

Abstract
The ability to rationally increase the stability and solubility of recombinant proteins has long been a goal of biotechnology and has significant implications for biomedical research. Poorly soluble enzymes, for example, result in the need for larger reaction volumes, longer incubation times, and more restricted reaction conditions, all of which increase the cost and have a negative impact on the feasibility of the process. Rational design is achieved here by means of the PoPMuSiC program, which performs in silico predictions of stability changes upon single-site mutations. We have used this program to increase the stability of the tobacco etch virus (TEV) protein. TEV is a 27-kDa nuclear inclusion protease with stringent specificity that is commonly used for the removal of solubility tags during protein purification protocols. However, while recombinant TEV can be produced in large quantities, a limitation is its relatively poor solubility (generally ∼1 mg/mL), which means that large volumes and often long incubation times are required for efficient cleavage. Following PoPMuSiC analysis of TEV, five variants predicted to be more stable than the wild type were selected for experimental analysis of their stability, solubility, and activity. Of these, two were found to enhance the solubility of TEV without compromising its functional activity. In addition, a fully active double mutant was found to remain soluble at concentrations in excess of 40 mg/mL. This modified TEV appears thus as an interesting candidate to be used in recombinant protein technology.
Keywords: TEV protease, stability, protein engineering, protein purification, in silico design
Recombinant proteins and peptides have an almost endless list of applications in biotechnology, biomedicine, and drug development. However, the limited stability and solubility of recombinant proteins can severely reduce their usefulness (Cabrita and Bottomley 2004; Chow et al. 2006). Thus a primary objective and a significant challenge in the biotechnology sector is the ability to increase through mutagenesis, the stability and solubility of proteins and peptides without affecting their activity. Two types of approaches can be used to guide the selection of appropriate mutations. The first is directed evolution experiments, where the protein is subjected to random mutagenesis and then screened for the chosen property (Roodveldt et al. 2005; van den Berg et al. 2006). The second approach, used here on the protease TEV, exploits bioinformatics programs to rationally design mutants presenting the required properties.
The 27-kDa C-type cysteine protease of the tobacco etch virus (TEV) is commonly used to remove solubility tags from either the N or C termini of recombinant proteins. TEV displays very high sequence specificity, targeting the recognition sequence ENLYFQG/S (Parks et al. 1995). This exquisite specificity allows TEV to be used at relatively high concentrations in a reaction without inflicting nonspecific proteolytic activity. Moreover, it can be recombinantly produced in Escherichia coli and is adaptable to a range of different buffering conditions. It is for these reasons combined that TEV has become a very popular protease. However, one drawback is that purified TEV suffers from limited solubility, and thus its production and storage can be problematic. At present, recombinant TEV is generally soluble at concentrations below ∼1 mg/mL and is typically stored with up to 50% (v/v) glycerol (Kapust et al. 2001). The addition of TEV in large amounts can complicate downstream processes, owing to the high concentration of glycerol (up to 25% [v/v]) that can be present. While comparatively small amounts of TEV are required for cleavage reactions in solution, some experiments such as on-column cleavage can require many more units of the protease for appreciable cleavage in a reasonable time frame. In addition, the ability to add large amounts of concentrated TEV would effectively accelerate such cleavage reactions, which can then proceed overnight at 4°C or at room temperature. This would be particularly useful for proteins that have a severe temperature dependency.
We thus sought to improve the stability and the solubility of TEV such that it would (1) maintain its structural integrity and activity as well as (2) remain soluble at high concentrations. The selection of mutations that could increase the stability of TEV was made with PoPMuSiC, a program that uses database-derived potentials to predict changes in folding free energy upon mutation (Gilis and Rooman 2000; Kwasigroch et al. 2002; Gilis et al. 2003). Based on the folding free energy estimations of PoPMuSiC, five point mutations predicted to increase the native-state stability of TEV were selected. Two of these mutations increase the surface polarity and conformational entropy of the protein and were therefore expected to enhance the solubility, with the assumption that they do not stabilize the aggregated state as strongly as they would the native state. The three other mutations decrease the surface polarity and are therefore not likely to improve the solubility. The five mutant proteins were produced and characterized experimentally. It was found that they all enhanced, or at least do not destabilize, the native-state stability, as predicted. In addition, the two variants that increase the surface polarity were demonstrated to significantly improve the solubility of TEV without compromising functional activity. A double-point mutation was thus generated and was found to have the ability to be concentrated at least 40 times more than what can be achieved with wild type. In addition to presenting an improved TEV variant, we illustrate the effectiveness of applying the PoPMuSiC algorithm to a common biotechnological problem.
Results
Search for TEV variants with enhanced stability and solubility
The aim of our study consisted of producing TEV variants that were easier to handle during biotechnology experiments. We focused on two parameters: the thermodynamic stability characterized by the folding free energy, ΔG, and the solubility of the variants. Initially, we searched for mutations that increase the thermodynamic stability of TEV.
With this in mind, we applied the PoPMuSiC program (see Materials and Methods) to the X-ray structure of the TEV protease, in order to identify mutations that were likely to stabilize the native form. We used the TEV structure complexed with its peptide substrate, with PDB code 1LVM (Phan et al. 2002). With the help of PoPMuSiC, all possible single-site mutations were introduced in silico in this structure (amounting to 4351 mutations), and their changes in folding free energy (ΔΔG) were computed. To reduce the errors in the ΔΔG values due to limitations of database-derived potentials, two different ΔΔG values were calculated for each mutation, using potentials derived from two different structure sets (see Materials and Methods). In what follows, the ΔΔG values refer to the average of these two values. Mutations involving prolines were ignored for they are likely to provoke conformational rearrangements in the protein, which are not taken into account by the PoPMuSiC program. Similarly, we did not consider mutations of residues that interact with the catalytic triad (Fig. 1) to avoid alterations to the activity of the protease. Among all tested mutations, the highly stabilizing mutations were identified, presenting a ΔΔG value less than or equal to −1 kcal/mol.
Figure 1.

Structure of the TEV protease. (Red) The residues subject to mutation (K45, L56, Q58, E106, S135). The four pictures correspond to the neighborhood of these five residues. (Blue) The catalytic triad H46, D81, and C151. The figure was generated with the PyMOL viewer (http://sourceforge.pymol.org).
Eight stabilizing mutations were selected according to these criteria. They are listed in Table 1 and presented in Figure 1. They are located at five different positions, one in the core (residue K45), two near the surface (E106 and S135), and two in between (L56 and Q58).
Table 1.
Stability, solubility, and structural parameters of TEV protease and its variants

Among these mutations, one mutation per position was selected for experimental tests. As we were also interested in improving TEV's solubility, we tried to determine which among the selected mutations at each of the five positions were most likely to enhance the solubility.
The solubility of proteins is determined by the relative stabilities of their solvated and solid (aggregated) forms. However, not much is known about the aggregated form. Does the protein undergo a conformational change upon aggregation, or does the solid state consist of packed proteins in native form? The answer to this issue is moreover likely to depend on the type of protein and its environmental conditions. If the protein maintains its structure upon aggregation, the solubility should be determined solely by the surface residues, because their interactions and entropies differ in the two states. In contrast, if the protein structure changes, information about the two structures is needed. In the two cases, the stabilization of the native structure through mutations of surface residues optimizing the interactions with the solvent is likely to increase the solubility, except if the aggregated state is equally stabilized. If there is a conformational change upon aggregation, some mutations of core residues may also improve the solubility.
Here, we do not have any indications about the structure of the aggregated TEV. A possibility to identify solubility-improving mutations was to select the mutations that optimize the free energy contributions related to interactions with the solvent, assumed to vanish in the aggregated form. The ΔΔG values in PoPMuSiC were computed using statistical mean force potentials that mix inter-residue interactions, entropic effects, and solvent contributions. It was therefore impossible to optimize the interactions with the solvent independently of the others. To solve this problem, when several stabilizing mutations were proposed by PoPMuSiC at one position, we selected the most polar of the mutated residues. We used, for that purpose, several amino acid polarity scales (Fauchere and Pliska 1983; Eisenberg et al. 1984; Wimley and White 1996).
Initially we retained the mutations L56V, E106G, and S135G, where only one mutated residue was identified per position. L56V increased the polarity of the protein, E106G decreased it, and S135G slightly increased or decreased it according to the hydrophobicity scale used. At the other two positions, 45 and 58, polar residues were substituted by hydrophobic ones. We kept the mutations K45F and Q58F, which present the smallest losses in polarity. Note that position K45, with its solvent accessibility of only 6%, can only be expected to play a part in solubility if aggregation involves structural modifications.
In summary, we selected five mutations stabilizing the native form for experimental tests. Two of them, the mutations L56V and S135G, tend to increase the overall surface polarity and were thus most likely to enhance the protein's solubility.
Experimental characterization of the selected mutations
The two mutants assumed to enhance the solubility (L56V and S135G), as well as three other stabilizing mutants (K45F, Q58F, and E106G), were introduced into a soluble TEV expression system and produced (Kapust and Waugh 1999). For each of these variants, the protein concentration was measured using a BCA assay, assuming that the protease had 100% activity. Circular dichroism was used to assess the effect of the mutations on the stability of the structure. Each variant was thermally denatured (25°–90°C), and the change in secondary structure was measured at 230 nm. The midpoint of denaturation, which is a measure of the thermostability of the structure, was found to be 52.1°C for wild type (WT). According to our measurements, all mutants except K45F were more thermostable than WT TEV (Table 1). We also determined the thermodynamic stability for each of the proteins (Fig. 2; Table 1). All of the proteins unfolded in a reversible two-state manner in accord with a previous study on TEV (Kapust et al. 2001). All the proteins were at least as stable as wild-type TEV, in keeping with the thermal stability data (Table 1). Q58F and E106G displayed the free energy of unfolding, 5.2 and 6.0 kcal/mol, respectively. In accord with the thermal stability data, L56V and S135G had little effect on the thermodynamic stability. In addition, the double mutant L56V/S135G had similar thermodynamic stability to wild-type TEV and the variants L56V and S135G (Table 1; Fig. 2). There is reasonable agreement between the ΔΔG values calculated using PoPMuSiC and those observed experimentally.
Figure 2.

Equilibrium denaturation of TEV and its variants. The unfolding of TEV was followed by monitoring the change in the center of spectral mass as a function of denaturant concentration.
The variants were subsequently assessed for their solubility using a centrifugal approach. Each of the variants was concentrated, while samples were collected at regular intervals, and the protein concentration was measured. Figure 3A compares the concentration profiles for wild-type TEV and the variant L56V. As illustrated, the plateau observed in the absorbance reading indicated the solubility threshold, beyond which the protein no longer remains in solution. The point mutations L56V and S135G induced a marked improvement in TEV solubility (Fig. 3B), with a maximum concentration of 6.2 mg/mL and 5.77 mg/mL, respectively. The other variants (K45F, Q58F, E106G) were found to have similar solubilities to the WT and thus were not studied further.
Figure 3.
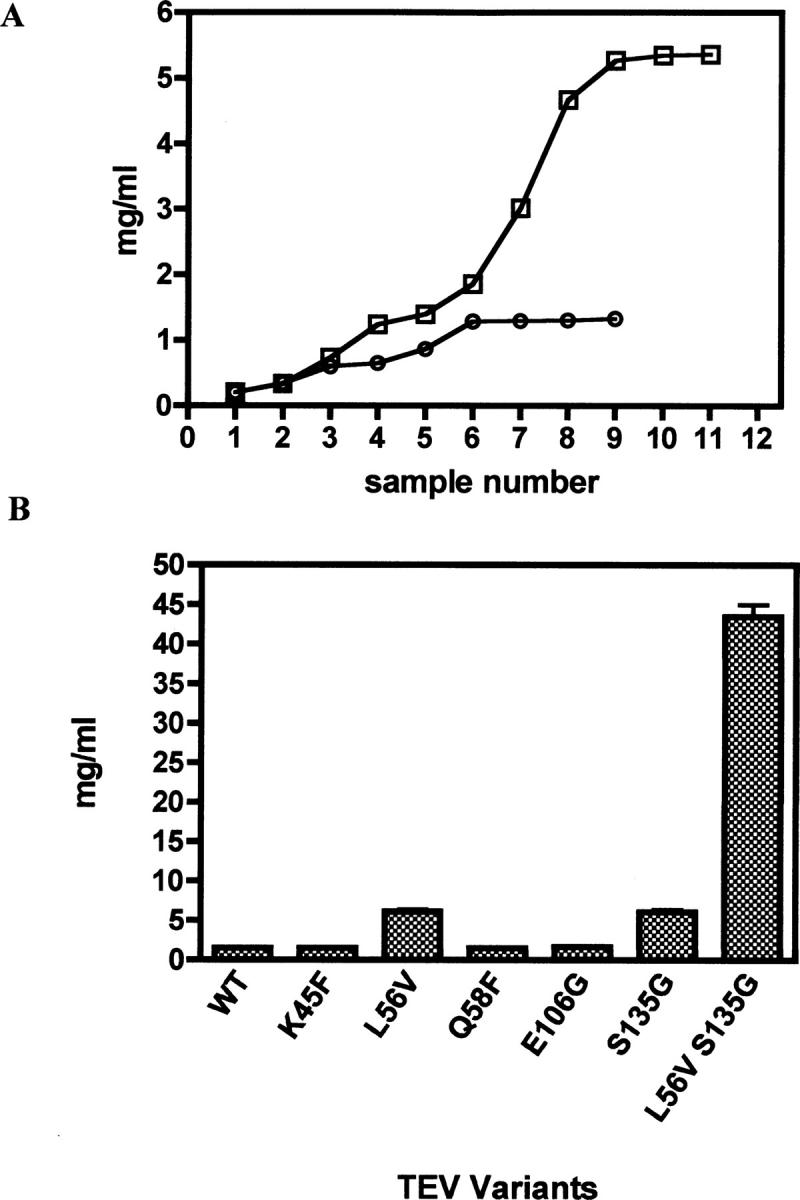
Solubility of the TEV variants. (A) Solubility curve for (○) WT and (□) L56V. The “maximum” concentration is obtained from the plateau observed in concentration. (B) Comparison of maximum concentrations obtained for each of the variants relative to WT TEV; the data shown represent the average of five separate experiments.
To verify the integrity of the solubility-enhancing variants, the activity was measured using a fluorescence-quenched substrate assay. Wild-type TEV followed simple Michaelis-Menten kinetics as determined previously (Kapust et al. 2001, 2002) and under the conditions tested, was found to have the characteristics as presented in Table 1. The variants L56V and S135G were assayed under the same conditions and were found to be either similar or slightly better than WT. This was the case for L56V, for instance, which demonstrated a slightly improved k cat/K m value compared to wild type (6.98 vs. 6.09 for WT) (Table 2).
Table 2.
Kinetic parameters of TEV protease and its variants

To complement the fluorescence-based kinetics, the ability of the TEV variants to cleave a protein subject was investigated (Fig. 3). The substrate used was UBL-GST, a 38-kDa fusion between GST and the UBL domain of the protein HHR23A, and it contains the TEV recognition sequence between the two domains (Cabrita et al. 2006). The reaction was performed at a temperature of 30°C and a ratio of 1:10 (TEV to substrate), which was used to allow for the time course to be followed over a reasonable time frame. In addition, it was more practical to monitor the degree of cleavage by the “loss” of the 38-kDa fusion as opposed to the appearance of the 8-kDa UBL domain. As can be seen in Figure 3, cleavage of the UBL-GST by TEV was found to be >90% complete after 2 h. A similar trend was also followed for each of the variants, which again indicates that there was no compromise in the functionality of TEV. In addition, a time course also completed at 4°C and 20°C illustrated that the mutants retained their activity at the common temperatures used for proteolytic cleavage. Under these conditions (1:10 ratio), on average, TEV requires ∼2–3 h to cleave at least 90% of the substrate at room temperature and a minimum of 6–8 h at 4°C (data not shown).
A double mutant of TEV with a greatly enhanced solubility
Of the five variants that were selected, two mutations (L56V, S135G) were found to enhance the thermal stability and the solubility of TEV without compromising the activity. These two mutations were thus introduced simultaneously to produce the double variant L56V/S135G. As these two mutations belonged to very different regions of the protein (Fig. 1), the double variant was expected to present larger thermal stability and solubility than the single mutants.
The double variant showed an increase in the thermal stability compared to WT, but not larger than the single-point mutation S135G (Table 1). On the other hand, the solubility was greatly enhanced, allowing the variant to be concentrated in excess of 40 mg/mL (Fig. 2B).
The activity of the double variant was assessed using both the fluorescence-based and PAGE assays and found to be slightly better than wild type (Table 2; Fig. 4). The activity of the mutant relative to its concentration was also investigated, in order to gauge whether there was any concentration-dependent inactivation. The activity was also assessed when the double variant was concentrated to a concentration of 5, 10, 20, and 40 mg/mL and stored for 24 h at 4°C. The activity of the TEV variant at each concentration was identical, indicating that no aggregation was occurring upon concentration and storage (data not shown).
Figure 4.

Protease activity. SDS-PAGE analysis of TEV cleavage of the protein substrate UBL-GST as a function of time at 30°C.
Discussion
For the removal of solubility tags from a purified protein, TEV is a popular protease because of its high specificity, although it presents a challenge in its production and storage because of its low solubility. To overcome this bottleneck, we performed a rational search for point mutations that could improve TEV's native-state stability and solubility. The PoPMuSiC algorithm was used to introduce, in silico, all possible single-site mutations in TEV, with the aim of selecting the stabilizing ones. Among these mutations, one increased the polarity of the protein surface, while another kept it constant. They were therefore suspected to have a positive impact on the solubility. When tested experimentally, these two mutations were, indeed, shown to lead to an improvement in the native-state thermal stability coupled to an enhancement of the TEV's solubility.
The principles governing protein solubility are complex and are a function of the amino acid sequence, pH, salt concentration, and temperature. In fact, solubility is a competing phenomenon between solvation of the native structure of the protein and its aggregation. At present, much attention is being geared toward elucidating the molecular determinants of protein aggregation. One school of thought is that aggregation is governed by electrostatic repulsions, and thus by altering the net charge of a protein, the solubility could be improved (Nishimura et al. 2001; Zhang et al. 2004; Calloni et al. 2005). The mutations (L56V and S135G) found here to improve the solubility do not modify the net charge, but they alter the surface interactions in a manner that would favor solubility against aggregation. Thus the results of the present support the idea that solubility can, indeed, be enhanced by stabilizing the native state, especially through substitutions of solvent-accessible residues into more polar amino acids. Of course, this cannot be expected to be a perfectly general law: Some mutations of core residue may improve solubility, for example, through structural rearrangements upon aggregation. On the other hand, it should not be excluded that some stabilizing surface mutations may lead to a deteriorated solubility, for instance, due to a larger stabilization of the aggregated form. However, our criteria allow us to select, for further experimental tests, a limited number of candidate mutants that are very likely to display an improved solubility.
The solubility of TEV was shown to increase by a factor of 4 upon substitution of L56 into V. Although V is significantly more polar than L, such a large increase was, indeed, surprising. The analysis of the spatial neighborhood of L56 in the native structure gives additional hints that explain the solubility improvement. Using the PyMOL viewer (http://sourceforge.pymol.org), we built the 19 rotamers of K65, the closest residue to L56. Of these, only four do not lead to steric clashes with L56 in the wild-type structure, against nine in the L56V mutant. Moreover, some of the additional rotamers provoke a decrease of the solvent accessibility of V56 and an increase in the solvent accessibility of K65. As a consequence, the large increase in the solubility of the L56V variants can be explained by the larger polarity of V relative to L and by the larger number of degrees of freedom of K65 present in the variant. As a consequence, the large increase of the solubility of the L56V variant can be explained by the larger polarity of V relative to L, the larger number of degrees of freedom of K65 in the variant and thus its larger conformational entropy, and the resulting side-chain rearrangements that increase the solvent accessibility of the polar K65 and decrease that of the hydrophobic V56. All these free energy contributions are expected to be favorable to the native structure but not to the aggregated one.
The S135G variant was shown to multiply the solubility of WT by a factor of 4. This significant increase was not obvious to explain, given that the polarities of S and G can be considered as roughly equal, even though they vary among the different hydrophobicity scales. It can, however, be argued that replacing an S by a G in a surface loop increases the flexibility of the chain and thus the conformational entropy of the native state, whereas the entropy of the aggregated structure is unlikely to be affected, owing to protein–protein interactions.
In conclusion, the major goal of our study, designing a fully active TEV variant with improved stability and high solubility, was successfully reached. This is an important achievement since the relatively poor solubility of TEV was a strong limitation to its otherwise valuable role in biotechnology. We would also like to highlight that this was accomplished through a rational approach combining in silico and in vitro techniques. Such an approach is applicable to any protein for which a structure (X-ray, NMR, or even modeled) is available and is thus likely to become more of a powerful tool, particularly in the biotechnological area.
Materials and Methods
Generation of the TEV variants using PoPMuSiC
The PoPMuSiC program proceeds by introducing all the possible single-site mutations in a protein structure and predicting the resulting folding free energy changes (ΔΔG computed = ΔG mutant − ΔG wild-type) (Gilis and Rooman 2000; Kwasigroch et al. 2002). It requires as input the protein structure in PDB format (Berman et al. 2000) and uses a simplified structural representation consisting of the Cartesian coordinates of the C, N, Cα, and O backbone atoms; the side-chain atom Cβ; and the pseudoatom Cμ corresponding to the side-chain geometric center averaged over all rotamers of a given residue in a protein set (Kocher et al. 1994). Mutations are assumed to keep the backbone structure unchanged. The calculations were performed in the absence of the substrate.
The ΔΔG values are computed in PoPMuSiC by means of database-derived potentials. These potentials are obtained from frequencies of association between residues or residue pairs and structural descriptors in a data set of known protein structures. They implicitly take the solvent into account, as they are derived from experimental structures in solution. Two data sets are used here: one consisting of 141 proteins with resolution ≤2.5 Å and presenting <25% sequence identity (Wintjens et al. 1996), and the other of 735 chains with resolution ≤2.0 Å and at most 20% sequence identity (Hobohm et al. 1992).
Two types of potentials are derived from these data sets. The torsion potential describes local interactions along the sequence and is obtained from frequencies of association of residues or residue pairs with backbone torsion angle domains. The distance potential is computed from propensities of residue pairs to be separated by a spatial distance evaluated between Cμs. The stability changes upon mutation are evaluated by linear combinations of these potentials:
where ΔΔG torsion and ΔΔG distance are the folding free energy changes upon mutation, computed with the torsion and the distance potentials, respectively. α, β, and γ are weighting factors depending on the solvent accessibility of the mutated residue, A (Gilis and Rooman 2000).
The good performances of PoPMuSiC have been attested to previously. Correlation coefficients between computed and measured ΔΔGs are equal to 0.87 on surface residues and 0.80 on buried residues of a test set (Gilis and Rooman 1996, 1997). Another study showed an excellent agreement between blind predictions on a protein from the serpin family and the experimental characterization of the mutations (Gilis et al. 2003).
Production of the TEV variants
The following point mutations were introduced into TEV-S219V to produce five variants: K45F, L56V, Q58F, E106G, and S135G. Two point mutations were introduced to produce the double mutant L56V/S135G. The mutations were introduced using the QuikChange Site Directed Mutagenesis kit (Stratagene). The proteins were expressed in BL21pRIPL E. coli (Stratagene), typically 1 L of culture, following isopropyl β-D-thiogalactopyranoside (IPTG) (0.5 mM) induction. The cells were then lysed using sonication. The sample was then applied to 1 mL of Ni-NTA agarose (GE Healthcare) pre-equilibrated with 25 mM sodium phosphate (pH 8.0), 10% (v/v) glycerol, 0.2 M NaCl, and 25 mM imidazole after which the protein was eluted with 25 mM sodium phosphate (pH 8.0), 10% (v/v) glycerol, 0.2 M NaCl, 500 mM imidazole. The eluted protein was then applied to a Superdex 75 (16/10) column in the following buffer: 25 mM Na2PO4, 200 mM NaCl, 10% (v/v) glycerol, 5 mM β-mercaptoethanol (pH 8.0). The eluted fractions containing TEV were pooled, flash-frozen in liquid nitrogen, and stored at −80°C.
Thermal stability
The thermal stability of the TEV variants was measured using circular dichroism. 0.2 mg/mL TEV in 25 mM Na2PO4, 200 mM NaCl, and 10% (v/v) glycerol was heated at a rate of 1°C/min between 20°C and 90°C, and the secondary structural changes were monitored at 230 nm.
Solubility assay
The solubility of TEV was assayed by concentrating the protein and taking aliquots at various stages, pelleting any precipitate, and assaying the concentration using the BCA assay. The maximum concentration was obtained when an equilibrium was reached, as determined in a plateau in absorbance readings.
Enzyme kinetics
Enzyme kinetics were performed at 30°C, in 25 mM Na3PO4, 200 mM NaCl, 10%(v/v) glycerol, and 5 mM BME (pH 8.0). The fluorescence quenched substrate Abz-E-N-L-Y-F-Q-G-A-A-Lys(DNP)-OH was synthesized to >90% purity (Auspep), and the concentration was determined using the relation 104 M−1 cm−1 at 360 nm, which monitors the absorbance of the DNP moiety. Fluorescence changes were monitored on a BMG Technologies FluorStar Galaxy fluorescent plate reader using an excitation wavelength of 320 nm and an emission wavelength of 420 nm. The initial reaction rate was determined at a single concentration of enzyme over a substrate range from 20 μM to 2 mM, and the data were fitted to the Michaelis-Menten equation to derive the kinetic values: V max, K m, k cat, and k cat/K m.
Activity of TEV using gel electrophoresis
Samples containing 10 μL of 0.5 mg/mL UBL-GST and 10 μL of 0.1 mg/mL TEV were incubated at 30°C and removed at 20-min intervals over the time range of 0–120 min. The samples were mixed with Laemmli buffer and boiled. The samples were then loaded onto a 12% (v/v) SDS gel.
Equilibrium unfolding analysis
Fluorescence emission spectra were recorded on a Perkin-Elmer LS50B spectrofluorimeter at 25°C in a 1-cm path length quartz cell. Excitation and emission slits were set at 5 nm for all spectra, and a scan speed of 10 nm/min was used. The absorbance at the excitation wavelength was monitored in all experiments and remained below 0.03. We followed unfolding by monitoring the change in center of spectral mass wavelength (COSM) as described previously (Kapust et al. 2001; Tew and Bottomley 2001). COSM is calculated as follows:
where F(λi) is the fluorescence intensity measured at the emission wavelength λi.
Stock solutions of GdnHCl in 25 mM Na2PO4, 200 mM NaCl, and 10% (v/v) glycerol were prepared and filtered through 0.22-μm membranes before use. Equilibrium unfolding curves were determined by adding a concentrated solution of native protein to a series of denaturant solutions. These solutions were incubated for 2 h at 25°C before analysis by measuring the fluorescence emission spectra from 300 to 450 nm. No differences were observed in experiments where spectroscopic measurements were made after more extensive equilibration. All unfolding curves were found to be fully reversible, and the data were fit to a two-state unfolding model using a nonlinear least-squares fitting algorithm. In determining the stability of TEV and its variants, we used an average m value of 3.6 kcal/mol per M in the global fitting procedure.
Acknowledgments
We thank David Waugh (National Cancer Institute at Frederick) for his gift of the wild-type TEV expression plasmid. This work was supported by grants from the National Health and Medical Research Council and the Australian Research Council to S.P.B. and D.G. Y.D. and M.R. acknowledge support from the Communauté Française de Belgique through the Action de Recherche Concertée 02/07-289, from the Belgian State Science Policy Office through an Interuniversity Attraction Poles Programme (DYSCO), and from the Belgian Fund for Scientific Research (FRS) through an FRFC project. M.R. is Research Director at the FRS. S.P.B. is a NHMRC Senior Research Fellow. L.D.C. is a CJ Martin Fellow of the NHMRC.
Footnotes
Reprint requests to: Stephen Bottomley, Department of Biochemistry and Molecular Biology, School of Biomedical Sciences, Monash University, Clayton 3800, Australia; e-mail: steve.bottomley@med.monash.edu.au; fax: 61-3-99054699.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072822507.
References
- Berman H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28: 235–242. doi: 10.1002/047001153X.g406303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita L.D. and Bottomley, S.P. 2004. Protein expression and refolding—A practical guide to getting the most out of inclusion bodies. Biotechnol. Annu. Rev. 10: 31–50. [DOI] [PubMed] [Google Scholar]
- Cabrita L.D., Dai, W., and Bottomley, S.P. 2006. A family of E. coli expression vectors for laboratory scale and high-throughput soluble protein production. BMC Biotechnol. 6: 12. doi: 10.1186/1472-6750-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloni G., Zoffoli, S., Stefani, M., Dobson, C.M., and Chiti, F. 2005. Investigating the effects of mutations on protein aggregation in the cell. J. Biol. Chem. 280: 10607–10613. [DOI] [PubMed] [Google Scholar]
- Chow M.K., Amin, A.A., Fulton, K.F., Whisstock, J.C., Buckle, A.M., and Bottomley, S.P. 2006. REFOLD: An analytical database of protein refolding methods. Protein Expr. Purif. 46: 166–171. [DOI] [PubMed] [Google Scholar]
- Eisenberg D., Schwarz, E., Komaromy, M., and Wall, R. 1984. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179: 125–142. [DOI] [PubMed] [Google Scholar]
- Fauchere J.L. and Pliska, V. 1983. Hydrophobic parameters—π of amino-acid side-chains from the partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem. 18: 369–375. [Google Scholar]
- Gilis D. and Rooman, M. 1996. Stability changes upon mutation of solvent-accessible residues in proteins evaluated by database-derived potentials. J. Mol. Biol. 257: 1112–1126. [DOI] [PubMed] [Google Scholar]
- Gilis D. and Rooman, M. 1997. Predicting protein stability changes upon mutation using database-derived potentials: Solvent accessibility determines the importance of local versus non-local interactions along the sequence. J. Mol. Biol. 272: 276–290. [DOI] [PubMed] [Google Scholar]
- Gilis D. and Rooman, M. 2000. PoPMuSiC, an algorithm for predicting protein mutant stability changes: Application to prion proteins. Protein Eng. 13: 849–856. [DOI] [PubMed] [Google Scholar]
- Gilis D., McLennan, H.R., Dehouck, Y., Cabrita, L.D., Rooman, M., and Bottomley, S.P. 2003. In vitro and in silico design of α1-antitrypsin mutants with different conformational stabilities. J. Mol. Biol. 325: 581–589. [DOI] [PubMed] [Google Scholar]
- Hobohm U., Scharf, M., Schneider, R., and Sander, C. 1992. Selection of representative protein data sets. Protein Sci. 1: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapust R.B. and Waugh, D.S. 1999. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8: 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapust R.B., Tozser, J., Fox, J.D., Anderson, D.E., Cherry, S., Copeland, T.D., and Waugh, D.S. 2001. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14: 993–1000. [DOI] [PubMed] [Google Scholar]
- Kapust R.B., Tozser, J., Copeland, T.D., and Waugh, D.S. 2002. The P1′ specificity of tobacco etch virus protease. Biochem. Biophys. Res. Commun. 294: 949–955. [DOI] [PubMed] [Google Scholar]
- Kocher J.P., Rooman, M.J., and Wodak, S.J. 1994. Factors influencing the ability of knowledge-based potentials to identify native sequence–structure matches. J. Mol. Biol. 235: 1598–1613. [DOI] [PubMed] [Google Scholar]
- Kwasigroch J.M., Gilis, D., Dehouck, Y., and Rooman, M. 2002. PoPMuSiC, rationally designing point mutations in protein structures. Bioinformatics 18: 1701–1702. [DOI] [PubMed] [Google Scholar]
- Nishimura C., Uversky, V.N., and Fink, A.L. 2001. Effect of salts on the stability and folding of staphylococcal nuclease. Biochemistry 40: 2113–2128. [DOI] [PubMed] [Google Scholar]
- Parks T.D., Howard, E.D., Wolpert, T.J., Arp, D.J., and Dougherty, W.G. 1995. Expression and purification of a recombinant tobacco etch virus NIa proteinase: Biochemical analyses of the full-length and a naturally occurring truncated proteinase form. Virology 210: 194–201. [DOI] [PubMed] [Google Scholar]
- Phan J., Zdanov, A., Evdokimov, A.G., Tropea, J.E., Peters III, H.K., Kapust, R.B., Li, M., Wlodawer, A., and Waugh, D.S. 2002. Structural basis for the substrate specificity of tobacco etch virus protease. J. Biol. Chem. 277: 50564–50572. [DOI] [PubMed] [Google Scholar]
- Roodveldt C., Aharoni, A., and Tawfik, D.S. 2005. Directed evolution of proteins for heterologous expression and stability. Curr. Opin. Struct. Biol. 15: 50–56. [DOI] [PubMed] [Google Scholar]
- Tew D.J. and Bottomley, S.P. 2001. Probing the equilibrium denaturation of the serpin α(1)-antitrypsin with single tryptophan mutants; evidence for structure in the urea unfolded state. J. Mol. Biol. 313: 1161–1169. [DOI] [PubMed] [Google Scholar]
- van den Berg S., Lofdahl, P.A., Hard, T., and Berglund, H. 2006. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 121: 291–298. [DOI] [PubMed] [Google Scholar]
- Wimley W.C. and White, S.H. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3: 842–848. [DOI] [PubMed] [Google Scholar]
- Wintjens R.T., Rooman, M.J., and Wodak, S.J. 1996. Automatic classification and analysis of α α-turn motifs in proteins. J. Mol. Biol. 255: 235–253. [DOI] [PubMed] [Google Scholar]
- Zhang Y.B., Howitt, J., McCorkle, S., Lawrence, P., Springer, K., and Freimuth, P. 2004. Protein aggregation during overexpression limited by peptide extensions with large net negative charge. Protein Expr. Purif. 36: 207–216. [DOI] [PubMed] [Google Scholar]