealed using a coverslip and vacuum grease to prevent evaporation over the course of the experiment. The fluorescence of single complexes diffusing in solution was collected using a one photon single molecule confocal setup with a 100-μm pinhole based on an IX-70 inverted microscope (Olympus) as previously described (Dahan et al. 1999; Liu et al. 2006). Data from the avalanche photodiode detectors were collected at 24 MHz using a two-channel data acquisition card and associated software (ISS).
Data from the donor and acceptor channels were binned at 1 kHz, and a threshold of 40 total, acceptor plus donor, photon counts was used to identify photon bursts arising from complexes diffusing through the illuminated volume. The photon counts were corrected for background fluorescence and for leakage of donor fluorescence in to the acceptor channel. The spFRET efficiency, E, was calculated using (Dahan et al. 1999):
 |
 |
where I is the corrected photon counts and D and A denote the donor and acceptor channels, respectively. The γ factor corrects for differences in the probability of detecting a photon from the donor or acceptor due to differences in the quantum yield, φ, or the detection efficiency of the APDs in different regions of the spectrum, η. For singly labeled samples with identical optical densities at the excitation wavelength, 520 nm, γ is the ratio of the fluorescence intensity of the acceptor-only samples to that of the donor-only samples measured on the single molecule fluorescence setup (Schuler et al. 2002). Values of the γ factor were measured using labeled free trypsin and labeled free α1PI as well as singly labeled trypsin-α1PI covalent complexes. The γ factor for BTryp-α1PI covalent complexes is 1, while that for RTryp-α1PI covalent complexes is 2 (Liu et al. 2006). The change in γ arises from an increase in Atto-610 fluorescence intensity in the RTryp–α1PI covalent complexes. SpFRET histograms for RTryp-α1PI show the same number of peaks and the same peak areas when calculated using γ values of 1 or 2, although the value of γ alters the location of the peaks.
Histograms of the spFRET data were generated and analyzed using Matlab (Mathworks) and Origin (Originlab). The spFRET histogram peak areas, half-widths, and maxima were determined using Gaussian fits to the data.
Determining the pKa for conversion of EpartI* to EfullI*
If the protonation of EpartI* affects the rate of conversion from EpartI* to EfullI*, then conversion may be modeled as:
where k 1c and k 2c are the conversion rate constants for the protonated and deprotonated EpartI* species, respectively, and conversion is irreversible. Based on this Scheme and the apparent conversion rate constant, kc, experimentally determined from the rate of increase in the EfullI* population at a variety of pH, the pKa for deprotonation can be determined according to:
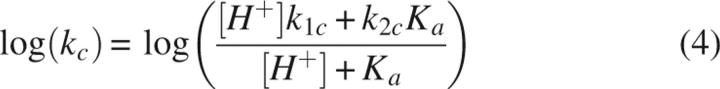 |
where k 1c, k 2c and Ka are fitting parameters. Fits were performed in Origin.
Determining the pKa for EpartI* dissociation
EpartI* can either convert to EfullI* or dissociate to form reactivated trypsin, E, and loop inserted α1PI, I*. Thus, the apparent rate constant for disappearance of EpartI*, k app,part, is dependent on kc and the EpartI* dissociation rate constant, k dp, where both k app,part and kc are experimentally determined. At neutral and higher pH, the dissociation rate constant mediated by water molecules, k H2O, is negligible, and the possible pathways for dissociation are mediated either by hydroxide ions, k OH, or, if the protonation of EpartI* affects the dissociation rate, by a deprotonated base in the protease or serpin with rate constant kB:
Similar to Scheme 3 for conversion, the pH dependence of k dp, and thus the pKa for B can be derived from Equation 5 and the following scheme:
 |
where k 1 and k 2, the base-mediated dissociation rate constants for protonated and deprotonated EpartI*, respectively, are fitting parameters along with Ka and k OH.
Scheme 4.

Electronic supplemental material
A table of peak centers and half-widths for BTryp and RTryp covalent complexes from pH 4.5–9.1.
Acknowledgments
This work was funded by NSF Grant MCB-0446220, and N.M. was supported by NIH Grant GM07956.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Anne Gershenson, Department of Chemistry, MS 015, Brandeis University, 415 South Street, Waltham, MA 02454, USA; e-mail: gershenson@brandeis.edu; fax: (781) 736-2516.
Abbreviations: BTryp, bovine trypsin; FRET, Förster resonance energy transfer; α1PI, human α1-proteinase inhibitor; PAI-1, plasminogen activator inhibitor-1; RTryp, rat anionic trypsin; RCL, reactive center loop; SI, stoichiometry of inhibition; spFRET, single pair Förster resonance energy transfer; tPA, tissue type plasminogen activator.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073111207.
References
- Cabrita L.D., Dai, W., and Bottomley, S.P. 2004. Different conformational changes within the F-helix occur during serpin folding, polymerization, and proteinase inhibition. Biochemistry 43: 9834–9839. [DOI] [PubMed] [Google Scholar]
- Calugaru S.V., Swanson, R., and Olson, S.T. 2001. The pH dependence of serpin–proteinase complex dissociation reveals a mechanism of complex stabilization involving inactive and active conformational states of the proteinase which are perturbable by calcium. J. Biol. Chem. 276: 32446–32455. [DOI] [PubMed] [Google Scholar]
- Carrell R. and Travis, J. 1985. α1-Antitrypsin and the serpins: Variation and countervariation. Trends Biochem. Sci. 10: 20–24. [Google Scholar]
- Chase T.J. and Shaw, E. 1967. p-Nitrophenyl-p′-guanidinobenzoate HCl: A new active site titrant for trypsin. Biochem. Biophys. Res. Commun. 29: 508–514. [DOI] [PubMed] [Google Scholar]
- Dahan M., Deniz, A.A., Ha, T., Chemla, D.S., Schultz, P.G., and Weiss, S. 1999. Ratiometric measurement and identification of single diffusing molecules. Chem. Phys. 247: 85–106. [Google Scholar]
- Dementiev A., Simonovic, M., Volz, K., and Gettins, P.G.W. 2003. Canonical inhibitor-like interactions explain reactivity of α1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J. Biol. Chem. 278: 37881–37887. [DOI] [PubMed] [Google Scholar]
- Dementiev A., Dobo, J., and Gettins, P.G.W. 2006. Active-site distortion is sufficient for proteinase inhibition by serpins: Structure of the covalent complex of α1-proteinase inhibitor with porcine pancreatic elastase. J. Biol. Chem. 281: 3452–3457. [DOI] [PubMed] [Google Scholar]
- Fulton K.F., Buckle, A.M., Cabrita, L.D., Irving, J.A., Butcher, R.E., Smith, I., Reeve, S., Lesk, A.M., Bottomley, S.P., Rossjohn, J., et al. 2005. The high-resolution crystal structure of a native thermostable serpin reveals the complex mechanism underpinning the stressed to relaxed transition. J. Biol. Chem. 280: 8435–8442. [DOI] [PubMed] [Google Scholar]
- Gettins P.G.W. 2002a. Serpin structure, mechanism, and function. Chem. Rev. 102: 4751–4803. [DOI] [PubMed] [Google Scholar]
- Gettins P.G.W. 2002b. The F-helix of serpins plays an essential, active role in the proteinase inhibition mechanism. FEBS Lett. 523: 2–6. [DOI] [PubMed] [Google Scholar]
- Griffiths S.W. and Cooney, C.L. 2002. Development of a peptide mapping procedure to identify and quantify methionine oxidation in recombinant human α1-antitrypsin. J. Chromatogr. A. 942: 133–143. [DOI] [PubMed] [Google Scholar]
- Hashimoto C., Kim, D.R., Weiss, L.A., Miller, J.W., and Morisato, D. 2003. Spatial regulation of developmental signaling by a serpin. Dev. Cell 5: 945–950. [DOI] [PubMed] [Google Scholar]
- Hedstrom L. 2002. Serine protease mechanism and specificity. Chem. Rev. 102: 4501–4523. [DOI] [PubMed] [Google Scholar]
- Hedstrom L., Szilagyi, L., and Rutter, W.J. 1992. Converting trypsin to chymotrypsin: The role of surface loops. Science 255: 1249–1253. [DOI] [PubMed] [Google Scholar]
- Hedstrom L., Lin, T.-Y., and Fast, W. 1996. Hydrophobic interactions control zymogen activation in the trypsin family of serine proteases. Biochemistry 35: 4515–4523. [DOI] [PubMed] [Google Scholar]
- Huber R. and Carrell, R.W. 1989. Implications of the three-dimensional structure of α1-antitrypsin for structure and function of serpins. Biochemistry 28: 8951–8966. [DOI] [PubMed] [Google Scholar]
- Huntington J.A. and Carrell, R.W. 2001. The serpins: Nature's molecular mousetraps. Sci. Prog. 84: 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington J.A., Read, R.J., and Carrell, R.W. 2000. Structure of a serpin–protease complex shows inhibition by deformation. Nature 407: 923–926. [DOI] [PubMed] [Google Scholar]
- Ibarra C.A., Blouse, G.E., Christian, T.D., and Shore, J.D. 2004. The contribution of the exosite residues of plasminogen activator inhibitor-1 to proteinase inhibition. J. Biol. Chem. 279: 3643–3650. [DOI] [PubMed] [Google Scholar]
- Im H., Seo, E.J., and Yu, M.-H. 1999. Metastability in the inhibitory mechanism of human α1-antitrypsin. J. Biol. Chem. 274: 11072–11077. [DOI] [PubMed] [Google Scholar]
- Kaslik G., Kardos, J., Szabó, E., Szilágyi, L., Závodszky, P., Westler, W.M., Markley, J.L., and Gráf, L. 1997. Effects of serpin binding on the target protease: Global stabilization, localized increased structural flexibility and conserved hydrogen bonding at the active site. Biochemistry 36: 5455–5464. [DOI] [PubMed] [Google Scholar]
- Kvassman J.O., Verhamme, I., and Shore, J.D. 1998. Inhibitory mechanism of serpins: Loop insertion forces acylation of plasminogen activator by plasminogen activator inhibitor-1. Biochemistry 37: 15491–15502. [DOI] [PubMed] [Google Scholar]
- Kwon K.-S., Kim, J., Shin, H.S., and Yu, M.-H. 1994. Single amino acid substitutions of α1-antitrypsin that confer enhancement in thermal stability. J. Biol. Chem. 269: 9627–9631. [PubMed] [Google Scholar]
- Kwon K.-S., Lee, S., and Yu, M.-H. 1995. Refolding of α1-antitrypsin expressed as inclusion bodies in Escherichia coli: Characterization of aggregation. Biochim. Biophys. Acta 1247: 179–184. [DOI] [PubMed] [Google Scholar]
- Laska M.E. 2001, Massachusetts Institute of Technology, Cambridge, MA.
- Lee C., Maeng, J.-S., Kocher, J.-P., Lee, B., and Yu, M.-H. 2001. Cavities of α1-antitrypsin that play structural and functional roles. Protein Sci. 10: 1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Mushero, N., Hedstrom, L., and Gershenson, A. 2006. Conformational distributions of protease–serpin complexes: A partially translocated complex. Biochemistry 45: 10865–10872. [DOI] [PubMed] [Google Scholar]
- Madison E.L., Goldsmith, E.J., Gerard, R.D., Gething, M.-J.H., and Sambrook, J.F. 1989. Serpin-resistant mutants of human tissue-type plasminogen activator. Nature 339: 721–724. [DOI] [PubMed] [Google Scholar]
- Madison E.L., Goldsmith, E.J., Gerard, R.D., Gething, M.H., Sambrook, J.F., and Bassel-Duby, R.S. 1990a. Amino acid residues that affect interaction of tissue-type plasminogen activator with plasminogen activator inhibitor 1. Proc. Natl. Acad. Sci. 87: 3530–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison E.L., Goldsmith, E.J., Gething, M.J., Sambrook, J.F., and Gerard, R.D. 1990b. Restoration of serine protease–inhibitor interaction by protein engineering. J. Biol. Chem. 265: 21423–21426. [PubMed] [Google Scholar]
- Mellet P., Mély, Y., Hedstrom, L., Cahoon, M., Belorgey, D., Srividya, N., Rubin, H., and Bieth, J.G. 2002. Comparative trajectories of active and S195A inactive trypsin upon binding to serpins. J. Biol. Chem. 277: 38901–38914. [DOI] [PubMed] [Google Scholar]
- Olson S.T., Swanson, R., Day, D., Verhamme, I., Kvassman, J., and Shore, J.D. 2001. Resolution of Michaelis complex, acylation, and conformational change steps in the reactions of the serpin, plasminogen activator inhibitor-1, with tissue plasminogen activator and trypsin. Biochemistry 40: 11742–11756. [DOI] [PubMed] [Google Scholar]
- Pasternak A., Liu, X., Lin, T.-Y., and Hedstrom, L. 1998. Activating a zymogen without proteolytic processing: Mutation of Lys15 and Asn194 activates trypsinogen. Biochemistry 37: 16201–16210. [DOI] [PubMed] [Google Scholar]
- Peterson F.C., Gordon, N.C., and Gettins, P.G.W. 2001. High-level bacterial expression and 15N-alanine labeling of bovine trypsin. Application to the study of trypsin-inhibitor complexes and trypsinogen activation by NMR spectroscopy. Biochemistry 40: 6275–6283. [DOI] [PubMed] [Google Scholar]
- Plotnick M.I., Samakur, M., Wang, Z.M., Liu, X., Rubin, H., Schechter, N.M., and Selwood, T. 2002. Heterogeneity in serpin–protease complexes as demonstrated by differences in the mechanism of complex breakdown. Biochemistry 41: 334–342. [DOI] [PubMed] [Google Scholar]
- Ray C.A., Black, R.A., Kronheim, S.R., Greenstreet, T.A., Sleath, P.R., Salvesen, G.S., and Pickup, D.J. 1992. Viral inhibition of inflammation: Cowpox virus encodes an inhibitor of the interleukin-1β converting enzyme. Cell 69: 597–604. [DOI] [PubMed] [Google Scholar]
- Rubin H., Wang, Z.M., Nickbarg, E.B., McLarney, S., Naidoo, N., Schoenberger, O.L., Johnson, J.L., and Cooperman, B.S. 1990. Cloning, expression, purification and biological activity of recombinant native and variant human α1-antichymotrypsins. J. Biol. Chem. 265: 1199–1207. [PubMed] [Google Scholar]
- Schuler B., Lipman, E.A., and Eaton, W.A. 2002. Probing the free-energy surface of protein folding with single-molecule fluorescence spectroscopy. Nature 419: 743–747. [DOI] [PubMed] [Google Scholar]
- Seo E.J., Im, H., Maeng, J.-S., Kim, K.E., and Yu, M.-H. 2000. Distribution of the native strain in human α1-antitrypsin and its association with protease inhibitor function. J. Biol. Chem. 275: 16904–16909. [DOI] [PubMed] [Google Scholar]
- Seo E.J., Lee, C., and Yu, M.-H. 2002. Concerted regulation of inhibitory activity of α1-antitrypsin by the native strain distributed throughout the molecule. J. Biol. Chem. 277: 14216–14220. [DOI] [PubMed] [Google Scholar]
- Shin J.-S. and Yu, M.-H. 2006. Viscous drag as the source of active site perturbation during protease translocation: Insights into how inhibitory processes are controlled by serpin metastability. J. Mol. Biol. 359: 378–389. [DOI] [PubMed] [Google Scholar]
- Silverman G.A., Bird, P.I., Carrell, R.W., Church, F.C., Coughlin, P.B., Gettins, P.G.W., Irving, J.A., Lomas, D.A., Luke, C.J., Moyer, R.W., et al. 2001. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions and a revised nomenclature. J. Biol. Chem. 276: 33293–33296. [DOI] [PubMed] [Google Scholar]
- Tachias K. and Madison, E.L. 1997. Variants of tissue-type plasminogen activator that display extraordinary resistance to inhibition by the serpin plasminogen activator inhibitor type 1. J. Biol. Chem. 272: 14580–14585. [DOI] [PubMed] [Google Scholar]
- Tew D.J. and Bottomley, S.P. 2001. Intrinsic fluorescence changes and rapid kinetics of proteinase deformation during serpin inhibition. FEBS Lett. 494: 30–33. [DOI] [PubMed] [Google Scholar]
- Ye S., Cech, A.L., Belmares, R., Bergstrom, R.C., Tong, Y., Corey, D.R., Kanost, M.R., and Goldsmith, E.J. 2001. The structure of a Michaelis serpin–protease complex. Nat. Struct. Biol. 8: 979–983. [DOI] [PubMed] [Google Scholar]
- Zhou A., Carrell, R.W., and Huntington, J.A. 2001. The serpin inhibitory mechanism is critically dependent on the length of the reactive center loop. J. Biol. Chem. 276: 27541–27547. [DOI] [PubMed] [Google Scholar]