

Abstract
Cd36 is a small-molecular-weight integral membrane protein expressed in a diverse, but select, range of cell types. It has an equally diverse range of ligands and physiological functions, which has implicated Cd36 in a number of diseases including insulin resistance, diabetes, and, most notably, atherosclerosis. The protein is reported to reside in detergent-resistant microdomains within the plasma membrane and to form homo- and hetero-intermolecular interactions. These data suggest that this class B scavenger receptor may gain functionality for ligand binding, and/or ligand internalization, by formation of protein complexes at the cell surface. Here, we have overexpressed Cd36 in insect cells, purified the recombinant protein to homogeneity, and analyzed its stability and solubility in a variety of nonionic and zwitterionic detergents. Octylglucoside conferred the greatest degree of stability, and by analytical ultracentrifugation we show that the protein is monomeric. A solid-phase ligand-binding assay demonstrated that the purified monomeric protein retains high affinity for acetylated and oxidized low-density lipoproteins. Therefore, no accessory proteins are required for interaction with ligand, and binding is a property of the monomeric fold of the protein. Thus, the highly purified and functional Cd36 should be suitable for crystallization in octylglucoside, and the in vitro ligand-binding assay represents a promising screen for identification of bioactive molecules targeting atherogenesis at the level of ligand binding.
Keywords: Cd36, scavenger receptor, ligand binding, protein purification, low-density lipoprotein, oxidized LDL, atherosclerosis, membrane protein
Cd36 is a multiligand membrane glycoprotein, highly expressed on the surface of platelets, monocyte/macrophages, endothelial cells of the microvasculature, adipose tissue, skeletal muscle, and heart. Cd36 function is complex and has been associated with diverse normal physiological processes (for example, uptake of modified lipid and apoptotic cells, long-chain fatty acid transport, adhesion, and angiogenesis), and its expression or deficiency has been linked with several pathological conditions, such as atherosclerosis, diabetes, and cardiomyopathy. For example, Cd36 is a high-affinity receptor for oxidatively modified low-density lipoprotein (oxLDL) and has accordingly been classified as a class B scavenger receptor (Endemann et al. 1993). This scavenger receptor function has been strongly implicated in the pathophysiology of atherosclerosis in which accumulation of oxLDL generated at sites of injury or inflammation at the arterial wall attracts macrophages, which in turn bind and internalize oxLDL, giving rise to lipid-laden foam cells. Fatty streaks or plaques formed from these foam cells in the arterial wall are a key stage in early atherogenesis. Cd36 has been identified as the primary scavenger receptor for oxLDL in macrophages, and loss of expression in the Cd36-null mouse, in a pro-atherogenic apoE-null background, showed a significant reduction in the development of plaques (Febbraio et al. 2000). Atherosclerosis is the principal cause of heart attack and stroke and accounts for 50% of all mortality in the Western world. Yet, given the role of Cd36 in early atherogenesis, the molecular mechanism underlying the binding and uptake of modified LDL by Cd36 is not fully understood.
Cd36 is a membrane-bound protein of 471 amino acids with a large extracellular loop (ECL) and, in all likelihood, two transmembrane α-helices, one each at the amino and carboxy termini (Tao et al. 1996; Gruarin et al. 2000). The protein is modified extensively post-translationally, and evidence showing palmitoylation of each of the four cysteines at positions 3, 7, 464, and 466 of the two short putative intracellular termini (Tao et al. 1996) supports the hairpin topology model. Recent evidence suggests that the palmitoylation is important for targeting of the protein to detergent-resistant microdomains in the plasma membrane (J. Wharton, C.A. Martin, and K.J. Linton, unpubl.). A further six cysteines are clustered in the C-terminal half of the ECL, and in bovine Cd36, which is 83% identical to rat Cd36, all six have been shown to be involved in disulfide bridging (Rasmussen et al. 1998). The disulfide bridging appears to be necessary for proper folding and trafficking of the protein to the cell surface (Gruarin et al. 2000). The ECL is also heavily glycosylated, and there are nine consensus glycosylation sites in rat Cd36 (this number varies between eight and 10 in mammalian orthologs). Seven of these sites are conserved in bovine Cd36 where all eight have been shown to be utilized (Berglund et al. 1996), although only eight of the 10 consensus sites in the human form appear to be utilized (S.J. Hoosdally, E. Andress, C. Wooding, C.A. Martin, and K.J. Martin, unpubl.).
The multiligand binding activity of Cd36 is mediated by the ECL. Specific sites implicated in this process have been mapped to the amino-terminal half of this domain, principally through the use of a variety of antibodies whose epitopes lie within an “immunodominant” domain (amino acids 155–183), to block specific Cd36 functions (Barnwell et al. 1985; Leung et al. 1992; Savill and Hogg 1992; Nicholson et al. 1995). The ligand-binding domain was further defined to suggest the presence of distinct but overlapping sites for ligand from competition studies with synthetic or recombinant Cd36 peptides (Asch et al. 1993; Frieda et al. 1995; Pearce et al. 1998). However, nothing is known of the shape of the binding pocket(s) or how the Cd36 molecule interacts with structurally diverse ligands. This has been due in large part to the absence of crystal structure data for Cd36. As there are no bacterial homologs of scavenger receptors, progress in this area is dependent on the study of the target protein directly and must overcome the difficulties associated with the attainment of structure data for mammalian membrane proteins. It has been suggested that Cd36 is homo- or hetero-multimeric, and indirect evidence has indicated that intermolecular disulfide bridging involving cysteines in the ECL are necessary for these interactions (Rhinehart-Jones and Greenwalt 1996; Thorne et al. 1997), and that localization of Cd36 to cholesterol-rich membrane microdomains is necessary for uptake of modified LDL (Zeng et al. 2003) and long-chain fatty acids (Pohl et al. 2005). These detergent-resistant microdomains, a form of lipid raft, have been hypothesized to be important for protein–lipid and protein–protein interactions at the membrane (Simons and Toomre 2000). However, it is not clear from the previously published data whether the homo- or hetero-quarternary structural interactions are important for initial recognition of ligand, or for internalization of the bound ligand.
In this paper, we have adopted a novel approach to investigate the molecular requirements for Cd36 to bind modified LDL. This has enabled us to investigate whether the Cd36 protein, purified from the raft microenvironment in which it normally resides, can recognize ligand in vitro and if protein complex formation or homo-multimerization is necessary for initial recognition of ligand. Purified rat Cd36 can bind modified LDL with high affinity, which shows that localization to the lipid raft microdomain is not a requirement for ligand binding. Furthermore, having demonstrated that the subunit complexity of the purified protein is monomeric, it is concluded that the protein interacts with these ligands in the absence of accessory proteins, and the minimal functional unit to bind ligand is a monomer.
Results
Heterologous expression of Cd36 in insect cells
Cd36 engineered with a 6-histidine tag at the C terminus was overexpressed in Trichoplusia ni (High Five) insect cells following infection with recombinant baculovirus. Maximal protein expression was achieved 72 h post-infection. The overexpressed protein was detected in crude membranes by Western blotting using a monoclonal antibody directed against the C-terminal His-tag (Fig. 1) or Cd36 itself (data not shown). Two proteins, of different apparent molecular weights (75 kDa and 50 kDa) were detected, as shown in Figure 1, lane 1. Treatment of the crude membrane fraction with the endoglycosidase PNGase F affected only the 75 kDa species, which was reduced to ∼50 kDa (Fig. 1, lane 2). The 75 kDa species therefore represented glycosylated Cd36, with the lower molecular weight form likely to be nonglycosylated but full-length Cd36 (the protein backbone has a predicted molecular weight of 55 kDa).
Figure 1.

Expression of Cd36 in High Five insect cell membranes. Membranes (20 μg) were treated with (lane 2) or without (lane 1) PNGase F, as described in Materials and Methods. Samples were separated by SDS-PAGE, and Cd36 was detected using anti-His (C-terminal) monoclonal antibody.
Efficient solubilization of Cd36 from insect cells
In order to purify the recombinant Cd36 protein for characterization studies, it was first necessary to find a nondenaturing detergent to efficiently extract Cd36 from the membrane fraction. Different classes of nonionic detergents with varying head-groups and acyl-chain lengths were tested for efficacy to solubilize Cd36 and compared with the ionic detergent SDS, which has high protein extractability. For each detergent tested, the crude membrane fraction was solubilized with a 2% detergent solution at a detergent:protein ratio of 4:1 (w/w). Equal amounts of total soluble membrane protein were analyzed by SDS-PAGE and Western blotting (Fig. 2). It was immediately apparent that only SDS was able to solubilize both the 75 kDa and 50 kDa forms of the protein, suggesting that the nonglycosylated Cd36 represents misfolded protein, perhaps aggregated similar to bacterial inclusion bodies. The efficacy of each nonionic detergent to solubilize the 75 kDa protein was measured and compared with SDS, which was considered 100% efficient (Table 1; Fig. 2). The smallest nonionic detergent, octylglucoside (OG), was the most effective tested, with up to 90% of the 75 kDa form of Cd36 solubilized.
Figure 2.

Solubilization of Cd36 from High Five cells using a range of nonionic detergents. Crude membrane preparations were solubilized with a 2% solution of each detergent at a detergent to protein ratio of 4:1 (w/w). Insoluble material was pelleted by ultracentrifugation. Soluble proteins in the supernatant (25 μg of total protein loaded per lane) were separated by SDS-PAGE, and Cd36 was detected using anti-His (C-terminal) monoclonal antibody. (Lane 1) Untreated membranes, (lane 2) octylglucoside, (lane 3) decylmaltoside, (lane 4) dodecylmaltoside, (lane 5) TX-100, (lane 6) TX-114, (lane 7) tri-decylmaltoside, (lane 8) tetra-decylmaltoside, (lane 9) SDS. Relative amount of solubilized protein was determined by densitometric analysis of Cd36 bands.
Table 1.
Efficacy of different detergents to extract Cd36 from High Five membranes

Purification of his-tagged Cd36 to near homogeneity by Ni-NTA affinity chromatography
The recombinant Cd36 was engineered initially with a 6× histidine tag at the C terminus to enable facile purification using the readily available Ni-NTA technology. However, it was only possible to purify the Cd36–6His protein to 90% homogeneity using a single-step purification protocol (Fig. 3A). A protein that migrated with an apparent molecular weight of 50 kDa copurified with Cd36–6His. Western analysis (data not shown), probing with anti-His and anti-Cd36 antibodies, demonstrated that this protein was not an immature or degraded Cd36 but more likely a native High Five cellular protein. In order to increase the homogeneity of the purified Cd36, which would be necessary for analysis by analytical ultracentrifugation, a recombinant Cd36 was engineered with a 12-histidine tag at the C terminus. Introduction of a longer tag did not compromise the expression of Cd36 protein in insect cells, and the protein behaved identically to the 6-histidine-tagged version during solubilization with detergent. However, the longer his-tag did confer recombinant Cd36 with higher-affinity binding to the Ni-NTA resin, allowing washes of higher stringency to remove contaminant proteins during the purification protocol. This strategy proved very successful, and it has been possible to purify the protein to at least 99% homogeneity (Fig. 3B). A 200-mL culture of Cd36-expressing High Five cells yields ∼200 mg of total membrane protein, from which 300–500 μg of purified Cd36–12His was typically obtained. Solubilization with OG extracts ∼90% of the glycosylated protein; therefore, the glycosylated recombinant protein comprises at least 0.17% of the total membrane protein in the baculoviral-infected insect cell system.
Figure 3.

Purification of his-tagged Cd36 from High Five membranes by Ni-NTA affinity chromatography. Purification fractions for Cd36–6His (A) and Cd36–12His (B) were subjected to 10% SDS-PAGE and proteins visualized by colloidal blue staining. (Lane 1) Standards, (lane 2) membrane preparation, (lane 3) OG-solubilized material, (lane 4) flow-through, (lanes 5–8) imidazole washes, pH 8 (20–35 mM for Cd36–6His, 60–120 mM for Cd36–12His), (lane 9) wash at pH 6.8, (lanes 10–13) elutions at pH 6.8 with 120 mM imidazole (Cd36–6His) or 250 mM imidazole (Cd36–12His).
Octylglucoside is best to maintain Cd36 in solution
Prior to analyzing the oligomeric state of purified Cd36 and determining its competency to bind ligand, it was necessary to increase the concentration of the protein eluted during the purification protocol. To achieve this and limit the loss of purified Cd36, the ability of a variety of detergents to maintain the solubility and monodispersity of the concentrated pure protein was investigated. Cd36 was solubilized in the presence of OG, thus ensuring the highest yield of extraction from the membrane, and bound to the Ni-NTA resin via its C-terminal his-tag. OG, with its relatively high cmc (critical micelle concentration) (Table 1), was exchanged for a range of different nonionic and zwitterionic detergents during subsequent column washes and elution. Cd36 eluted in different detergent micelles was concentrated to 0.2–0.4 mg/mL and its solubility measured by high-speed centrifugal sedimentation using a modification of an assay developed by Gutmann et al. (2007), as described in Materials and Methods. The proportion of protein remaining in the supernatant, relative to the input protein solution, is shown in Figure 4A for each detergent investigated. A distinct reduction in the levels of Cd36 in the supernatant following centrifugation at 100,000g and 250,000g in the presence of TX114 and TX100, respectively, was observed, indicative of protein aggregation in this class of detergent. Significant aggregation of Cd36 was also apparent in the presence of the maltoside detergents DM, DDM, and tri-decylmaltoside, but only after centrifugation of protein samples at 350,000g, demonstrating that the maltoside head group or longer acyl chain length offers little advantage. The zwitterionic detergents LDAO and fos-choline-12 did offer greater solubility, with 75% of Cd36 remaining soluble after the highest centrifugation. However, OG conferred the greatest stability with no diminution in the amount of protein remaining in the supernatant at any of the speeds used. In fact, there was no aggregation of Cd36 in OG micelles at protein concentrations up to 2 mg/mL. This finding is consistent with the behavior of the purified protein sample when subjected to nondenaturing Blue-Native PAGE (Fig. 4C). The Cd36-OG micelle migrated predominantly as a single discrete species, supporting the finding from the high-speed ultracentrifugation assay that the protein is monodispersed in OG. When compared with soluble hydrophilic protein markers, the smallest Cd36-OG micelle (the sample migrates as a broad band on native-PAGE) had an apparent molecular weight close to 146 kDa. However, the bound detergent micelle and the extensive glycosylation of the extracellular domain of Cd36 will increase the Stoke's radius of the glycoprotein detergent complex and retard its migration through the native gel. The protein backbone of the expressed Cd36 has a theoretical molecular weight of 55 kDa, and the apparent molecular weight observed by Blue-Native PAGE is likely consistent with a monomer or dimer of Cd36, but rules out a higher-order multimer. To establish unequivocally the subunit complexity of the purified Cd36, analytical centrifugation was used.
Figure 4.

Effect of detergent on the solubility and monodispersity of purified rat Cd36. (A) Purified Cd36–12His, in different detergent micelles, was concentrated to 0.2–0.6 mg/mL immediately prior to successive high-speed ultracentrifugation as indicated. Equivalent aliquots of the supernatant were removed after each spin and added directly to Laemmli sample buffer for gel analysis and protein quantitation. Cd36 was visualized by staining with colloidal blue. Data are presented as the proportion of protein–detergent complex remaining in solution as a function of centrifugation speed. (B) Representative gel showing SDS-PAGE analysis of solubility of Cd36 in OG micelles at a protein concentration of 0.4 mg/mL following high-speed ultracentrifugation. (C) Purified Cd36–12His in 1% OG resolved on 4%–16% gradient gel by Blue-Native electrophoresis.
Analytical ultracentrifugation indicates that Cd36 in OG micelles is monomeric
Sedimentation velocity experiments were performed to determine the size distribution profile of sedimenting species. The sedimentation coefficient distribution of a 0.5 mg protein/mL Cd36/OG solution was determined, and one main species (>90% of the sample) was observed (Fig. 5A) for which a sedimentation coefficient s20,w (10−13 S) value of 5.19S was determined after correcting the experimentally derived values to ideal conditions, using the program SEDNTERP (Reynolds and Tanford 1976). This is consistent with the data obtained by Blue-Native PAGE.
Figure 5.
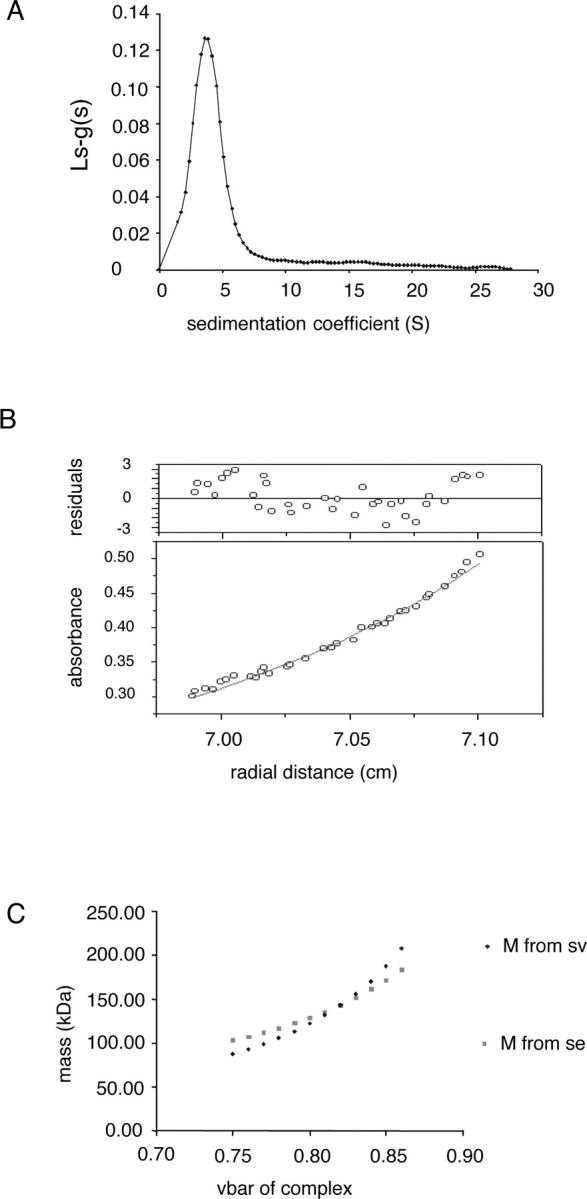
Analysis of the oligomeric state of Cd36 in OG micelles by analytical ultracentrifugation. (A) Sedimentation velocity analysis conducted with a single concentration of Cd36 (0.5 mg/mL). Data are plotted showing the apparent sedimentation coefficient distribution obtained from the ls-g*(s) model of SEDFIT, as described in Materials and Methods. (B) Equilibrium distribution for protein–detergent molecular mass obtained from sedimentation equilibrium analysis of Cd36 in OG, with the residuals shown in the upper panel. (C) Molecular weights for Cd36 protein–detergent complex derived from sedimentation velocity and equilibrium data, at different partial specific volumes, are plotted.
In order to determine the molecular mass of this protein species in solution and establish whether it represents monomeric or dimeric Cd36, sedimentation equilibrium (SE) measurements were taken. Data were acquired after a 0.72 mg/mL Cd36/OG sample had attained equilibrium at a rotor speed of 9000 rpm. Absorbance was plotted versus the radial position of the protein–detergent complex in the centrifuge cell and was best described by a single species fit, as shown in Figure 5B. This, taken together with the sedimentation velocity data, indicates that the Cd36–OG complex behaves as a homogeneous species. An apparent molecular mass of 94,500 Da was determined for the Cd36–detergent complex from the SE data and can be corrected for the presence of detergent using Equation 3 (Materials and Methods). However, as the number of protein molecules per protein–detergent complex was unknown, it was not possible to assign a value to the vbarmicelle (partial specific volume of protein–detergent complex) term in Equation 3. Therefore, molecular weights derived from both SV and SE data (using Equations 3 and 4) were plotted as a function of different theoretical partial specific volumes of the protein–detergent complex (Fig. 5C). A vbar of 0.820 mL/g was derived from the SV and SE data, which is very close to the theoretical vbar of the protein–detergent complex if one Cd36 molecule is present per micelle (0.795 mL/g). This value indicates that the protein–detergent micelle contains 47% protein and 53% detergent. Forty-seven percent of the 94,500 Da molecular mass calculated for the protein–detergent complex indicated a protein component of 44,400 Da, consistent only with purification of monomeric Cd36 (Cd36–12His has a theoretical polypeptide backbone of 54,587 Da).
The purified Cd36 in OG detergent micelles retains affinity for ligand
Having established that the subunit complexity of purified Cd36 in OG micelles is monomeric, it was of interest to determine whether monomeric Cd36 was sufficient to bind ligand. A solid-phase binding assay was developed where the C-terminal 12× histidine tag was exploited to achieve oriented immobilization of Cd36 molecules onto Ni-NTA-coated 96-well plates. The affinity of Cd36 for acLDL was determined by measuring equilibrium binding to increasing concentrations of Alexa488 or BODIPY-conjugated ligand. There was no binding of ligand to Ni-NTA plates in the absence of immobilized Cd36, indicating the specificity of ligand interaction with the Cd36 protein (data not shown). Data for specific binding of acLDL to Cd36 was best fitted by a binding isotherm, which describes interaction of ligand with a single class of binding site from which a value for Kd, a measure of binding affinity, was determined (Fig. 6A). The affinity of purified Cd36 in OG detergent micelles for acLDL had a mean Kd ± s.e.m. = 2.98 ± 0.82 μg/mL (n = 7). This correlates closely with the ligand-binding affinity of a fully glycosylated version of the same recombinant protein expressed on the surface of mammalian cells, Kd = 5 μg/mL (Fig. 6B), and is within the range reported in the literature for native protein ex vivo (Acton et al. 1994). In addition, the binding of ligand to protein/detergent micelles is reversible and specific, as binding of subsaturating concentrations of fluorophore-conjugated ligand was displaced by unlabelled acLDL with a mean EC50 = 150 ± 66 μg/mL (n = 4), as shown in Figure 6C. The displacement binding curves had a slope of unity, again indicative of ligand interaction at a single class of binding site. The ligand-binding competency of monomeric Cd36 in OG micelles was further confirmed by high-affinity binding to the physiological ligand DiI-oxLDL with a mean Kd ± s.e.m. = 10.44 ± 0.08 μg/mL (n = 2) (Fig. 6D).
Figure 6.

Interaction of Cd36–12His with modified low-density lipoproteins. (A) In vitro binding of increasing concentrations of Alexa Fluor 488 or Bodipy acLDL to purified Cd36 in OG detergent micelles as described in Materials and Methods. A representative binding curve is depicted, with relative fluorescence units for ligand binding plotted as a function of ligand concentration. Curve fitting was conducted as described in Materials and Methods from which values for Kd were determined. Fluorophore-conjugated acLDL bound with a mean Kd ± s.e.m. = 2.98 ± 0.82 μg/mL (n = 7). (B) In vivo flow-cytometric analysis of the interaction of Cd36–12His, expressed transiently in HEK 293T cells, with Bodipy-acLDL over a range of concentrations. Median fluorescence of ligand bound to cells expressing Cd36–12His was plotted as a function of ligand concentration as described in Materials and Methods. A value for Kd of 5 μg/mL was obtained. (C) Displacement of fluorophore-conjugated acLDL (5 μg/mL) binding to purified Cd36 in OG detergent micelles by increasing concentrations of unlabelled acLDL. Representative data are presented as the proportion of bound fluorophore-conjugated acLDL remaining in the presence of increasing concentrations of unlabelled acLDL. Fluorophore-conjugated ligand was displaced by unlabelled acLDL with a mean EC50 = 150 ± 66 μg/mL (n = 4). (D) Saturation binding of DiI-oxLDL to purified Cd36:OG micelles. A representative binding curve is shown, and curve fitting was conducted as described in Materials and Methods to obtain a mean Kd ± s.e.m. = 10.44 ± 0.08 μg/mL (n = 2).
Discussion
The class B scavenger receptor CD36 plays a significant pathological role in early atherogenesis by virtue of its ability to bind and internalize oxLDL. Yet, there are many questions concerning the molecular mechanism underlying this function that must be addressed in order to be able to design strategies to overcome this pro-atherogenic activity. We have used purified rat Cd36 to investigate a number of key issues, heretofore not satisfactorily addressed in the published literature, to better understand the molecular requirements for Cd36 to bind modified LDL. We chose Trichoplusia ni insect cells as a suitable heterologous host for expression of the rat class B scavenger receptor, Cd36, to levels that provide material sufficient for purification and subsequent functional and biophysical analysis. Insect cells are known to recognize mammalian glycosylation sites efficiently but only to core glycosylate (Jarvis 2003). The less elaborate glycosylation is not likely to compromise the functional integrity of the expressed protein, as human CD36 expressed in Spodopteran insect cells reached the cell surface and retained binding affinity for its ligands oxLDL and acLDL (Calvo et al. 1998). By exploiting the 12-histidine tag introduced to the recombinant protein and following efficient solubilization from the insect cell membranes with the nonionic detergent OG, milligram quantities of highly pure Cd36 were achieved using a single-step chromatographic procedure.
However, a major problem associated with the use of purified membrane proteins to study function and activity is the requirement for detergent during protein solubilization, purification, and downstream analysis to maintain protein fold and solubility in the absence of the membrane lipid bilayer. Screening for detergent compatibility is therefore a necessary prerequisite to conducting functional analysis, and a range of nonionic and zwitterionic detergents were tested for their ability to maintain stability and solubility of purified Cd36 to eliminate detergent-related artifacts. The nonionic detergent OG, which had been used in the purification protocol, conferred the highest degree of solubility on Cd36. Blue-Native PAGE confirmed that Cd36 does not aggregate in OG micelles and that the Cd36–detergent complex represents a single species. However, it was not possible to accurately determine the molecular mass of the protein by this method because the effect of bound detergent and extensive glycosylation on migration of the protein through the nondenaturing gel cannot be accounted for. Analytical ultracentrifugation was used to address this question and ascertain the molecular mass of the protein, because it can more readily correct for the effect of bound detergent and glycosylation on the mass of the sedimenting species at equilibrium. We were able to show unequivocally that the subunit complexity of purified Cd36 in OG detergent micelles is monomeric. This conclusion apparently contradicts several reports in the literature (Rhinehart-Jones and Greenwalt 1996; Daviet et al. 1997; Thorne et al. 1997). Thorne et al. (1997) used a variety of techniques to conclude that Cd36, isolated from platelets or COS-7 cells transfected transiently, formed dimers and higher-order multimers. The oligomerization of the protein was reversed in the presence of reducing agent, suggesting a covalent association, and following mutagenesis that ruled out a contribution from the intracellular cysteines, the investigators concluded that cysteines in the ECL were responsible. However, Rasmussen et al. (1998) showed clearly that there are no free cysteines in bovine Cd36 and defined the cross-linking pattern (C242–C310, C271–C333, C312–C321), but the analysis was unable to discriminate between intra- and intermolecular disulfide bonds. Our data, showing that Cd36 purified from the insect cell membrane is monomeric, is consistent with the simplest interpretation that the three disulfide bridges are intramolecular, possibly defining a ∼100 amino acid domain within the protein. This is similar to the arrangement in the cysteine-rich domains of Mac2BP (Hohenester et al. 1999) and Hepsin (Herter et al. 2005), neither of which share a significant level of primary sequence identity with each other or with Cd36, but where three intramolecular disulfide bridges stabilize a tertiary fold with a β-roll around a single α-helix.
It has been established previously that raft localization of Cd36 is important for function. Zeng et al. (2003) showed that Cd36, localized to a noncaveolar lipid raft microdomain, internalized oxLDL by a novel receptor-mediated endocytic pathway. Furthermore, Rahaman et al. (2006) showed the involvement of a Cd36-specific signaling complex, likely to be formed in the lipid raft, in the internalization of oxLDL and subsequent foam cell formation. However, it is not known whether cofactors are required for initial binding of modified LDL. Using a solid-phase binding assay, we were able to show that purified monomeric Cd36 in OG-detergent micelles is competent to bind acLDL and the physiologic oxLDL with, in the case of acLDL, an affinity similar to the fully glycosylated protein expressed transiently in mammalian cells, and to Cd36 in native membranes (Acton et al. 1994). Thus, the initial interaction of Cd36 with its atherogenic ligand oxLDL is not a function of membrane localization and does not require any protein co-receptor. This is entirely consistent with our observation that nonpalmitoylated CD36 retains affinity for ac- and ox-LDL despite failing to target to “lipid raft microdomains” in the plasma membrane (unpublished work).
It seems clear, therefore, that interaction of Cd36 with ligand is a property of a binding domain defined by the fold of the monomer. This does not rule out the possibility that Cd36 forms part of a homo- or heterocomplex to internalize ligand or for signaling purposes. Indeed, Daviet et al. (1997) suggested that membrane-bound Cd36 was monomeric, but saw evidence for protein homodimerization upon binding with thrombospondin-1. Our data (unpublished work) also suggest that palmitoylation-dependent targeting to lipid rafts, while irrelevant for ligand binding, is important for internalization. Taken together with the data from Thorne et al. (1997), these quaternary interactions, likely to be formed in lipid raft microdomains and possibly dependent on ligand binding, will rely on intramolecular disulfide bridging to stabilize the tertiary structure of the cysteine-rich domain, but will not involve intermolecular disulfide bridges. The major finding of this paper, which shows clearly that Cd36 can bind oxLDL as a monomer in the absence of associating cofactors, has implications for future strategies to identify or design pharmacological inhibitors of the pro-atherosclerotic activity of Cd36. The purified protein, characterized extensively for its compatibility with different nonionic and zwitterionic detergents in this paper, is suitable for use in high-throughput screens to identify novel pharmacological agents to inhibit Cd36 at the level of ligand binding and for use in crystallization studies.
Materials and Methods
Materials
The detergents n-Octyl-β-D-glucopyranoside, n-Dodecyl-β-D-maltoside, ULTROL Grade, Triton-X-100 (hydrogenated), Triton-X-114 PROTEIN GRADE, and C12E8 were purchased from Merck Biosciences. n-Decyl-β-D-maltopyranoside, n-tridecyl-β-D-maltopyranoside, n-tetra-β-D-maltopyranoside, Fos-choline-10, and Fos-choline-12 (all SOL-GRADE) were from Anatrace Inc. LDAO was purchased from Sigma-Aldrich. Ni-NTA Agarose and Ni-NTA-coated 96-well microtiter plates were from Qiagen Ltd. Acetylated-LDL, Bodipy FL acetylated-LDL, Alexa Fluor 488 acetylated-LDL, mouse monoclonal against Anti-His (C terminus), and Colloidal Blue Staining Kit were supplied by Invitrogen. 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI) oxidized-LDL was purchased from Biomedical Technologies Inc. The protease inhibitors benzamidine and leupeptin, and fatty-acid-free BSA, fraction V, were all from Sigma-Aldrich. Pepstatin A was supplied by Merck Biosciences. Amicon Ultra 15 (MWCO 50) and microcon (MWCO 50) centrifugal devices were purchased from Millipore (UK) Ltd. PNGase F was purchased from New England Biolabs. NativePAGE Novex Bis-Tris Gel System, and NativeMark Unstained Protein Standard were from Invitrogen.
Generation of recombinant baculovirus encoding Cd36 with a poly-histidine, carboxy-terminal tag
The translational stop codon of the cDNA encoding rat Cd36 with a carboxy-terminal, 6-histidine tag in pciCd36 (Lepretre et al. 2004) was replaced with an AgeI restriction site using the primer pair 5′-GGTCACCATCACCATCACCACACCGGTAGAGCGGCCGCTTCCC-3′ and 5′-GGGAAGCGGCCGCTCTACCGGTGTGGTGATGGTGATGGTGACC-3′, as directed by the QuikChange XL protocol (Stratagene). The modified cDNA was excised from the plasmid using restriction enzymes SacI and AgeI and subcloned into similarly digested pIZ/V5-His (Invitrogen). This fused, in frame, the coding sequences for the 6-histidine tag of the cDNA with the 6-histidine tag of the pIZ/V5-His vector, thus encoding a protein of full-length Cd36 with a carboxy-terminal tag of GHHHHHHTGHHHHHH. The cDNAs for Cd36-12His and Cd36-6His were excised from their respective plasmids and subcloned into the baculoviral transfer vector BlueBac4.5 (Invitrogen). The transfer vector was used to engineer recombinant baculoviruses using the Bac-N-Blue system (Invitrogen), as directed. For expression in mammalian cells, the modified Cd36-12His cDNA was subcloned into pCI-neo (Promega) using the restriction enzymes NheI and XbaI to generate pciCd36-12His.
Expression in Trichoplusia ni cells
Suspension cultures of Trichoplusia ni (High Five) insect cells were grown in serum-free Excell 405 media at 27°C with shaking at 100 rpm. Cells were infected at a cell density of 2 × 106 cells/mL with recombinant Cd36 baculovirus using a multiplicity of infection of at least three viruses per cell. After several hours, the culture was diluted to 106 cells/mL with fresh media and incubated for 72 h before harvesting.
Preparation of insect cell membranes
Infected cells (200 mL) were harvested by centrifugation (1000g, 10 min at 4°C) and rinsed with 30 mL of ice-cold buffer 1 (10 mM Tris-HCl pH 7.4, 0.25 M sucrose, 0.2 mM CaCl2, 2 mM benzamidine, 40 μM leupeptin, 1 μM pepstatin A). The washed pellet was resuspended in 10 mL of buffer 1 and frozen at −20°C, then thawed and homogenized over ice (10 × 20 sec bursts at 24,000 rpm, [Yellow Line DI 25 homogenizer]). Large organelles were removed by slow-speed centrifugation (500g, 10 min at 4°C). The crude membrane fraction was isolated from the supernatant by ultracentrifugation in a TLA100.2 rotor (Beckman) (100,000g, 50 min at 4°C). The pelleted membranes were resuspended in buffer 2 (buffer 1 plus 10% [v/v] glycerol) and stored at −80°C. The total membrane protein concentration was determined by DC Protein assay (Bio-Rad).
Deglycosylation of membrane protein fraction
Proteins in the insect cell membrane fraction (20 μg) were denatured and incubated with PNGase F for 2 h at 37°C, as directed (New England Biolabs). Membrane proteins pre- and post-deglycosylation were separated by SDS-PAGE, and Cd36 was detected by Western analysis.
Solubilization and purification of Cd36 from insect cell membrane fraction
In a typical small-scale purification, 200 mg of membrane protein were pelleted (100,000g, 20 min, 4°C) then resuspended at 10 mg/mL in buffer 3 (20 mM Tris-HCl pH 6.8, 4% [w/v] octylglucoside, 150 mM NaCl, 1.5 mM MgCl2, 5% [v/v] glycerol, 2 mM benzamidine, 40 μM leupeptin, 1 μM pepstatin A), homogenized over ice by extrusion five times through a 21G needle, and incubated with constant mixing for 90 min at 4°C. Insoluble material was pelleted by ultracentrifugation in a TLA100.2 rotor (Beckman Coulter Ltd.) at 100,000g for 20 min at 4°C and discarded.
Ni-NTA resin was pre-equilibrated in buffer 4 (20 mM Tris-HCl, pH 6.8, 150 mM NaCl, 1.5 mM MgCl2, 5% [v/v] glycerol, 2% [w/v] OG, 2 mM benzamidine, 1 μM pepstatin A, 40 μM leupeptin) in the presence of 20 mM imidazole. OG-solubilized membrane protein was added at a protein:resin ratio of 1:8 (w/w) and mixed for 1 h at 4°C. Proteins bound nonspecifically were eluted by four washes with 20 bed volumes and a stepwise gradient of imidazole (20–35 mM for the 6-histidine tag; 60–120 mM for the 12-histidine tag) in buffer 5 (20 mM Tris-HCl, pH 8, 150 mM NaCl, 1.5 mM MgCl2, 5% [v/v] glycerol, 1% [w/v] OG, 2 mM benzamidine, 1 μM pepstatin A, 40 μM leupeptin). Cd36-His was eluted in 4 × 2 bed volumes of imidazole (120 mM for the 6-histidine tag; 250 mM for the 12-histidine tag) in buffer 4, but with OG reduced to 1%. Aliquots (∼1%) of the purification fractions were subjected to SDS-PAGE and proteins were visualized by staining with colloidal blue, to follow the efficiency of purification. Concentration and purity of the eluted Cd36-His were determined following densitometry of colloidal-blue-stained SDS-PAGE gels in comparison with internal BSA standards, using NIH image software. For protein characterization studies, eluted protein was concentrated at 4°C using centrifugal devices (Amicon Ultra 15) with a molecular weight cut-off of 50 kDa, from Millipore.
Ligand binding in vitro
Purified Cd36–12His (1–2 μg) was added per well of a 96-well Ni-NTA-coated plate (Qiagen) as a 150-μL aliquot in protein-binding buffer (20 mM Tris-HCl, pH 6.8, 150 mM NaCl, 1.5 mM MgCl2, 5% [v/v] glycerol, 0.5% [w/v] OG) and bound overnight at 4°C with gentle rocking. Unbound protein was aspirated and wells washed with 2 × 200 μL of ligand binding buffer (LBB) (PBS, 1 mM MgCl2, 1 mM CaCl2, 0.5% [w/v] OG, 0.2% [w/v] fatty-acid-free BSA). Fluorophore-conjugated ligand was added in LBB in the presence or absence of unlabelled acLDL in a total assay volume of 50 μL. The binding reaction was incubated for 90 min at room temperature, with gentle rocking. Unbound ligand was removed and the wells washed with 3 × 150 μL of ice-cold wash buffer (PBS, 1 mM MgCl2, 1 mM CaCl2, 0.5% [w/v] OG, 0.05% [w/v] BSA). PBS (50 μL) was added per well prior to determining the amount of bound fluorescence using a fluorescence plate-reader (Spectra Max Gemini EM, Molecular Devices; Ex485nm/Em538nm for Alexa488 and Bodipy fluorophores, and Ex514nm/Em550nm for DiI).
Data from all binding assays were analyzed using Graphpad Prism version 4.0 software. Saturation binding data that showed saturable binding of ligand to Cd36 over a range of ligand concentrations were best fitted by the Langmuir adsorption isotherm (Equation 1), which describes binding of ligand to a single class of binding site as defined below;
 |
where B is bound ligand (relative fluorescence units), B max is maximal binding (relative fluorescence units), [L] is concentration of ligand (μg/mL), and Kd is the concentration of ligand (μg/mL) giving half-maximal binding and is a measure of the affinity of the receptor–ligand interaction.
Homologous displacement binding assays, in which fluorophore-conjugated acetylated LDL was displaced by increasing concentrations of unlabelled acetylated LDL, were analyzed using the dose-response relationship described by De Lean and Rodbard (1978);
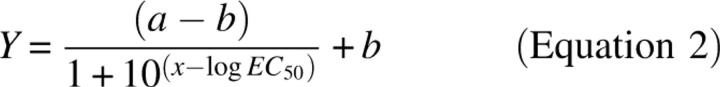 |
where Y represents the fraction of binding remaining, a is initial binding, b is maximal binding, log EC 50 is the concentration of unlabelled acLDL that displaced 50% of the bound labeled acLDL, and x is log10 ligand concentration (μg/mL).
Ligand binding in cells
Human embryonic kidney (HEK) 293T cells were transfected transiently with pciCd36–12His as described previously (Lepretre et al. 2004). Forty-eight hours post-transfection the cells were harvested using triple express (Invitrogen), washed, and resuspended at 1 × 107 cells/mL in buffer 6 (PBS with 1% fatty-acid-free BSA). To 350 μL of the cell suspension, 7 μg (saturating concentration; data not presented) of anti-Cd36 mAb1955 (R&D systems) were added and the suspension incubated for 30 min at 4°C. The cells were then pelleted by centrifugation (400g for 1 min at 4°C) and resuspended in 1 mL of buffer 6. This wash step was repeated twice more, before resuspension of the cells in 350 μL of buffer 6 and addition of a saturating amount (5.25 μg; data not presented) of rabbit, anti-rat IgG secondary antibody, conjugated to R-phycoerythrin (RPE; Oxford Biotechnology). After 30 min at 4°C in the dark, the cells were washed as before and resuspended in 350 μL of buffer 6. Aliquots (50 μL) of the antibody-labeled cells were incubated with Bodipy-acLDL (0–40 μg/mL) for 2 h at 4°C in the dark. The cells were washed as described above and analyzed by flow cytometry as follows: Ten thousand cells, gated for normal size and granularity, were analyzed for cell surface Cd36–12His expression (R-PE fluorescence; Ex565nm/Em578nm) and bound acLDL (Bodipy fluorescence; Ex485nm/Em530nm). Bodipy-acLDL bound to cells expressing a consistent level of Cd36–12His (gating on ∼4000 cells), was analyzed. Binding of acLDL to Cd36 is not affected by binding of mAb 1955 (data not shown). Median Bodipy-acLDL fluorescence binding was plotted versus ligand concentration and analyzed using Graph Pad Prism 4.0 software. Binding of acLDL to Cd36 was saturable and fitted well with the Langmuir adsorption isotherm, which describes ligand binding to a single class of binding site (Equation 1).
Protein stability and monodispersity in detergent
Detergent exchange was carried out during protein purification. Following binding of OG-solubilized Cd36–12His to Ni-NTA resin, bound protein was washed with 6 × 20 bed volumes of buffer 5 but with OG replaced by 2–4× cmc concentration of the detergent of interest. Following elution, Cd36 detergent micelles were concentrated to 0.2–0.4 mg/mL protein in an Amicon Ultra 15 (MWCO 50 kDa) centrifugal device. Samples in the different detergents were then subjected to increasingly high ultracentrifugation in a TLA100 rotor (100,000g, 250,000g, and 350,000g) for 30-min intervals. Aliquots of the supernatant were taken after each run, added directly to Laemmli sample buffer, and analyzed by SDS-PAGE and staining the gels with colloidal blue. The amount of protein remaining in the supernatant was determined from densitometry in comparison with internal BSA standards. Data were expressed as the proportion of the input protein remaining in the supernatant following each ultracentrifugation.
Blue-Native PAGE
Purified Cd36 in OG micelles was resolved by Blue-Native PAGE using the NativePAGE Novex Bis-Tris Gel System from Invitrogen, which is based on a modification of the method first described by Schägger and von Jagow (1991). Briefly, 1 μg of purified Cd36 in sample buffer (50 mM BisTris-HCl, 50 mM NaCl, 10% glycerol, 1% OG, 0.25% Coomassie G-250 dye [v/v]) was loaded onto 4%–16% NativePAGE Novex Bis-Tris gel. NativeMark protein standards (1236–20 kDa range) were run in an adjacent well. A BisTris/Tricine running buffer system was used (50 mM BisTris, 50 mM Tricine, pH 6.8), and Coomassie G-250 dye was included in the cathode buffer only (0.02% [v/v]). Protein was resolved after electrophoresis for 150 min at 4°C and protein bands visualized after fixing in 40% methanol/10% acetic acid for 15 min followed by destaining overnight in 8% acetic acid.
AUC measurements
Sedimentation velocity
Cd36 analytical ultracentrifugation (AUC) measurements were performed using a Beckman Coulter Optima XLI instrument. Sedimentation velocity was carried out at a rotor speed of 50,000 rpm, at 10°C, scanning at 2 min intervals. Detection was via absorbance optics set at a wavelength of 280 nm. A 400 μL volume of Cd36 in OG micelles was run at a concentration of 0.5 mg protein/mL in buffer 4, without imidazole. Data were analyzed using the ls-g*(s) model of SEDFIT (Schuck 2000) to produce an apparent sedimentation coefficient distribution profile.
Sedimentation equilibrium
Sedimentation equilibrium was performed at a single concentration (0.72 mg/mL) of Cd36 in OG micelles. A sample volume of 100 μL was loaded and subjected to a rotor speed of 9000 rpm at 10°C. Samples were scanned at four hourly intervals.
Data analysis and interpretation
Sedimentation equilibrium data were analyzed using the Origin software from Beckman to find the weight-average molecular weight for the protein–detergent complex. To correct this value for the presence of detergent, the flotation term described below was used:
where: Mapp = apparent molecular weight before correction (Da), vbarglyc–protein = partial specific volume of glycoprotein (mL/g), vbarmicelle = partial specific volume of micelle containing glycoprotein (mL/g), and ρ = solvent density.
For determination of the molecular weight of the complex from sedimentation velocity data, the following equation was applied:
The value for vbarglyc-protein determined by the program SEDNTERP from the protein sequence and glycosylation status (Laue 1992) is 0.7278 mL/g. The known vbar for octylglucoside is 0.6456 mL/g. In order to calculate vbarmicelle, it was necessary to know the glycoprotein content of the detergent micelle. Therefore, molecular weights for the glycoprotein derived from sedimentation equilibrium and sedimentation velocity data using equations 3 and 4, respectively, were plotted versus different theoretical partial specific volumes of the proteo–detergent complex. The best estimate for the complex (vbarmicelle) was derived at the partial specific volume where both data sets agreed.
Acknowledgments
This research was funded by a project grant (PG/03/044/15328) awarded to K.J.L. from the British Heart Foundation. We are indebted to Professor Arthur Rowe for invaluable help with the analysis and interpretation of the analytical ultracentrifugation data. We also thank the MRC for their support.
Footnotes
Reprint requests to: Kenneth Linton, MRC Clinical Sciences Centre, Imperial College, Hammersmith Hospital Campus, Du Cane Road, London W12 0NN, UK; e-mail: kenneth.linton@ic.ac.uk; fax: 44-(0)20-8383-8337.
Abbreviations: OG, octylglucoside; DM, decylmaltoside; DDM, dodecylmaltoside; Tri-DM, tri-decylmaltoside; Tetra-DM, tetra-decylmaltoside; MWCO, molecular weight cutoff; mAb, monoclonal antibody; LCFA, long-chain fatty acid; SE, sedimentation equilibrium; SV, sedimentation velocity; AcLDL, acetylated low-density lipoprotein; OxLDL, oxidized low-density lipoprotein; LDL, low-density lipoprotein.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073007207.
References
- Acton S.L., Scherer, P.E., Lodish, H.F., and Krieger, M. 1994. Expression cloning of SR-BI, a CD36-related Class B scavenger receptor. J. Biol. Chem. 269: 21003–21009. [PubMed] [Google Scholar]
- Asch A.S., Liu, I., Briccetti, F.M., Barnwell, J.W., Kwakye-Berko, F., Dokun, A., Goldberger, J., and Pernambuco, M. 1993. Analysis of CD36 binding domains: Ligand specificity controlled by dephosphorylation of an ectodomain. Science 262: 1436–1440. [DOI] [PubMed] [Google Scholar]
- Barnwell J., Ockenhouse, C., and Knowles, D. 1985. Monoclonal antibody OKM5 inhibits the in vitro binding of Plasmodium falciparum-infected erythrocytes to monocytes, endothelial, and C32 melanoma cells. J. Immunol. 135: 3494–3497. [PubMed] [Google Scholar]
- Berglund L., Petersen, T.E., and Rasmussen, J.T. 1996. Structural characterization of bovine CD36 from the milk fat globule membrane. Biochim. Biophys. Acta 1309: 63–68. [DOI] [PubMed] [Google Scholar]
- Calvo D., Gomez-Coronado, D., Suarez, Y., Lasuncion, M.A., and Vega, M.A. 1998. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J. Lipid Res. 39: 777–788. [PubMed] [Google Scholar]
- Daviet L., Malvoisin, E., Wild, T.F., and McGregor, J.L. 1997. Thrombospondin induces dimerization of membrane-bound, but not soluble CD36. Thromb. Haemost. 78: 897–901. [PubMed] [Google Scholar]
- De Lean A.M.P. and Rodbard, D. 1978. Simultaneous analysis of families of sigmoidal curves: Applications to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 235: E97–E102. [DOI] [PubMed] [Google Scholar]
- Endemann G.S.L., Madden, K.S., Bryant, C.M., White, R.T., and Protter, A.A. 1993. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 268: 11811–11816. [PubMed] [Google Scholar]
- Febbraio M.P.E., Smith, J.D., Hajjar, D.P., Hazen, S.L., Hoff, H.F., Sharma, K., and Silverstein, R.L. 2000. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Invest. 105: 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieda S.P.A., Wu, J., and Silverstein, R.L. 1995. Recombinant GST/CD36 fusion proteins define a thrombospondin binding domain. Evidence for a single calcium-dependent binding site on CD36. J. Biol. Chem. 270: 2981–2986. [DOI] [PubMed] [Google Scholar]
- Gruarin P., Thorne, R.F., Dorahy, D.J., Burns, G.F., Sitia, R., and Alessio, M. 2000. CD36 is a ditopic glycoprotein with the N-terminal domain implicated in intracellular transport. Biochem. Biophys. Res. Commun. 275: 446–454. [DOI] [PubMed] [Google Scholar]
- Gutmann D.A., Mizohata, E., Newstead, S., Ferrandon, S., Henderson, P.J., van Veen, H.W., and Byrne, B. 2007. A high-throughput method for membrane protein solubility screening: The ultracentrifugation dispersity sedimentation assay. Protein Sci. 16: 1422–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herter S., Piper, D.E., Aaron, W., Gabriele, T., Cutler, G., Cao, P., Bhatt, A.S., Choe, Y., Craik, C.S., Walker, N., et al. 2005. Hepatocyte growth factor is a preferred in vitro substrate for human hepsin, a membrane-anchored serine protease implicated in prostate and ovarian cancers. Biochem. J. 390: 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenester E., Sasaki, T., and Timpl, R. 1999. Crystal structure of a scavenger receptor cysteine-rich domain sheds light on an ancient superfamily. Nat. Struct. Biol. 6: 228–232. [DOI] [PubMed] [Google Scholar]
- Jarvis D.L. 2003. Developing baculovirus-insect cell expression systems for humanized recombinant glycoprotein production. Virology 310: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue T. 1992. Analytical ultracentrifugation in biochemistry and polymer science (eds. R.A. Harding et al.), pp. 90–125. Redwood Press, Cambridge, UK.
- Lepretre F., Vasseur, F., Vaxillaire, M., Scherer, P.E., Ali, S., Linton, K., Aitman, T., and Froguel, P. 2004. A CD36 nonsense mutation associated with insulin resistance and familial type 2 diabetes. Hum. Mutat. 24: 104. doi: 10.1002/humu.9256. [DOI] [PubMed] [Google Scholar]
- Leung L.L.K., Li, W.X., McGregor, J.L., Albrecht, G., and Howard, R.J. 1992. CD36 peptides enhance or inhibit CD36-thrombospondin binding. A two-step process of ligand–receptor interaction. J. Biol. Chem. 267: 18244–18250. [PubMed] [Google Scholar]
- Nicholson A.C., Frieda, S., Pearce, S.F., and Silverstein, R.L. 1995. Oxidized LDL binds to CD36 on human monocyte-derived macrophages and transfected cell lines. Evidence implicating the lipid moiety of the lipoprotein as the binding site. Arterioscler. Thromb. Vasc. Biol. 15: 269–275. [DOI] [PubMed] [Google Scholar]
- Pearce S.R.P., Nicholson, A.C., Hajjar, D.P., Febbraio, M., and Silverstein, R.L. 1998. Recombinant glutathione S-transferase/CD36 fusion proteins define an oxidized low density lipoprotein-binding domain. J. Biol. Chem. 273: 34875–34881. [DOI] [PubMed] [Google Scholar]
- Pohl J., Ring, A., Korkmaz, U., Ehehalt, R., and Stremmel, W. 2005. FAT/CD36-mediated long-chain fatty acid uptake in adipocytes requires plasma membrane rafts. Mol. Biol. Cell 16: 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahaman S.O., Lennon, D.J., Febbraio, M., Podrez, E.A., Hazen, E.L., and Silverstein, R.L. 2006. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 4: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen J.T., Berglund, L., Rasmussen, M.S., and Petersen, T.E. 1998. Assignment of disulfide bridges in bovine CD36. Eur. J. Biochem. 257: 488–494. [DOI] [PubMed] [Google Scholar]
- Reynolds J. and Tanford, C. 1976. Determination of molecular weight of the protein moiety in protein–detergent complexes without direct knowledge of detergent binding. Proc. Natl. Acad. Sci. 73: 4467–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinehart-Jones T. and Greenwalt, D.E. 1996. A detergent-sensitive 113-kDa conformer/complex of CD36 exists on the platelet surface. Arch. Biochem. Biophys. 326: 115–118. [DOI] [PubMed] [Google Scholar]
- Savill J. and Hogg, N. 1992. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Invest. 90: 1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H. and von Jagow, G. 1991. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199: 223–231. [DOI] [PubMed] [Google Scholar]
- Schuck P. 2000. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm modelling. Biophys. J. 78: 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. and Toomre, D. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1: 31–39. [DOI] [PubMed] [Google Scholar]
- Tao N., Wagner, S.J., and Lublin, D.M. 1996. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J. Biol. Chem. 271: 22315–22320. [DOI] [PubMed] [Google Scholar]
- Thorne R.F., Meldrum, C.J., Harris, S.J., Doraghy, D.J., Shafren, D.R., Berndt, M.C., Burns, G.F., and Gibson, P.G. 1997. CD36 forms covalently associated dimers and multimers in platelets and transfected COS-7 cells. Biochem. Biophys. Res. Commun. 240: 812–818. [DOI] [PubMed] [Google Scholar]
- Zeng Y., Tao, N., Chung, K.N., Heuser, J.E., and Lublin, D.M. 2003. Endocytosis of oxidized low density lipoprotein through scavenger receptor CD36 utilizes a lipid raft pathway that does not require caveolin-1. J. Biol. Chem. 278: 45931–45936. [DOI] [PubMed] [Google Scholar]