

Abstract
Periplasmic expression screening is a selection technique used to enrich high-affinity proteins in Escherichia coli. We report using this screening method to rapidly select a mutated D-glucose/D-galactose-binding protein (GGBP) having low affinity to glucose. Wild-type GGBP has an equilibrium dissociation constant of 0.2 μM and mediates the transport of glucose within the periplasm of E. coli. The protein undergoes a large conformational change on binding glucose and, when labeled with an environmentally sensitive fluorophore, GGBP can relay glucose concentrations, making it of potential interest as a biosensor for diabetics. This use necessitates altering the glucose affinity of GGBP, bringing it into the physiologically relevant range for monitoring glucose in humans (1.7–33 mM). To accomplish this a focused library was constructed using structure-based site-saturation mutagenesis to randomize amino acids in the binding pocket of GGBP at or near direct H-bonding sites and screening the library within the bacterial periplasm. After selection, equilibrium dissociation constants were confirmed by glucose titration and fluorescence monitoring of purified mutants labeled site-specifically at E149C with the fluorophore IANBD (N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylene-diamine). The screening identified a single mutation A213R that lowers GGBP glucose affinity 5000-fold to 1 mM. Computational modeling suggested the large decrease in affinity was accomplished by the arginine side chain perturbing H-bonding and increasing the entropic barrier to the closed conformation. Overall, these experiments demonstrate the ability of structure-based site-saturation mutagenesis and periplasmic expression screening to discover low-affinity GGBP mutants having potential utility for measuring glucose in humans.
Keywords: GGBP, glucose/galactose-binding protein, low affinity, structure-based site-saturation mutagenesis, screening, focused library
Combining targeted mutagenesis (Lutz and Patrick 2004; Chockalingam et al. 2005; Yuan et al. 2005; Reetz and Carballeira 2007) with a rapid screening method (Olsen et al. 2000; Arnold and Georgiou 2003; Hoogenboom 2005) can be an efficient way to identify affinity-modified proteins (Parikh and Matsumura 2005). However, screening even relatively small libraries can be demanding, making the choice of screening methods especially important. One method, periplasmic expression with cytometric screening (PECS), was used previously to select scFv antibodies with increased affinity to digoxigenin (Chen et al. 2001). For the selection scFv's were expressed in bacteria and secreted into the periplasmic space as soluble proteins then challenged with fluorescently tagged digoxigenin. In bacteria small molecules can freely equilibrate between the periplasm and the external environment via channels in the cell wall called porins. During the screening, bacteria that more extensively concentrated the ligand due to its interaction with the scFv were selected by flow. This technique continues to evolve, and more recently “E-clonal” screening was reported where full-length antibodies were secreted into the periplasm for selection (Mazor et al. 2007).
Like most other screening methods (Brannigan and Wilkinson 2002; Fernandez-Gacio et al. 2003) PECS was performed with the goal of enriching proteins having increased ligand affinity. Generally, increases in affinity are large enough for the generated signal to be easily detected over both binding to the recombinant parental protein and nonspecific binding. However, if screening is used to select a low-affinity mutant, then ligand uptake into the periplasm will be decreased compared to that of the recombinant parental protein. It is rarely desired to identify a low-affinity protein from a library, but, when it is, one solution is to eliminate the larger population of mutants having high affinity for the ligand, thus leaving behind the desired mutants. To make this strategy successful, the ability to quantify the signal generated by the binding event and the assay's sensitivity must be thoroughly characterized prior to selection. This was the challenge with selecting a low-affinity glucose/galactose-binding protein (GGBP) using periplasmic expression (PE) screening (Amiss et al. 2006).
GGBP is a glucose transport protein from the family malG of Escherichia coli (Boos and Gordon 1971). The wild-type (wt) protein has a glucose affinity of 0.2 μM (Vyas et al. 1988) and has been characterized as having simple kinetic behavior (Miller III et al. 1983). In nature GGBP exists in the periplasm of E. coli where it binds one molecule of D-glucose or D-galactose transporting it to the membrane-bound Trg receptor (Vyas et al. 1987). GGBP has been crystallized in both the closed ligand-bound (Vyas et al. 1988) and open ligand-free forms (Borrok et al. 2007) and is comprised of two globular domains connected by a hinge region (Fig. 1). On binding a large conformational change occurs (31° hinge movement) and the two domains close to entrap glucose (Boos et al. 1972; Vyas et al. 1987; Quiocho and Ledvina 1996; Borrok et al. 2007). When site-specifically labeled with an environmentally sensitive fluorophore, GGBP generates a signal that can be used to quantify glucose (Tolosa et al. 1999; Salins et al. 2001; De Lorimier et al. 2002). This makes the protein potentially useful as a detection chemistry for monitoring glucose in diabetics (Hellinga and Marvin 1998). However, because the wt protein would be saturated at blood glucose concentrations typically monitored for diabetes (1.7–33 mM), (Sacks et al. 2002), the glucose affinity of GGBP had to be significantly lowered.
Figure 1.

The structures of GGBP in the open (2FW0.pdb) and closed (2FVY.pdb) state. The 34-kDa protein has two large domains connected by a hinge region. On glucose binding a large conformational change (31°) occurs, bringing the two domains together and entrapping glucose (Borrok et al. 2007).
Our goal was to discover a GGBP with low affinity to glucose using structure-based site-saturation mutagenesis (Parikh and Matsumura 2005; Reetz and Carballeira 2007) to create a focused library that could be screened in the bacterial periplasm. This strategy enabled rapid evaluation of glucose affinity without the necessity of purifying and fluorescently labeling each mutant. During the experiments PE screening efficiently eliminated 98% of the population, selecting only mutants having apparent affinities near human physiological glucose concentrations. These mutants were then purified, fluorescently labeled, and characterized for glucose binding. This process successfully selected the A213R mutation that reduces glucose affinity 5000-fold, producing an equilibrium dissociation constant for E149C/A213R of 1 mM (Amiss et al. 2003). This mutant was then further modified to yield E149C/A213R/L238S (Kd = 10 mM). Overall, these results demonstrate the utility of structure-based site-saturation mutagenesis and PE screening to rapidly identify GGBP mutants with low affinity to glucose.
Results
Screening strategy to identify mutants with low affinity to glucose
The library was created by randomizing 16 amino acids in the binding pocket of GGBP. Screening parameters were developed resulting in a selection process where E. coli expressing mutants having strong glucose affinities were eliminated in the first round. The second round enriched E. coli near the detection limit (LOD) of the screening assay and in the third round only E. coli that were determined to be expressing recombinant GGBP were selected. These mutants were then DNA-sequenced, expressed, purified, labeled with the fluorophore IANBD (N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine), and fully characterized for glucose affinity and fluorescent response.
Determination of screening parameters using purified GGBP mutants
The ability to remove nonspecific binding was determined using purified mutants in a glucose-binding assay. The GGBP mutant H152C was immobilized through the His-tag onto a Co2+ surface. This protein, when labeled with IANBD, has a glucose affinity of 134 μM (De Lorimier et al. 2002) and, although the unlabeled affinity has not been reported, it is expected to be between the range of wt and labeled affinities (0.2–134 μM). The amount of protein bound to the resin was well below the resin's capacity; therefore, it was assumed that the H152C density was low enough to ensure that all the protein had equal access to glucose. Next, we challenged the immobilized H152C with a 10 μM glucose solution containing 0.1% tritiated (3H)-glucose. The total amount of glucose added was well in excess of the molar amount of GGBP immobilized and was 50-fold greater than the 0.2 μM wt equilibrium constant. This was done so that H152C would have ∼50% fractional occupancy. To remove nonspecific binding a single wash step was tested over the range of 0.2–0.5 mL. Then the GGBP-bound 3H-glucose was counted and graphed (Fig. 2A). This demonstrated that a single 0.4-mL wash was adequate to remove 81% of the 3H-glucose nonspecifically bound to H152C. The remaining 3H-glucose was believed to be specifically bound.
Figure 2.

Experiments were performed to establish screening parameters using purified proteins immobilized on Co2+ resin and challenged with a 10 μM glucose/3H-glucose solution. (A) By titrating the wash solution it was determined that 0.4 mL of DPBS removed 81% of the 3H-glucose from immobilized H152C. (B) The amount of purified protein needed to produce a large differential signal between the high-affinity H152C and W183C-acrylodan (Kd = 6 mM) was determined. The graph demonstrates that at 2 nmol of H152C there was a 13-fold signal increase over W183C-acrylodan. (C) Using the conditions above, immobilized H152C was challenged with 3H-glucose solution spiked with increasing amounts of cold glucose and determined a glucose affinity of 0.3 μM ± 0.05.
Next, the amount of protein needed to obtain a large differential signal between proteins having high and low affinity to glucose was determined. Increasing amounts (0.2–3 nmol) of either H152C or W183C-acrylodan were immobilized and challenged with the glucose/3H-glucose solution. When labeled with the fluorophore acrylodan, GGBP W183C has a glucose affinity of 6 mM (De Lorimier et al. 2002). During the experiment, as the amount of immobilized H152C increased, there was a large increase in the counts of bound 3H-glucose prior to a signal decline (Fig. 2B). We suspected this decline was due to the immobilized H152C becoming too concentrated on the resin and thereby interfering with glucose binding. Alternatively, a decline of this sort could have been caused by an exhaustion of the glucose supply; however, the glucose concentration was well in excess at all times during the experiment. In comparison, W183C-acrylodan demonstrated only a slight increase (6%) in bound glucose. This suggested that the weak glucose affinity of this mutant prevented specific binding to glucose at the 10 μM glucose concentration. The 6% signal increase for W183C-acrylodan was probably due to nonspecific glucose binding. The greatest signal difference between H152C and W183C-acrylodan was 13-fold (52% versus 4%) at 2 nmol of protein and this was chosen as the sample size for screening.
To test the ability of the assay parameters above to estimate the equilibrium dissociation constant for H152C, the protein (2 nmol) was immobilized and incubated with increasing amounts of cold glucose containing a constant 3H-glucose concentration (Fig. 2C). This experiment determined a glucose affinity of 0.3 ± 0.05 μM for H152C, which is within the range of 0.2–134 μM. Together these results suggested that the assay parameters as determined could be used to distinguish high- and low-affinity mutants in the purified form. This would enable the elimination of the high-affinity recombinant parental proteins from the library. However, our goal was to rapidly screen GGBP mutants when located within the periplasm. It was unclear whether mutants with strong glucose affinity expressed and unpurified within the periplasm could be selectively eliminated.
Determination of screening parameters in E. coli
Using purified proteins it was demonstrated that 2 nmol of GGBP was needed to produce a differential signal of 13-fold between mutants having high and low affinity to glucose. Using that information, the amount of E. coli Sg13009 cells needed to express 2 nmol of recombinant GGBP after induction was determined. First, bacterial culture was induced to express H152C, then 0.25–1 mL was pelleted and lysed using 100 μL lysis solution. A fraction of the lysate (5 μL), along with known amounts of purified H152C, was analyzed by SDS-PAGE (Fig. 3A). After staining, the protein signal was quantified and the data graphed. The purified H152C showed that 20 pmol (1/100 of 2 nmol) of H152C produced PhosphorImager counts of 303,678 (Fig. 3B). This number was then used to back-calculate the amount of E. coli needed to produce an equivalent amount of H152C (Fig. 3C). This suggested that, after a 2-h induction, a 0.5 mL (5 μL × 100-fold dilution) aliquot of culture at an optical density (OD600 = 1.5) would produce 2 nmol of H152C. For screening it was assumed that the library population would express protein with approximately the same yield, and 0.5 mL of culture with an OD600 = 1.5 was used as the sample size throughout screening.
Figure 3.

Determination of the amount of E. coli used for screening. (A) The Sg13009 strain of E. coli was induced and the lysate analyzed by SDS-PAGE along with known amounts of purified H152C (34 kDa). (B) PhosphorImager was used to quantify the H152C bands and the data were graphed. Arrows are drawn from the 20-pmol amount of H152C (1/100 of 2 nmol) to intersect the linear transformation and then to the corresponding number of counts on the Y-axis. (C) The amount of E. coli needed to produce 2 nmol of H152C was estimated. The counts produced by the purified protein were used to determine the corresponding volume of E. coli culture (0.5 mL) needed to produce 2 nmol of H152C.
The Sg13009 strain of E. coli used for protein expression produced endogenous GGBP and it was important to determine if the level of native protein expressed would interfere with the screening process. Both induced and noninduced bacteria were grown, then 0.5 mL fractions from both cultures were removed and analyzed by SDS-PAGE (Fig. 4A). The amount of H152C protein expressed in each bacterial sample was compared by visual inspection to purified H152C run side-by-side. By visual inspection no distinct 34-kDa band was apparent in the noninduced sample. When quantified (Fig. 4B) the noninduced signal (145,054 with background correction) had at least 24-fold less counts than the induced sample (3,430,761). This suggested that there was insignificant endogenous GGBP protein expressed in this E. coli strain and implied any wt GGBP expressed would have a negligible effect on screening and selection.
Figure 4.
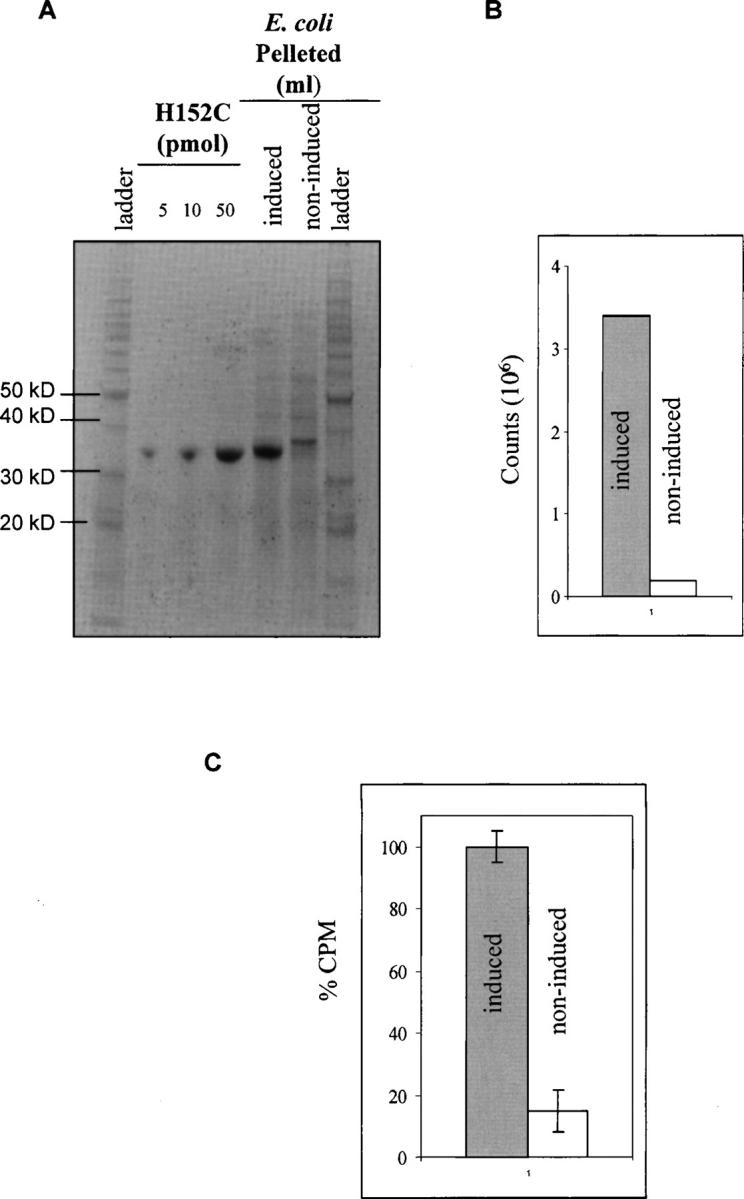
Testing GGBP binding of a 3H-glucose solution in bacteria. (A) Known amounts of purified H152C were analyzed by SDS-PAGE and compared to a 5 μL aliquot from 0.5 mL of bacteria that had been lysed into 100 μL. The bacteria from both induced and noninduced samples were run side-by-side. (B) With background correction the noninduced sample signal was ∼24-fold less than the induced. (C) Induced and noninduced E. coli samples were run through the screening protocol. The induced sample demonstrated 5.6-fold greater signal than the noninduced.
Next, it was determined whether the expressed H152C in the periplasm could bind glucose and be detected under screening conditions. Samples (0.5 mL) from both induced and noninduced bacteria were challenged with 10 μM glucose/3H-glucose, washed, and the remaining 3H counts were quantified. The results demonstrated that E. coli expressing a mutant having high affinity to glucose such as H152C generated a significantly larger (5.6-fold) signal than noninduced E. coli (Fig. 4C). The LOD of 3422 3H counts generated by the noninduced bacterial sample was used for screening. Setting the LOD to this level would conservatively select all colonies expressing mutants having low affinity for glucose. During induction antibiotic selection was used, and bacteria without an expression plasmid were expected to be eliminated.
Library creation using structure-based site-saturation mutagenesis
To generate the library, structure-based site-saturation mutagenesis was used to randomize amino acids in the binding pocket of GGBP at or near the glucose contact sites as identified by X-ray crystallography (Vyas et al. 1994). In total 16 amino acids were mutated and the library was designed so that there would be only 2–3 amino acids randomized in the GGBP open reading frame (ORF) at any one time. This created a focused library having a diversity of ∼32,200 and, because of the small number of mutations per gene, should produce mostly functional GGBP variants. In addition to the randomized amino acids, mutation E149C was placed in each mutant for site-specific fluorophore conjugation after protein purification. The E149C mutant exhibits a wild-type glucose affinity (De Lorimier et al. 2002; Amiss et al. 2003; Hsieh et al. 2004) and did not interfere with screening for low glucose affinity.
Library screening to enrich low-affinity GGBP
Initially, a small number of transformants (120) from the library were challenged with glucose/3H-glucose, then washed and counted. In the first round, colonies having counts near the H152C control sample, and thus considered high-affinity binders, were eliminated. Those remaining went to the second round where colonies were selected near the LOD (3422 CPM) for the assay. Overall, in two rounds of screening, 90% (108) of the colonies were eliminated due to strong glucose affinity. The 12 remaining colonies were analyzed by SDS-PAGE to confirm GGBP expression. This identified eight colonies that did not express a strong 34-kDa GGBP band, and these colonies were eliminated from further analysis. Additionally, of the four remaining candidates, one mutant ran at a slightly higher molecular weight and was also eliminated from further consideration. Of the three remaining colonies, DNA sequencing confirmed that two expressed mutant E149C/N256S, and a single colony expressed mutant E149C/A213R. Next, the glucose equilibrium dissociation constants were determined for these two mutants.
Determination of glucose affinity of the selected GGBP mutants
The selected mutants E149C/A213R and E149C/N256S were purified and labeled at the E149C site with the fluorophore IANBD. A binding curve was generated by glucose titration confirming that E149C/A213R had a glucose affinity equal to 1 mM and produced a sevenfold increase in fluorescence at saturation (Fig. 5A). Mutant E149C/N256S did not reach saturation at 100 mM glucose and had only a twofold increase in fluorescence at 100 mM glucose (data not shown). This mutant was eliminated from further testing due to its very weak glucose affinity.
Figure 5.
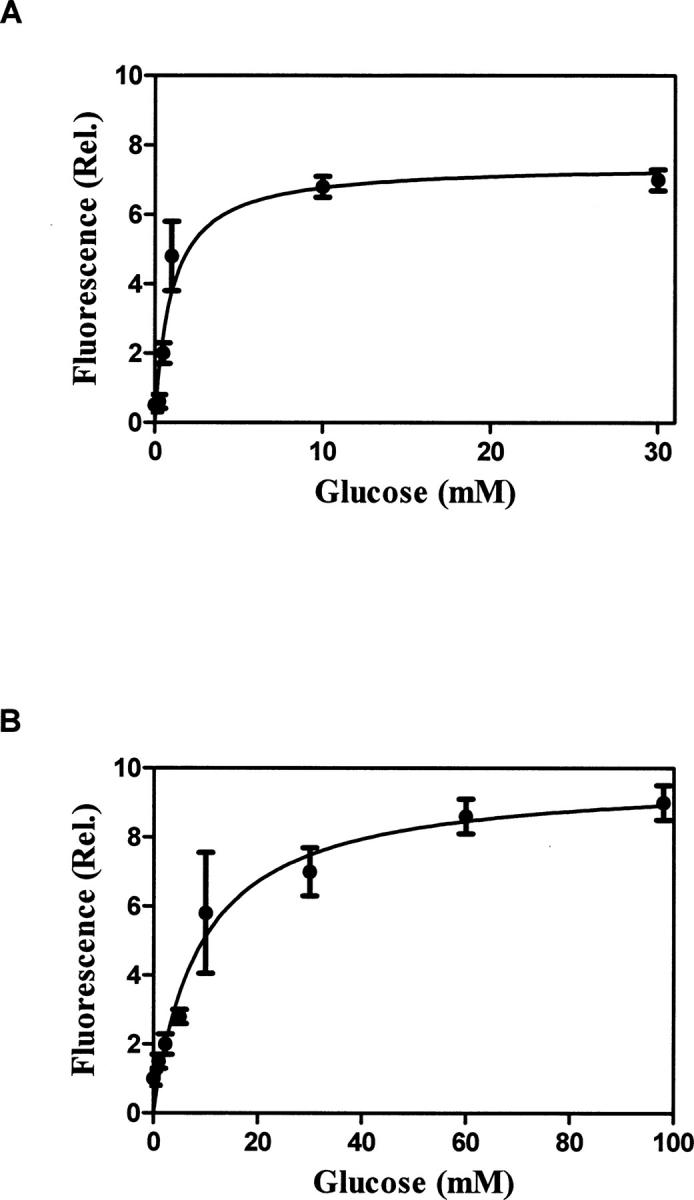
Determination of glucose affinity for the selected mutant. (A) Purified and IANBD-labeled E149C/A213R was challenged with increasing concentrations of glucose, and the fluorescence response was graphed. This demonstrated a Kd equal to 1 mM and a sevenfold fluorescent response. (B) The glucose affinity of the E149C/A213R/L238S mutant was determined in the same manner and demonstrated an eightfold increase in fluorescence and Kd equal to 10 mM.
Overall, the screening had successfully identified the A213R mutation as having a glucose affinity near the human physiologic range. To decrease the glucose affinity further, a third mutation was added to E149C/A213R. During previous experiments to identify dye conjugation sites, a mutation L238S was shown to produce a moderate glucose affinity (Kd = 0.08 mM). By adding the L238S mutation to E149C/A213R, the triple mutant E149C/A213R/L238S having a glucose affinity of 10 mM (Fig. 5B) was created (Amiss et al. 2003). This mutant retained the large increase in fluorescence on binding, making it a good candidate for monitoring glucose concentration by tracking the fluorescent signal.
Discussion
The purpose of this study was to identify GGBP mutants having low affinity to glucose. This was accomplished by constructing a library using structure-based site-saturation mutagenesis and PE screening. This screening method was considered ideal for a GGBP library since this protein functions in nature in the bacterial periplasm and there was little concern over proper functioning or transport into the space via the N-terminal signal sequence. Overall, the method rapidly identified a GGBP mutant having low affinity to glucose.
The GGBP mutant A213R was identified by screening only 120 bacteria on the benchtop without the aid of flow cytometry. To enable flow a fluorescently labeled ligand was needed but was difficult to obtain due to the simple molecular structure of glucose. Even if available, glucose with a conjugated fluorophore may have prevented the molecule from comfortably fitting into the binding pocket of GGBP and could have interfered with screening. Another alternative to enable automated screening was the construction of a GGBP-fluorescent fusion protein. These fusions have been shown to produce fluorescent resonance energy transfer on binding glucose (Ye and Schultz 2003; Amiss et al. 2005; Deuschle et al. 2005), which could have conceivably been used in sorting. However, the straightforward approach using 3H-glucose to detect binding was attempted first without the added complexity of constructing fusions.
During screening several measures were taken to increase the chance of success. First, screening parameters such as the ability to detect specific binding and the assay's sensitivity were rigorously established. Second, few mutations were made in the GGBP ORF. The intent was to retain the ability to bind glucose in the majority of library components since, generally, as the average number of mutations increases, function decreases exponentially (Drummond et al. 2005). Remarkably, a single mutation at A213R reduced the affinity of GGBP 5000-fold. It is interesting to note that the A213R is near the glucose-GGBP H-bonding site at M214. The majority of the mutations constructed (13/18 or 72%) including the N256 were direct H-bonding sites, while the rest (28%) including A213 were residues adjacent to H-bonds. The mutation at N256S almost entirely abolished glucose binding, and a homology search (BLASTP) of glucose or putative glucose-binding proteins from other prokaryotic organisms revealed that N256 is a highly conserved asparagine. A consensus alignment of 34 homologous proteins showed that conservation of N256 site extends over all 34 proteins examined, whereas the A213 residue was conserved only through the first 14 most homologous proteins and then changed to a glycine, serine, or glutamine. Overall, this indicated that the conserved nature of targeted residues is also an important consideration during the library construction.
The reason for E149C/N256S mutant's loss of glucose binding even at 100 mM glucose was not pursued. It was suspected that the protein may have been incorrectly folded or had lost specificity to glucose. Although not completed, nondenaturing electrophoresis or an abnormal thermal melting temperature by either circular dichroism (CD) or isothermal calorimetry could have provided evidence for improper folding of this mutant. Loss of glucose specificity was also untested in E149C/N256S as an alternative explanation for the inability to reach glucose saturation. However, glucose specificity was thoroughly characterized in the E149C/A213R/L238S mutant and it was found to be highly specific for glucose with little interference caused by sugars such as fructose or sucrose or sugar-alcohols like sorbitol (M. Sistare, unpubl.). Additionally, the mutant could specifically bind glucose in human sera and, although GGBP still binds galactose, negligible amounts of this carbohydrate are found in humans.
Due to the desirable characteristics of E149C/A213R/L238S no further screening was attempted. Overall, the selection identified a single low-affinity mutant in the first 120 mutants tested. In contrast, the percent of selected mutants not expressing GGBP (8/120 or 6.6%) was significantly greater. The selection of these bacteria was related to how the LOD was set, since there was no differentiation between very low-affinity mutants and bacteria that did not express GGBP. To attempt to minimize the occurrence of non-GGBP-expressing clones, antibiotic selection was used during induction. However, since several were still selected, these transformants may have suffered from premature termination of transcription, a mutation in the T5 promoter of the expression plasmid, or alternatively the bacteria may have experienced segregation and compensatory mutations (Dahlberg and Chao 2003).
In addition to the unexpected selection of non-GGBP-expressing clones, it is interesting to note that DNA sequencing of the A213R mutation revealed that the codon for the arginine was an AGA which has only 4.3% usage in gram-negative bacteria (Ellington and Cherry 1997). Replacement of this amino acid with a more frequently used arginine codon, such as CGC, may have made the protein expression more efficient. However, at no time during the testing of this mutant was it determined that the highly purified protein (≥90% pure by high-performance liquid chromatography) contained truncated or large amounts of nonfunctional protein. Regardless, codon bias in E. coli as well as the presence of possible stop codons should also be considered during library design. Another notable aspect of the selection was that primers producing the A213R and N256S mutations were expected to produce clones having three and two randomizations, respectively. However, each of the selected clones contained only a single mutation produced by randomization. The efficiency of the randomizations for all primers was determined by sequencing 60 clones from the library. Out of the 60 sequences, six clones had only two mutations and one clone had a single mutation. This indicated that overall ∼11.7% (7/60) of the clones had fewer than the expected number of randomizations. Since Taq polymerase was used with no error-correcting ability, it was suspected that loss of codon randomization was probably due to improper primer annealing. It was also possible that clones having greater numbers of mutations may have been deselected at the stage of E. coli transformation and colony picking from agar plates. If the larger, more robustly growing colonies were picked from the plate, it was conceivable these may have been clones with fewer mutations.
Although site-saturation mutagenesis and library construction can be somewhat challenging, the process did successfully identify the A213R mutation by screening a relatively small number of clones. It would have been interesting to test the ability of “alanine scanning” (Cunningham and Wells 1989) to produce a mutant having low glucose affinity yet high glucose specificity. If successful, the systematic replacement of the H-bonds with alanine would have been the most efficient method of reaching our goal. The randomization, however, did allow for the incorporation of residue side chains and it was suspected that the side chain of the arginine at A213R played a significant role in affinity modification. Computer modeling was used to investigate how A213R altered glucose binding. Models of the open and closed forms of GGBP were analyzed using a genetic algorithm conformational search. The analysis of the A213R substitution in the open form (glucose-free) model produced 20 conformers, and in the majority of these A213R formed salt bridges or H-bonds to adjacent residues. In contrast, only a single conformer was obtained for A213R in the closed form where the arginine side chain adopts a relatively linear conformation and is constrained by adjacent residues. Together this suggests that entropic barriers must be overcome going from the open to the closed form with this residue. In effect, the bulky arginine side chain is inhibiting the GGBP from closing as compared to the native alanine residue and shifts the equilibrium between the open and closed forms in favor of the open form. This in turn weakens binding affinity, a mechanism first proposed by Hellinga (Marvin and Hellinga 2001).
In conclusion, these experiments demonstrated that structure-based site-saturation mutagenesis combined with periplasmic expression screening can be used to efficiently select for lower affinity. The selection process readily identified mutation A213R that was the basis for the triple mutant E149C/A213R/L238S. Overall, this is one of the few examples of performing screening where significantly decreased ligand affinity is desired. Additionally, this work contributed to the development of a glucose biosensor based on GGBP sensing.
Materials and Methods
GGBP cloning
Plasmid pTZ18R has the MglB gene from E. coli strain JM109. The 1.0-kb fragment of the GGBP gene was amplified from pTZ18R and ligated into pQE70 (Qiagen). This created a C-terminal histidine-tag that is wt in sequence except for a lysine-to-arginine change at amino acid 309 and the addition of a serine at position 310. This construct was designated pGGBP. All proteins used in the study were histidine-tagged, contained an E149C mutation for dye conjugation, and were confirmed by DNA sequencing at the University of North Carolina Sequencing Facility.
GGBP protein expression and purification
GGBP was expressed from E. coli strain Sg13009 following standard protocols (Qiagen). After induction, bacteria were lysed using Bugbuster protein extraction reagent (Novagen) and purified using Talon Co2+ resin (BD Clontech). The purified protein in solution was filtered through a 100 kDa cutoff filter (Amicon Bioseparations) to remove aggregates and then concentrated using a 10-kDa cutoff filter (Amicon Bioseparations). The protein (1–2 mg/mL) was dialyzed at 4°C into a phosphate-buffered saline solution (pH 7.4) containing calcium and magnesium (DPBS), then stored at 20°C. Under these conditions the protein was active for at least six months. The yield from the purification was ∼140 mg/L.
Site-saturation mutagenesis
The GGBP library was constructed by randomizing codons at or near the H-bonding sites at the protein-glucose interface. Mutations were generated in the pGGBP construct by a modified QuikChange method (Stratagene) (Hogrefe et al. 2002; Kretz et al. 2004; Zheng et al. 2004). Using a standard reaction mixture, PCR was performed using primer pairs having randomized nucleotides NNK (where N = A, C, G, or T, and K = G or T) at the appropriate locations. After PCR, the product was digested with DpnI and then a second PCR reaction was performed directly on the first PCR product using primers that placed a cysteine at position 149, creating the mutant E149C. This second PCR reaction corrected base-pairing for the randomized amino acid. Using the standard reaction mixture, randomization was performed using primer pairs (forward primers are given): 5′-TGTAACAATCVNNAAGTACGACVNNAACVNNATGTCTGTAGTGC-3′, 5′-TGGTTTTCTTCVNNVNNGAACCGTCTCG-3′, 5′-TACTACGTTVNNVNNGACVNNAAAGAGTCCG-3′, 5′-TTGTCCGGGCVNNCCGVNNGCAGAAGCAVNNACCACTTAGTG-3′, 5′-TGGTTATCGCCVNNVNNGATVNNATGGCAATGGG-3′, 5′-GGTGTTTGGCVNNVNNGCGVNNCCAGAAGCGC-3′, 5′-ACCGTACTGVNNGATGCTAACAACVNNGCGAAAGCG-3′ (where N = A, T, C, or G and V = A, C, or G).
Immobilized GGBP binding assay
A competitive binding assay using 68 μg (2 nmol) purified GGBP proteins immobilized on 100 μL Co2+ resin (BD Clontech) was used in the development of the periplasmic screening assay. Mutants were immobilized and challenged with 0.1 mL of a 10 μM glucose solution containing 0.01 μCi of 3H-glucose (New England Nuclear). After incubation (5 min) the excess glucose solution was removed and protein was rapidly washed with 0.4 mL of the wash buffer (DPBS). The immobilized protein was eluted using 0.03 mL of imidazole solution (0.2 M imidazole in DPBS) placed in scintillation fluid and quantified (LS 6500 Scintillation Counter, Beckman).
SDS-PAGE analysis
Proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) following standard procedures (Ausubel et al. 1998) and then stained using SYPRO Orange (Amersham Biosciences). After staining, protein was quantified using a Typhoon PhosphorImager (Molecular Dynamics).
Periplasmic screening assay
To quickly screen libraries individual colonies were grown and induced to express the GGBP mutants. Then 0.5 mL of each colony was pelleted and washed twice with 1 mL DPBS for 15 min to remove any growth media that might contain glucose. The bacteria were then resuspended in 10 μM glucose solution containing 0.01 μCi of 3H-glucose and incubated for 5 min. The bacteria were pelleted, quickly rinsed with 0.4 mL of DPBS, and re-pelleted. The bacteria were resuspended in 0.03 mL of DPBS placed in scintillation fluid and counted.
Fluorescence assay
For mutants selected by periplasmic screening, a fluorescence assay (Zhou and Cass 1991) was used to determine glucose affinity. Conjugation of the protein to the fluorophore N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine (IANBD amide) was performed as described by the manufacturer (Molecular Probes). Briefly, 0.5 mL of purified protein (1–2 mg/mL) was treated with 2.5 molar excess of dithiothreitol for 30 min. Then a 10-fold molar excess of freshly prepared solution of IANBD in DMSO (0.5 mg/100 uL) was added. While protected from light, the protein and dye were gently mixed for 4 h at 25°C before the unreacted dye was removed by NAP-5 size exclusion column chromatography (Amersham Biosciences). The efficiency of the coupling was determined by absorbance:
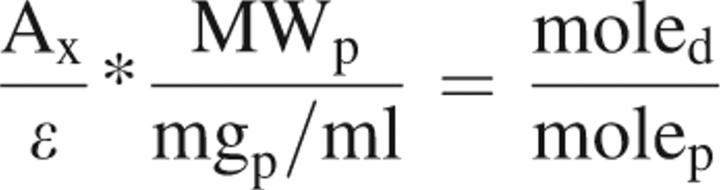 |
where Ax is the absorbance value of the dye at the absorption maximum wavelength, ɛ is the molar extinction coefficient of the dye at the absorption maximum. MWp, mgp/mL, and molep are the molecular weight, concentration, and molar amount of the protein. The molar amount of dye is moled. Using this labeling method, the coupling efficiency should range from 0.95 to 1.1 moles dye to moles protein. An efficiency of >1 indicates that either there is free-dye in the solution or nonspecific labeling occurred. Generally, efficiencies were near 1.0 and the more common issue was free-dye, which could be removed by dialysis. Binding constants were determined by titration of increasing concentrations of glucose into a solution of 0.1 μM protein in PBS (phosphate-buffered saline) and monitoring the change in fluorescence. The samples comprising the standard curve were each made separately and read (without polarizers) in a cuvette at excitation 478 nm and emission 541 nm at room temperature (25°C). The Kd was determined from the following relationships as adapted from Pisarchick and Thompson (1990):
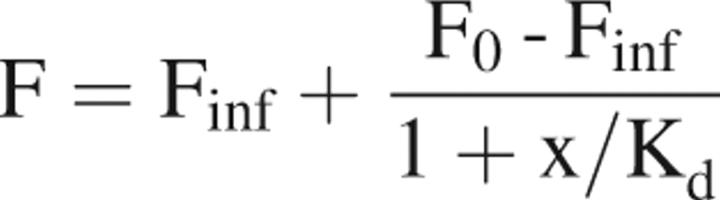 |
where F is fluorescence intensity, Finf is fluorescence at infinity, F0 is fluorescence at zero glucose, and x is the free concentration of glucose ([Glc]free) as determined by the relationship
 |
where [Glc]tot and [Prot]tot are the total concentrations of glucose and protein, respectively. Additionally, fold-change in fluorescence was determined by dividing the fluorescence emission at glucose saturation (100 mM) by the fluorescence emission in the absence of glucose at a constant protein concentration (0.1 μM).
Conformational analysis
Genetic algorithm conformational searches were performed in Sybyl (Tripos) on mutant GGBP structures based on 2GBP.pdb (Protein Data Bank) for the closed form and an open form homology model based on ribose-binding protein. Bonds in the side chains at the mutation sites were defined as rotatable while the rest of the protein was treated as a rigid structure. Explicit hydrogens were used and Gasteiger charges were applied. The default settings in Sybyl were employed for the energy calculation, i.e., nonbonded cutoff = 16 Å, H-bond radius scaling = 0.07, dielectric distance function, hydrogen van der Waals (vdW) radius = 1.5 Å, hydrogen vdW force constant = 0.042. For the genetic algorithm, a population of 100 was employed for 5000 generations with a selection pressure of 1.2. The duplicate window was set to 20° instead of 60°.
Footnotes
Reprint requests to: Terry J. Amiss, BD Technologies, 21 Davis Drive, Research Triangle Park, NC 27709, USA; e-mail: Terry_Amiss@bd.com; fax: (919) 597-6400.
Abbreviations: GGBP, glucose/galactose-binding protein; IANBD, N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine; wt, wild type; ORF, open reading frame; LOD, limit of detection; DPBS, Dulbecco's phosphate-buffered saline; PBS, phosphate-buffered saline.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073119507.
References
- Amiss, T.J. Nycz, C.M. Pitner, J.B. Sherman, D.B. and Wright, D.J. 2003. Binding protein as biosensors. U.S. Patent Application July 17, 2003/0134346A1. http://www.freepatentsonline.com/20030134346.html.
- Amiss, T.J. Pitner, J.B. Freitas, T.C. and Giel, J.L. 2005. Compositions and methods for measuring analyte concentrations. U.S. Patent Application May 26, 2005/0112685A1. http://www.freepatentsonline.com/20050112685.html.
- Amiss, T. Snowden, E. and Pitner, J.B. 2006. Methods of screening proteins. U.S. Patent Application Febuary 23, 2006/0040327A1. http://www.freepatentsonline.com/20060040327.html.
- Arnold F.H. and Georgiou, G. 2003. Directed enzyme evolution: Screening and selection methods, Vol. Vol. 230. Humana Press, New Jersey.
- Ausubel F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., and Struhl, K. 1998. Current protocols in molecular biology, Vol. Vol. 1. John Wiley, New York.
- Boos W. and Gordon, A.S. 1971. Transport properties of the galactose-binding protein of Escherichia coli. Occurrence of two conformational states. J. Biol. Chem. 246: 621–628. [PubMed] [Google Scholar]
- Boos W., Gordon, A.S., Hall, R.E., and Price, H.D. 1972. Transport properties of the galactose-binding protein of Escherichia coli. Substrate-induced conformational change. J. Biol. Chem. 247: 917–924. [PubMed] [Google Scholar]
- Borrok M.J., Kiessling, L.L., and Forest, K.T. 2007. Conformational changes of glucose/galactose-binding protein illuminated by open unliganded, and ultra-high-resolution ligand-bound structures. Protein Sci. 16: 1032–1041. doi: 10.1110/ps.062707807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan J.A. and Wilkinson, J.A. 2002. Protein engineering 20 years on. Nat. Rev. Mol. Cell Biol. 3: 964–970. [DOI] [PubMed] [Google Scholar]
- Chen G., Hayhurst, A., Thomas, J.G., Harvey, B.R., Iverson, B.L., and Georgiou, G. 2001. Isolation of high-affinity ligand-binding proteins by periplasmic expression with cytometric screening (PECS). Nat. Biotechnol. 19: 537–542. [DOI] [PubMed] [Google Scholar]
- Chockalingam K., Chen, Z., Katzenellenbogen, J.A., and Zhao, H. 2005. Directed evolution of specific receptor-ligand pairs for use in the creation of gene switches. Proc. Natl. Acad. Sci. 102: 5691–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham B.C. and Wells, J.A. 1989. High-resolution mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244: 1081–1085. [DOI] [PubMed] [Google Scholar]
- Dahlberg C. and Chao, L. 2003. Amelioration of the cost of conjugative plasmid carriage in Eschericha coli K12. Genetics 165: 1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lorimier R.M., Smith, J.J., Dwyer, M.A., Looger, L.L., Sali, K.M., Paavola, C.D., Rizk, S.S., Sadigov, S., Conrad, D.W., Loew, L., et al. 2002. Construction of a fluorescent biosensor family. Protein Sci. 11: 2655–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuschle K., Okumoto, S., Fehr, M., Looger, L.L., Kozhukh, L., and Frommer, W.B. 2005. Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Sci. 14: 2304–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond D.A., Iverson, B.L., Georgiou, G., and Arnold, F.H. 2005. Why high-error-rate random mutagenesis libraries are enriched in functional and improved proteins. J. Mol. Biol. 350: 806–816. [DOI] [PubMed] [Google Scholar]
- Ellington A. and Cherry, J.M. 1997. Characteristics of amino acids. In Current protocols in molecular biology (eds. F.M. Ausubel et al.), pp. A.1C.1–A.1C.12. John Wiley, New York. [DOI] [PubMed]
- Fernandez-Gacio A., Uguen, M., and Fastrez, J. 2003. Phage display as a tool for the directed evolution of enzymes. Trends Biotechnol. 21: 408–414. [DOI] [PubMed] [Google Scholar]
- Hellinga H.W. and Marvin, J.S. 1998. Protein engineering and the development of generic biosensors. Trends Biotechnol. 16: 183–189. [DOI] [PubMed] [Google Scholar]
- Hogrefe H.H., Cline, J., Youngblood, G.L., and Allen, R.M. 2002. Creating randomized amino acid libraries with the QuikChange Multi Site-Directed Mutagenesis Kit. Biotechniques 33: 1158–1160, 1162. [DOI] [PubMed] [Google Scholar]
- Hoogenboom H.R. 2005. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 23: 1105–1116. [DOI] [PubMed] [Google Scholar]
- Hsieh H.V., Pfeiffer, Z.A., Amiss, T.J., Sherman, D.B., and Pitner, J.B. 2004. Direct detection of glucose by surface plasmon resonance with bacterial glucose/galactose-binding protein. Biosens. Bioelectron. 19: 653–660. [DOI] [PubMed] [Google Scholar]
- Kretz K.A., Richardson, T.H., Gray, K.A., Robertson, T.E., Tan, X., and Short, J.M. 2004. Gene site saturation mutagenesis: A comprehensive mutagenesis approach. Methods Enzymol. 388: 3–11. [DOI] [PubMed] [Google Scholar]
- Lutz S. and Patrick, W.M. 2004. Novel methods for directed evolution of enzymes: Quality not quantity. Curr. Opin. Biotechnol. 15: 291–297. [DOI] [PubMed] [Google Scholar]
- Marvin J.S. and Hellinga, H.W. 2001. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat. Struct. Biol. 8: 795–798. [DOI] [PubMed] [Google Scholar]
- Mazor Y., Blarcom, T.V., Mabry, R., Iverson, B.L., and Georgiou, G. 2007. Isolation of engineered, full-length antibodies from libraries expressed in Escherichia coli . Nat. Biotechnol. 25: 563–565. [DOI] [PubMed] [Google Scholar]
- Miller D.M., Olson, J.S., Pflugrath, J.W., and Quiocho, F.A. 1983. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J. Biol. Chem. 258: 13665–13672. [PubMed] [Google Scholar]
- Olsen M., Iverson, B., and Georgiou, G. 2000. High-throughput screening of enzyme libraries. Curr. Opin. Biotechnol. 11: 331–337. [DOI] [PubMed] [Google Scholar]
- Parikh M.R. and Matsumura, I. 2005. Site-saturation mutagenesis is more efficient than DNA shuffling for the directed evolution of β-fucosidase from β-galactosidase. J. Mol. Biol. 352: 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarchick M.L. and Thompson, N.L. 1990. Binding of a monoclonal antibody and its Fab fragment to supported phospholipid monolayers measured by total internal reflection fluorescence microscopy. Biophys. J. 58: 1235–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiocho F.A. and Ledvina, P.S. 1996. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: Variation of common themes. Mol. Microbiol. 20: 17–25. [DOI] [PubMed] [Google Scholar]
- Reetz M.T. and Carballeira, J.D. 2007. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protoc. 2: 891–903. doi: 10.1038/nprot.2007.72. [DOI] [PubMed] [Google Scholar]
- Sacks D.B., Bruns, D.E., Goldstein, D.E., Maclaren, N.K., McDonald, J.M., and Parrott, M. 2002. Guidelines and recommendations for laboratory analysis in the diagnosis and management of Diabetes Mellitus. Clin. Chem. 48: 436–472. [PubMed] [Google Scholar]
- Salins L.L., Ware, R.A., Ensor, C.M., and Daunert, S. 2001. A novel reagentless sensing system for measuring glucose based on the galactose/glucose-binding protein. Anal. Biochem. 294: 19–26. [DOI] [PubMed] [Google Scholar]
- Tolosa L., Gryczynski, I., Eichhorn, L.R., Dattelbaum, J.D., Castellano, F.N., Rao, G., and Lakowicz, J.R. 1999. Glucose sensor for low-cost lifetime-based sensing using a genetically engineered protein. Anal. Biochem. 267: 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas N.K., Vyas, M.N., and Quiocho, F.A. 1987. A novel calcium binding site in the galactose-binding protein of bacterial transport and chemotaxis. Nature 327: 635–638. [DOI] [PubMed] [Google Scholar]
- Vyas N.K., Vyas, M.N., and Quiocho, F.A. 1988. Sugar and signal-transducer binding sites of the Escherichia coli galactose chemoreceptor protein. Science 242: 1290–1295. [DOI] [PubMed] [Google Scholar]
- Vyas M.N., Vyas, N.K., and Quiocho, F.A. 1994. Crystallographic analysis of the epimeric and anomeric specificity of the periplasmic transport/chemosensory protein receptor for D-glucose and D-galactose. Biochemistry 33: 4762–4768. [DOI] [PubMed] [Google Scholar]
- Ye K. and Schultz, J.S. 2003. Genetic engineering of an allosterically based glucose indicator protein for continuous glucose monitoring by fluorescence resonance energy transfer. Anal. Chem. 75: 3451–3459. [DOI] [PubMed] [Google Scholar]
- Yuan L., Kurek, I., English, J., and Keenan, R. 2005. Laboratory-directed protein evolution. Microbiol. Mol. Biol. Rev. 69: 373–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L., Baumann, U., and Reymond, J.L. 2004. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32: e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.Q. and Cass, A.E.G. 1991. Periplasmic binding protein based biosensors. 1. Preliminary study of maltose binding protein as sensing element for maltose biosensor. Biosens. Bioelectron. 6: 445–450. [Google Scholar]