

Abstract
The transparency of the eye lens depends on the high solubility and stability of the lens crystallin proteins. The monomeric γ-crystallins and oligomeric β-crystallins have paired homologous double Greek key domains, presumably evolved through gene duplication and fusion. Prior investigation of the refolding of human γD-crystallin revealed that the C-terminal domain folds first and nucleates the folding of the N-terminal domain. This result suggested that the human N-terminal domain might not be able to fold on its own. We constructed and expressed polypeptide chains corresponding to the isolated N- and C-terminal domains of human γD-crystallin, as well as the isolated domains of human γS-crystallin. Both circular dichroism and fluorescence spectroscopy indicated that the isolated domains purified from Escherichia coli were folded into native-like monomers. After denaturation, the isolated domains refolded efficiently at pH 7 and 37°C into native-like structures. The in vitro refolding of all four domains revealed two kinetic phases, identifying partially folded intermediates for the Greek key motifs. When subjected to thermal denaturation, the isolated N-terminal domains were less stable than the full-length proteins and less stable than the C-terminal domains, and this was confirmed in equilibrium unfolding/refolding experiments. The decrease in stability of the N-terminal domain of human γD-crystallin with respect to the complete protein indicated that the interdomain interface contributes 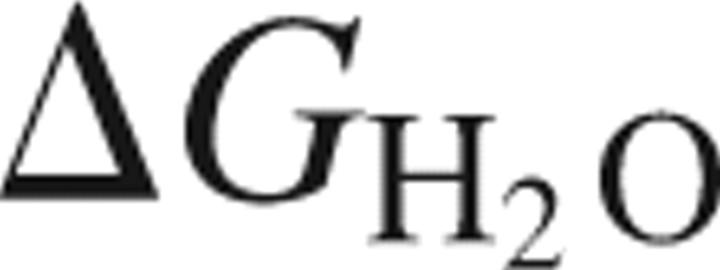 of 4.2 kcal/mol to the overall stability of this very long-lived protein.
of 4.2 kcal/mol to the overall stability of this very long-lived protein.
Keywords: human γD-crystallin, human γS-crystallin, domain interface, cataract, equilibrium unfolding/refolding transitions, refolding kinetic intermediates, protein stability
Crystallins are the major proteins in the elongated fiber cells of vertebrate eye lenses, present at concentrations of 200–450 mg/mL (Fagerholm et al. 1981; Siezen et al. 1988; Slingsby and Clout 1999). The crystallins are responsible for both the transparency and high refractive index of the lens (Delaye and Tardieu 1983; Fernald and Wright 1983). The β- and γ-crystallins are thought to be primarily structural proteins, while α-crystallin possesses an additional chaperone activity. Terminally differentiated fiber cells in the lens are enucleated and devoid of all other organelles and are unable to degrade damaged crystallins or to synthesize new ones (Oyster 1999). Thus, the crystallins must remain soluble for decades despite high concentrations of protein, continual UV exposure, and potential oxidative stress. This is particularly true of the crystallins in the central lens nucleus, which are synthesized in utero (Harding and Crabbe 1984; Aarts et al. 1989; Lampi et al. 2002a).
Cataracts, which interfere with light transmission, represent aggregated, precipitated, or other insoluble states of the crystallins. Mature-onset cataract affects more than 40% of people who experience some form of blindness, regardless of gender, race, or economic status. The protein composition of the insoluble fraction of aged and cataractous lenses includes the α-, β-, and γ-crystallins, with many carrying a variety of modifications such as deamidation, oxidation, glycation, methylation, disulfide bond formation, and truncations (Lampi et al. 1998; Hanson et al. 2000; Lapko et al. 2005; Searle et al. 2005; Wilmarth et al. 2006).
Models for the causes of mature-onset cataract include oxidative or UV-induced protein damage leading to protein unfolding, aberrant activation of fiber proteases, and saturation of α-crystallin chaperone function. Any or all of these could result in protein precipitation, insolubility, or aggregation. The remarkable stability of the crystallins and chaperone function in the lens is presumably to prevent the onset of these aggregated states. The biochemical basis of the very high stability of the crystallins and the nature of the misfolded, modified, or aggregated states are thus of considerable importance in understanding the etiology of cataract.
All β- and γ-crystallin polypeptide chains have two homologous double Greek key domains. They are believed to have evolved by gene duplication and fusion from an ancestral single-domain βγ-crystallin (Lubsen et al. 1988; Piatigorsky 2003). A candidate for this precursor has been found recently in the urochordate sea squirt, Ciona intestinalis. Gene and structural alignment comparisons suggest that β- and γ-crystallins evolved from this common ancestral protein (Shimeld et al. 2005).
Each domain of the β- and γ-crystallins consists of two intercalated antiparallel β-sheet Greek key motifs. The two highly symmetrical domains interact through a hydrophobic interface and are connected by an interdomain linker. The γ-crystallin proteins are monomeric, whereas the β-crystallins can form dimers, homo-oligomers, and in many cases hetero-oligomers (Werten et al. 1999; Bateman et al. 2001, 2003; Lampi et al. 2001). The major differences between these two groups are attributed to N- and/or C-terminal extensions found primarily in the β-crystallins (Bloemendal et al. 2004; Hejtmancik et al. 2004). The N- and/or C-terminal extensions and slight variations in the interface side-chain interactions are implicated in the formation of the β-crystallin multimers (Norledge et al. 1997; Smith et al. 2007).
It has been suggested that the high stabilities of the β- and γ-crystallin proteins are due to the complex topology of the double Greek key (MacDonald et al. 2005), in which strand d of the first motif is paired with strand c of the second motif (Fig. 1). Although the majority of β- and γ-crystallins have these features, there are differences in the thermodynamic and kinetic stabilities among the crystallins and their individual domains.
Figure 1.

Crystal structure of γDWT and NMR structure of murine γSWT. Both full-length proteins are ∼20 kDa in size. (A) A ribbon diagram of the γDWT X-ray crystal structure (PDB ID: 1HK0) (Basak et al. 2003). The isolated γDN protein ends at Pro82 (highlighted in dark gray) not including the short interdomain linker. The isolated γDC protein begins at Arg 89 (highlighted in dark gray) at the beginning of the β-sheet. (B) A ribbon diagram representing the NMR structure of the murine γS-crystallin protein (PDB ID: 1ZWO) (Wu et al. 2005). The His 86 position is the same amino acid in the human sequence, while the Ala 93 position is replaced with Tyr in the human sequence. Sequence alignment between human and murine γS-crystallin shows 89% identity and 96% similarity (bl2seq, Blosum 62 matrix).
An additional distinctive feature of the vertebrate β- and γ-crystallin family is the interface between the Greek key domains. In human γD-crystallin, this is composed of a hydrophobic patch of six residues, three from each domain, plus a pair of glutamines shielding the hydrophobic domain from solvent, and an arginine and methionine at the base of the interface near the linker (Basak et al. 2003). These residues are highly conserved among vertebrate β- and γ-crystallins, except for the peripheral methionine, which is unique to human γD-crystallin and is usually a charged residue in other vertebrate β- and γ-crystallins. The significance of this amino acid difference in human γD-crystallin is unknown. The contribution of these residues to overall stability has been assessed by characterizing proteins with alanine substitutions of the interface residues. Substitutions of both the hydrophobic and the polar residues of the interface significantly destabilized the native protein (Flaugh et al. 2005a,b). Substitutions in the interface of bovine γB-crystallin (BγB-Crys) have also been found to be destabilizing (Palme et al. 1997). Furthermore, Liu and Liang (2006) have shown that polar substitutions of hydrophobic residues in the β-strands of the domain interfaces of human βB2-crystallin significantly destabilized the protein.
Human γD-crystallin and human γS-crystallin are two of the most abundant γ-crystallins in the human lens. These proteins share 69% sequence similarity and 50% sequence identity and are ∼21 kDa in size. γD-Crystallin is found at its highest levels in the central nucleus and is primarily synthesized in utero. Thus the long-term solubility and stability of this protein are particularly important for maintaining lens transparency. Families carrying single amino acid substitutions in this protein exhibit juvenile-onset cataracts (Heon et al. 1999; Stephan et al. 1999; Pande et al. 2000; Smith et al. 2000; Santhiya et al. 2002). One of these mutations, P23T, reduces the stability and solubility of the mutant protein in vitro (Evans et al. 2004; Pande et al. 2005). On the other hand, γS-crystallin is more prevalent in the outer regions of the lens, primarily the cortex, which continues to grow throughout life (Cook et al. 1994; Bron et al. 2000; Wistow et al. 2002). Both protein synthesis and turnover are thought to occur in these regions.
The crystal structure of wild-type human γD-crystallin has been solved as well as the C-terminal domain of wild-type human γS-crystallin in isolation (Purkiss et al. 2002; Basak et al. 2003). The full-length murine γS-crystallin structure has been resolved by NMR methods (Wu et al. 2005). There are few noticeable differences between the two proteins, although γS-crystallin has a four-amino-acid N-terminal extension and possibly two additional amino acids in its interdomain linker (Fig. 1). Crystal structure and modeling studies have shown that both γD-crystallin domains have high structural similarity (Blundell et al. 1981; Basak et al. 2003). The NMR solved structure of murine γS-crystallin also demonstrated high structural similarity among domains (Wu et al. 2005). Therefore, both γ-crystallins have similar structures, and their individual domains display high structural similarity between each other.
Human γD-crystallin (γDWT) has been cloned, expressed, purified, and characterized with respect to its folding and unfolding in vitro (Kosinski-Collins and King 2003; Kosinski-Collins et al. 2004). Similarly, the unfolding and refolding of human γS-crystallin (γSWT) in vitro has also been characterized (Wenk et al. 2000).
The unfolding and refolding of γDWT is a three-state process. Both kinetic and equilibrium studies have identified a major partially folded intermediate on its unfolding and refolding pathway, in vitro. This species has its C-terminal domain folded and its N-terminal domain unfolded, or at least disordered. From the three-state melting transitions, it was clear that the N-terminal domain was much less stable than the C-terminal domain. Further information on the interaction between the domains was obtained from site-specific mutations of the residues forming the domain interface. Not surprisingly, substitutions of the N-terminal domain residues contributing to the interface destabilized the N terminus. However, unexpectedly, substitutions of the C-terminal domain residues in the interface had little effect on the C terminus itself but also destabilized the N-terminal domain (Flaugh et al. 2005b). This suggested that the C-terminal domain stabilized the N-terminal domain and provided a template for its refolding. This raised the possibility that the N-terminal domain of γDWT could not fold independently.
Recently, it has been also shown that Gln → Glu mutations mimicking deamidation in the γDWT interface decreased thermodynamic and kinetic stability, indicating the importance of the interface in the unfolding barrier (Flaugh et al. 2006). Deamidation in the interface of human βB2-crystallin, human βB1-crystallin, and the predicted interface of human βA3-crystallin also destabilizes the intact protein, supporting an important role for the domain interface (Kim et al. 2002; Lampi et al. 2006; Takata et al. 2007).
Wenk et al. (2000) showed that the unfolding and refolding of γSWT followed a two-state transition, implying that the two domains of γS-crystallins have similar stabilities. Stabilities of the isolated domains were also studied, and the results revealed that the γS N-terminal domain (γSN) and the C-terminal domain (γSC) had slightly different stabilities. This result suggested that the domain interface may not be important for the stability of the protein and that this protein does not fold sequentially like γDWT but instead more cooperatively. Previously, in studies of bovine γB-crystallin, differential domain stability was observed at acidic pH (20°C) but not at neutral pH, suggesting sequential domain folding and important domain interface interactions under these conditions (Rudolph et al. 1990; Mayr et al. 1997). Other studies have shown that the isolated domains of human and rat βB2-crystallin are not as stable as the full-length protein (Wieligmann et al. 1999; Fu and Liang 2002).
To explore more deeply the question of the contribution of domain interactions to overall protein stability, we have prepared and investigated the properties of the isolated N- (γDN) and C-terminal (γDC) domains of γDWT. We have included in these experiments the analogous isolated N- and C-terminal domains of γSWT. Efforts to crystallize γSN have been unsuccessful, raising the possibility that its N terminus in isolation was also not in the native fold. The studies below attempt to address the question of the importance of the domain interface in the evolution of the two domain vertebrate crystallins.
Results
Protein purification and characterization
The specific residues that would comprise the isolated domains of γDWT were chosen by examining the crystal structure and selecting those sequences of the protein not including the linker. The γD N-terminal domain construct included residues Gly 1–Pro 82, which is the last residue at the end of the β-strand before the linker. The γDN construct was created by introducing a stop codon at residue 83 (His 83 stop) into the γDWT sequence. The γD C-terminal domain construct was created by cloning into the pQE1 vector (Qiagen), sequences corresponding to Arg 89 (the first residue after the linker and at the beginning of the β-strand), continuing to Ser 174 at the end of the protein (Fig. 2).
Figure 2.

Amino acid sequence alignment of γDWT and γSWT. The regions of the proteins included in the isolated domain proteins are highlighted, (blue) γDN G1–P82, (red) γDC R89–S174, (green) γSN S1–H86, (gray) γSC Y93–E177. Upper numbers represent the residues in γDWT, and lower numbers represent the residues in γSWT.
At the time that the γS isolated domain constructs were created, there was no structure available of the γS N-terminal domain or linker. Thus, it was difficult to predict what sequences should be included in the constructs. The γS N-terminal domain construct was created by introducing a stop codon at residue 87 (Leu 87 stop) into the γSWT sequence. Recently, the NMR structure of the murine γS predicted a linker region of γSWT from residues 85 to 93 according to the Entrez Protein Database (#NP_060011). If the domain structures of human γSWT are consistent with the murine structure, the γSN protein constructed here starts with Ser 1 and includes one residue within the proposed linker (His 86). The γS C-terminal domain was created by cloning sequences corresponding to Tyr 93 to Glu 177 at the end of the protein into the pQE1 vector (Fig. 2). The X-ray crystal structure of the γS C-terminal domain also began with Tyr 93 in the C-terminal domain (Purkiss et al. 2002).
All full-length and isolated domain proteins were expressed by inducing Escherichia coli cell cultures with IPTG and incubating them for several hours at 37°C. The full-length recombinant crystallins were soluble, accumulating in the cell supernatant. The isolated domains also behaved as soluble subunits, accumulating in the cell supernatants. The isolated domain recombinant crystallins were purified using Ni-NTA affinity chromatography, following the same protocols used for the full-length γD- and γS-crystallins. The elution of the isolated domains was similar to that of the full-length proteins during column purification. γSWT and its isolated domains eluted from the Ni-NTA column with lower concentrations of imidazole than γDWT and its isolated domains. In general, the isolated domains behaved similarly to the full-length crystallins during protein expression and purification. All proteins were >90% pure based on SDS-PAGE gel electrophoresis and Coomassie Blue staining.
Analysis of N-terminally His-tagged γDWT revealed no difference in the kinetic, equilibrium, and secondary structural characteristics compared to recombinant protein without the His-tag (Kosinski-Collins and King 2003; Kosinski-Collins et al. 2004). In addition, analysis of N-terminally His-tagged γSWT compared to no His-tag recombinant γSWT detected no difference in the secondary structure as analyzed by CD spectroscopy or in kinetic and equilibrium data as analyzed by fluorescence spectroscopy (data not shown).
Analytical size exclusion chromatography
Previous studies of the isolated bovine βB2 N-terminal domain showed the possibility of homodimerization (Wieligmann et al. 1999). In addition, the microbial single domain protein Spherulin 3a, a structural homolog of the βγ-crystallins, formed dimers at physiological concentrations (Kretschmar et al. 1999). In order to determine if the isolated domain dimerized under experimental conditions used here, analytical size exclusion chromatography (SEC) was used. Protein was loaded onto the column at a concentration of 80 μg/mL. Elution volumes of molecular weight standards were determined to formulate an elution volume, protein size relationship. All of the isolated domain proteins eluted after the 13.7 kDa protein standard (Ribonuclease A), indicating that all proteins were in the monomer form (Fig. 3). Moreover, chromatographs of the isolated domains did not overlay with the wild-type proteins, confirming that the isolated domains did not form stable dimers in appreciable amounts.
Figure 3.
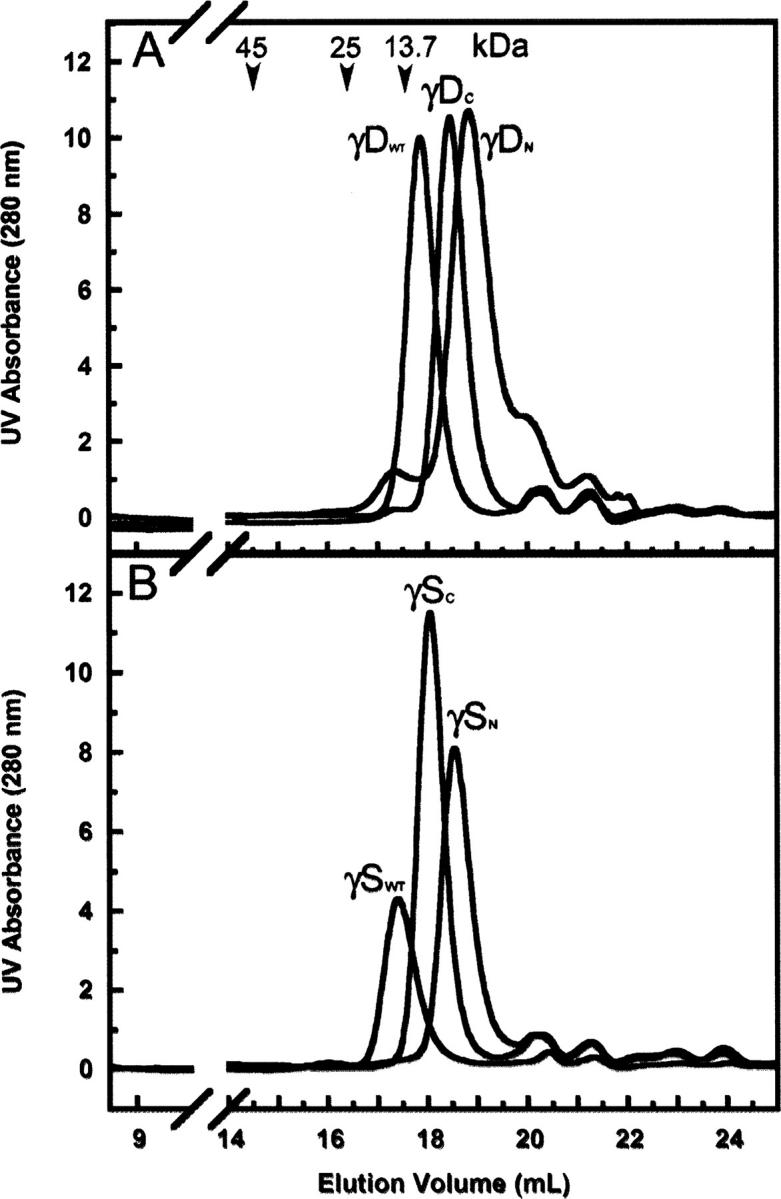
Analytical size exclusion chromatography profiles of the isolated domains and wild-type proteins. All samples were loaded onto a Superdex 200 10/300 GL column at a protein concentration of 80 μg/mL. The molecular weight standards were ovalbumin (45 kDa), chymotrypsinogen A (25 kDa), ribonuclease A (13.7 kDa), elution volumes of which are depicted by arrows. (A) γDWT, γDN, and γDC; (B) γSWT, γSN, and γSC. The secondary peak seen in the γDN sample was variable and usually less prominent and has not been identified.
γDWT protein (21.817 kDa) eluted from the column at a peak volume of 17.86 mL, while γSWT (22.157 kDa) eluted at 17.4 mL. This result is expected since γSWT is slightly larger than γDWT. The γDC (11.833 kDa) isolated domain eluted at a peak volume of 18.45 mL, and the γDN (10.972 kDa) isolated domain at a peak volume of 18.84 mL, confirming that γDC is larger than γDN (Fig. 3A). The isolated domains of γS eluted at similar volumes as the γD isolated domains (Fig. 3B). Eluted peak volumes of γSN (11.4 kDa) and γSC (11.5 kDa) were 18.54 mL and 18.04 mL, respectively. All of the γ-crystallin samples described below were predominately monomeric.
Circular dichroism (CD) spectroscopy
To determine if the isolated domains were folded into native-like conformations, we examined them by CD and fluorescence spectroscopy. The secondary structures of the isolated domains and the full-length proteins were analyzed by far-UV CD spectroscopy at 37°C (Fig. 4). The CD spectrum of the complete γDWT protein demonstrated the characteristic β-sheet ellipticity minimum at 218 nm. The spectrum of γDC also indicated the characteristic β-sheet structure as well and was not distinctly different from that of γDWT (Fig. 4A). In contrast, the γDN spectrum exhibited a possible increase in random-coil structure as seen by an increase in negative molar ellipticity at 204 nm. These CD spectra were further analyzed by deconvolution software, CDPro Suite, to determine quantitative percentages of secondary structure (Sreerama and Woody 2000). Deconvolution of the CD spectra agreed with qualitative observations that the γDWT and γDC β-sheet structures were not distinguishable (Table 1). γDWT analysis showed a ∼40% β-sheet, ∼6% α-helical, mostly 310 α-helical structure (consistent with 3D structure), ∼21% turn, and ∼31% unordered. γDC deconvolution was similar to γDWT with ∼41% β-sheet, ∼5% α-helical, ∼22% turn, and ∼32% unordered. The differences in percentages are probably within experimental error of one another. However, the CD deconvolution of γDN suggested it had a decrease in β-sheet (∼30%) and an increase in α-helix (∼11%), turns (∼24%), and unordered (∼35%) secondary structure (Table 1).
Figure 4.

Far-UV CD spectra of isolated domains and full-length proteins. Samples are at a protein concentration of 100 μg/mL in 10 mM sodium phosphate buffer, pH 7.0 at 37°C. (A) CD spectra recorded from 195–260-nm wavelengths for (black ♦) γDWT, (blue •) γDN, and (red ▪) γDC. (B) CD spectra recorded from 195–260-nm wavelengths for (blue ▴) γSWT, (green  ) γSN, and (orange
) γSN, and (orange  ) γSC.
) γSC.
Table 1.
Deconvoluted CD spectra and fluorescence emission spectra maximums for γD and γS wild-type and isolated domain proteins

γSWT also exhibited the characteristic β-sheet spectrum with a minimum at 218 nm. γSC had a similar spectrum to full-length γSWT (Fig. 4B). However, the spectrum of γSN was significantly different than the γSWT CD spectrum. The deconvolution of γSN yielded ∼29% β-sheet, ∼4% α-helical, ∼25% turns, and ∼40% unordered compared to ∼33% β-sheet, ∼6% α-helical, mostly 310 α-helical structure (consistent with 3D structure), ∼24% turns, and ∼36% unordered for γSWT. Again, γSC deconvolution was similar to γSWT with ∼32% β-sheet, ∼9% α-helical, ∼26% turns, and ∼34% unordered (Table 1). Secondary structure similarity between γSWT and γSC is consistent with the atomic structure similarity observed between the murine NMR γSWT structure and the crystal structure of human γSC (Purkiss et al. 2002; Wu et al. 2005).
Crystallin proteins consist of Greek key antiparallel β-sheets, a topology that contains a considerable amount of twisted β-sheets (Blundell et al. 1981; Bax et al. 1990). It has been observed previously that deconvolutions of these proteins' CD spectra are problematic to interpret because the twisted antiparallel β-sheets have similar optical dispersions as unordered peptides, as well as because of the lack of reference sets for these proteins (Sreerama and Woody 2003). This is supported by the high percentage of unordered structure in our deconvolution analysis.
Fluorescence spectroscopy
Fluorescence spectroscopy was used to monitor the tertiary structure of both crystallins and their domains. γDWT and γSWT both have four conserved tryptophans, two buried within the hydrophobic core of each domain. The tryptophans are at positions 42 and 68 in the γDN (46 and 72 in γSN) and 130 and 156 in the γDC (136 and 162 in γSC). The fluorescence of these tryptophans is highly quenched in the folded state (Kosinski-Collins et al. 2004; Chen et al. 2006). Trp 68 and Trp 156 are quenched through a charge transfer to the polypeptide chain backbone, while Trp 42 and Trp 130 undergo an energy transfer mechanism to Trp 68 and Trp 156, respectively (Chen et al. 2006). γSWT may be quenched in the native state by a similar mechanism. As a result, tryptophan fluorescence is a sensitive reporter of the native-like state of these proteins.
In addition to the four tryptophans, there are 14 semiconserved tyrosines (71% identity, 93% similarity based on 32 diverse γ-crystallin sequences) located throughout the protein. There are seven tyrosines in each domain of γDWT. For γSWT, there are eight tyrosines in γSN and six in γSC. In order to monitor preferentially the structure surrounding the tryptophan residues, we excited the proteins at 295 nm and recorded the fluorescence emission spectra from 310 to 400 nm.
The fluorescence emission spectra of the isolated domains at 37°C shown in Figure 5 was similar to those of the full-length proteins. γDWT had a quenched native emission maximum of ∼326 nm and a redshifted unfolded emission maximum of 350 nm (Table 1). The native maximum of γDC was similar to full-length and also had quenched fluorescence, indicating a native-like structure. The γDN domain fluorescence had a higher quantum yield than both γDWT and γDC, indicating that it was not as quenched in isolation possibly because of disruptions in structure around Trp 42. However, upon denaturation in GuHCl, the fluorescence intensity of γDN increased, indicating it was native-like in the absence of denaturant.
Figure 5.

Fluorescence emission spectra of native and unfolded isolated domains and full-length γDWT and γSWT. All proteins were excited at 295 nm and emissions were recorded from 310 to 400 nm. Samples consisted of 10 μg/mL protein in 100 mM sodium phosphate, 1 mM EDTA, 5 mM DTT, pH 7.0, and 5.5 M GuHCl for unfolded samples equilibrated at 37°C. (A) Native spectra for γDWT (♦) and unfolded spectra for γDWT (line). (B) γDN native (•) and γDN unfolded (line). (C) γDC native (▪) and γDC unfolded (line). (D) γSWT native (▴) and γSWT unfolded (line). (E) γSN native (◢) and γSN unfolded (line). (F) γSC native (◣) and γSC unfolded (line).
γSWT had a quenched native emission maximum of ∼329 nm and an unfolded maximum of 350 nm (Fig. 5D; Table 1). γSC had higher native fluorescence emission intensity, also indicating that the C-terminal domain had a higher quantum yield and is not as quenched in the native state (Fig. 5F). γSN domain had a similar peak and fluorescence intensity compared to γSWT, although the overall shape of the spectra indicates that there may be some differences in tertiary structure (Fig. 5E).
In conclusion, all domains in isolation, although exhibiting slight alterations in secondary and tertiary structures, are structurally similar to their respective full-length proteins as well as to each other.
Thermal denaturation indicates differential domain stability
Circular dichroism and fluorescence emission spectra indicated that the isolated domains were in native-like conformations. To assess the stability of these domains qualitatively, we examined their thermal denaturation. Although thermal denaturation is qualitative, it measures stability in a more physiological-relevant manner than chemical denaturant experiments.
Figure 6 shows the thermal denaturation behavior monitored by CD. All four of the isolated domains remained folded until 60°C or higher and exhibited a cooperative melting transition consistent with typical thermal denaturation unfolding transitions. Thermal denaturation was an irreversible reaction demonstrated by visible aggregation upon the completion of the experiment. Since the calculation of meaningful thermodynamic parameters is limited by the lack of reversibility, we report only the fraction native as a function of increasing temperature (Fig. 6).
Figure 6.

Thermal denaturation of γDWT, γSWT, and their individual domains. Samples were prepared at 100 μg/mL protein concentration in 10 mM sodium phosphate buffer (pH 7.0). CD signal at 218 nm was monitored as the temperature was increased from 25°C to 90°C. Data were normalized and the native fraction was calculated (see Materials and Methods). (A) (♦) γDWT, (•) γDN, and (▪) γDC. (B) (▴) γSWT, (◢)γSN, and (◣) γSC.
Both full-length γDWT and γSWT were extremely stable, with a T M of 84.5°C and 74.1°C, respectively. The differences in stability between the individual domains were evident in the thermal experiments; the T M of γDN was 64°C, while the T M of γDC was 77°C (Fig. 6A). The T M of γSN was 69.1°C and of γSC was 75.1°C (Fig. 6B). The T M difference between γD isolated domains was larger than that of the γS isolated domains, confirming the idea that the γDWT interface is more essential in increasing the thermodynamic stability of its N-terminal domain. In addition, the T M of γDWT is 7°C greater than the T M of γDC, reinforcing the idea that both domains are necessary for overall conformational stability. The interface of γSWT, even though essential for stabilizing its N-terminal domain, may not be as crucial in the overall stability of the protein, consistent with other studies (Zarina et al. 1994; Wenk et al. 2000).
Equilibrium unfolding and refolding in vitro
The polypeptide chains of the isolated domains described above folded into their native-like state within E. coli. To determine if the isolated domains would refold in vitro, we conducted equilibrium unfolding/refolding experiments. These results also allowed us to estimate quantitatively the stability of each domain. Previously, bovine γB-crystallin has been shown to be resistant to urea denaturation at neutral pH and requires GuHCl (Rudolph et al. 1990). Human γDWT is also resistant to unfolding in up to 8 M urea at neutral pH, and in order to best mimic physiological conditions, previous equilibrium unfolding/refolding experiments were performed at pH 7, 37°C (Kosinski-Collins and King 2003). Therefore, all of the crystallins were analyzed under the same conditions as described above for comparison purposes to human γDWT.
As seen in Figure 7, γDN and γDC refolded at high yield when diluted out of denaturant. As reported by Wenk et al. (2000), γSN and γSC also refolded at high yield when diluted out of denaturant. In contrast to γDWT, we observed no evidence for aggregated states competing with productive refolding for any of the isolated domains and γSWT (Figs. 7 and 8).
Figure 7.
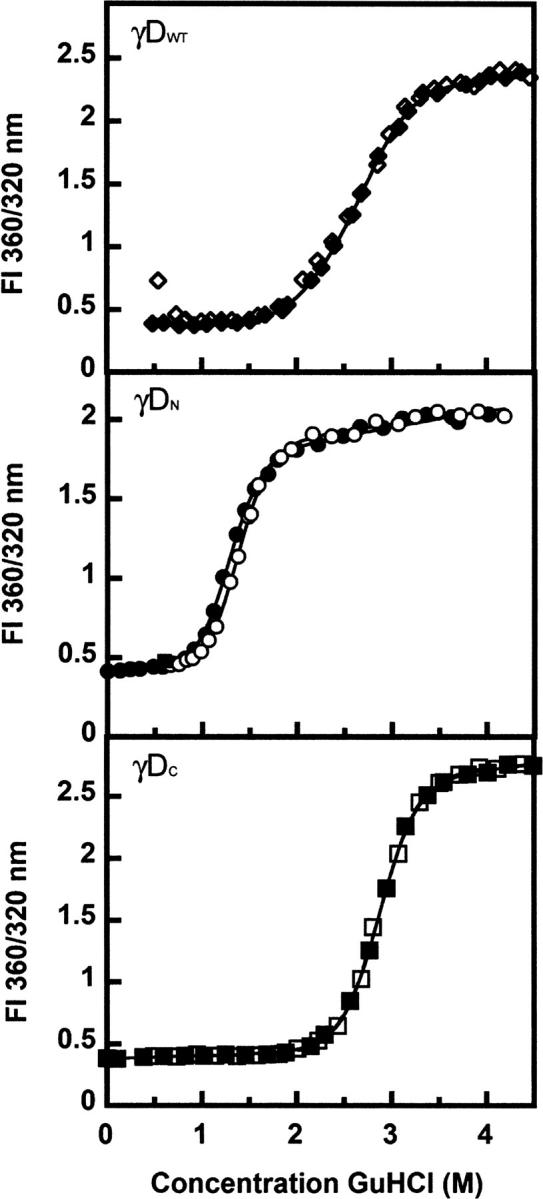
Equilibrium unfolding (closed symbols) and refolding (open symbols) transitions of (♦) γDWT, (•) γDN, and (▪) γDC. Samples contained 10 μg/mL protein, 100 mM sodium phosphate, 1 mM EDTA, 5 mM DTT (pH 7.0), and various concentrations of GuHCl at 37°C. The ratios of fluorescence emission at 360 nm and 320 nm were calculated. Equilibrium data fits are indicated by solid black lines.
Figure 8.
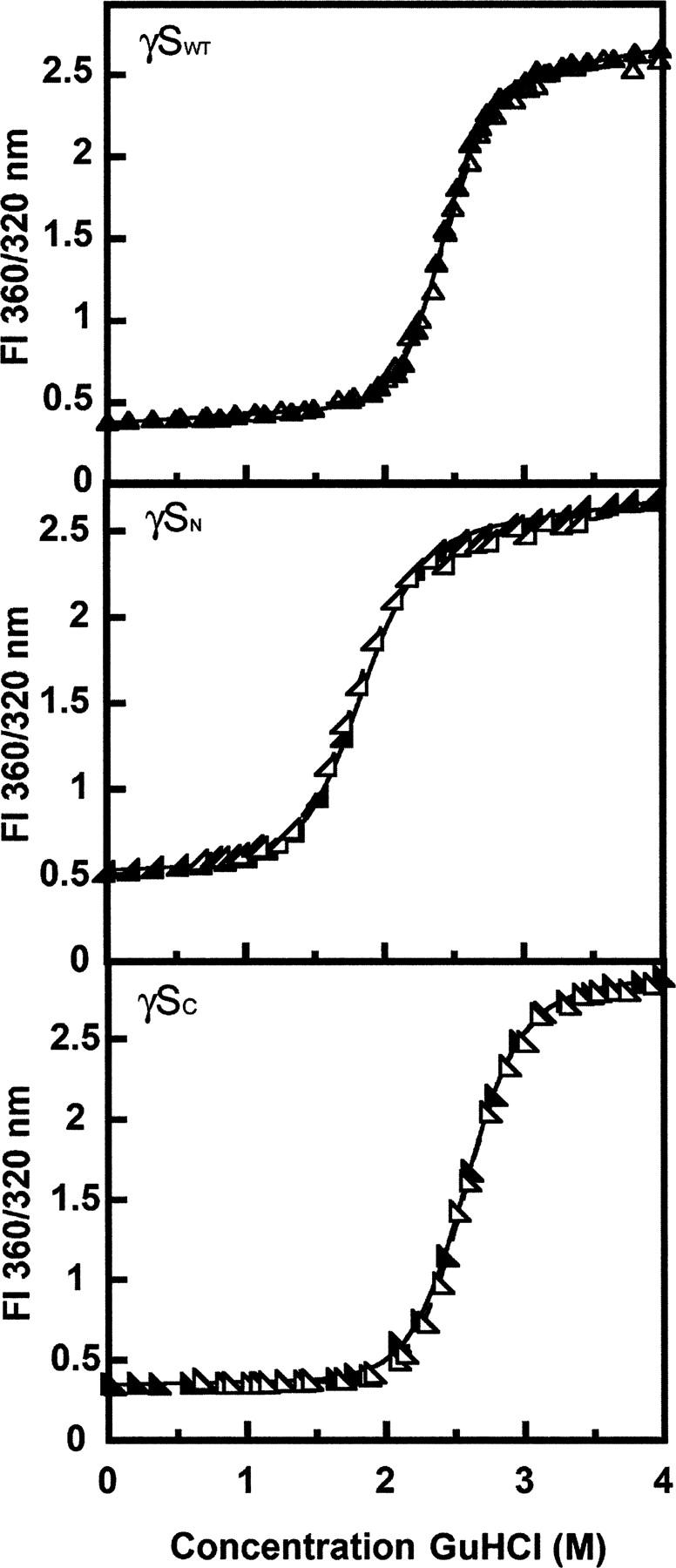
Equilibrium unfolding (closed symbols) and refolding (open symbols) transitions for (▴) γSWT, (◢) γSN, and (◣) γSC. Samples contained 10 μg/mL protein, 100 mM sodium phosphate, 1 mM EDTA, 5 mM DTT (pH 7.0), and various concentrations of GuHCl at 37°C. The ratios of fluorescence emission at 360 nm and 320 nm were calculated and plotted versus the concentration of GuHCl. Two-state fits of the equilibrium data are indicated by solid black lines.
The equilibrium unfolding/refolding curves were determined by calculating the ratio of the fluorescence emission at 360 nm and 320 nm versus denaturant concentration (Figs. 7 and 8). Since the irreversibility of γDWT can cause extrapolation errors in determining free energy (H2O), we used transition midpoints (C M) of the equilibrium unfolding/refolding curves, the GuHCl concentration at which 50% of the protein is denatured, as a measure of stability. However, since all of the other proteins exhibited full reversibility under these experimental conditions, we have extrapolated values and have included these and associated m-values in Table 2 for γD and Table 3 for γS. The thermodynamic parameters were calculated from both 360 nm emission data and 360/320 nm emission ratio data. Both data analyses were comparable and within standard error of one another. The 360/320 nm representation is shown for visual clarity of equilibrium transitions, and the single wavelength 360 nm data were used to calculate m and values for better accuracy. As expected, the relative and C M values of γD and γS were consistent.
Table 2.
Equilibrium unfolding/refolding at 37°C and thermal unfolding parameters of γD wild-type and isolated domain proteins

Table 3.
Equilibrium unfolding/refolding at 37°C and thermal unfolding parameters of γS wild-type and isolated domain proteins

The equilibrium unfolding/refolding curves for γDWT were best fit to three-state models (Flaugh et al. 2005b). The C M of the first transition was 2.2 M GuHCl, and the C M of the second transition was 2.8 M GuHCl. The corresponding values were 7.7 and 8.9 kcal mol−1, respectively (Flaugh et al. 2005b). The equilibrium unfolding/refolding transitions of γDN and γDC were best fit to two-state models with no detectable equilibrium intermediates along the pathways. The γDN domain was destabilized in isolation with a C M of 1.2 M GuHCl, and of 3.7 kcal mol−1. The γDC domain had a C M of 2.7 M GuHCl and of 8.7 kcal mol−1 in isolation, comparable to the C M of 2.8 M observed for the second transition of γDWT. Therefore, this result confirms that the second transition almost certainly corresponds to the unfolding of the γDC domain, while the first transition corresponds to the unfolding of the γDN domain. The overall ΔG of γDWT can be estimated by the addition of the ΔG of both individual domains plus the ΔG of the γDWT interface, depicted in the following equation:
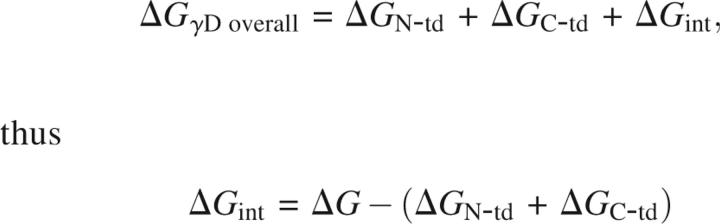 |
Using this equation, we estimated that the γDWT interface contributes ∼4.2 kcal mol−1 to the overall free energy of γDWT. In other words, in the absence of the γDC and its interface contacts, γDN was ∼4.2 kcal mol−1 less stable than the N-terminal domain in γDWT.
γSWT equilibrium unfolding/refolding analysis also demonstrates differential domain stability
Initially, we did not know if γSWT would refold as efficiently as γDWT under these conditions, pH 7 and 37°C. Previously, Wenk et al. (2000) analyzed the stability of the human and bovine γS-crystallin as well as their isolated N- and C-terminal domains at 20°C. Equilibrium unfolding/refolding experiments were performed to analyze the stability of γSWT and its individual domains. We were able to detect complete refolding of γSWT out of denaturant at pH 7 and 37°C as observed in the overlay of native and refolding equilibrium curves (Fig. 8). Therefore, we analyzed the stability of γSWT as well as its γSN and γSC under these previously described conditions. The results from these experiments also allowed us to compare γDWT and its isolated domains to γSWT and its isolated domains.
All of the unfolding and refolding transitions of full-length γS-crystallin and the γSWT isolated domains were best fit to a two-state model. The C M value observed for γSWT was 2.3 M GuHCl, and the was 10.5 kcal mol−1. The difference in stability between the γSWT isolated domains was significant but was not as great as the difference between stabilities of the γDWT isolated domains. The equilibrium unfolding experiments yielded a C M of 1.7 M GuHCl and a of 4.9 kcal mol−1 for γSN. The γSC C M and values were 2.3 M GuHCl and 8.2 kcal mol−1, respectively. Given the slight differences between γSN and γSC, we suggest that the C M of γSWT is likely a contribution from both domain stabilities. Thus, the unfolding/refolding of γSWT may populate intermediates that are not detectable here. Since there are differences between the stabilities of N- and C-terminal domains, there is a possibility that the one domain folded, one domain unfolded intermediate described for other γ-crystallins may also occur in the transitions of γSWT. Further experiments consisting of interface mutants such as those performed for γDWT will be essential in determining the stability of the interface as well as whether there is an equilibrium intermediate with one domain folded and one domain unfolded.
Comparison between the γDWT and γSWT isolated domains showed similar stability results as the thermal denaturation experiments. γSN was less stable than the γSC; however, the isolated γSN was more stable than the isolated γDN. In contrast, the isolated γDC was more stable than the isolated γSC. In general, from these experiments we detect consistent differential domain stability among both crystallins' N- and C-terminal domains. The sequence homology between both crystallins' N- and C-terminal domains is higher than the N- and C-terminal domains within the γD- or γS-crystallin.
Productive kinetic refolding
All full-length and single-domain proteins had the ability to refold after complete unfolding in GuHCl and subsequent dilution out of denaturant. Protein refolding kinetics were monitored using fluorescence spectroscopy to measure the quenching of the buried tryptophans as the proteins folded into their native states. To better resolve the intermediate steps, these experiments were performed at lower temperature (18°C). All proteins refolded in a time frame of ∼300 sec with the exception of γDWT and γSWT (Figs. 9 and 10). γDWT was productively refolded in 1 M GuHCl since it exhibited off-pathway aggregation when refolded in <1 M GuHCl (Kosinski-Collins and King 2003). The rest of the proteins were allowed to refold in 0.55 M GuHCl.
Figure 9.
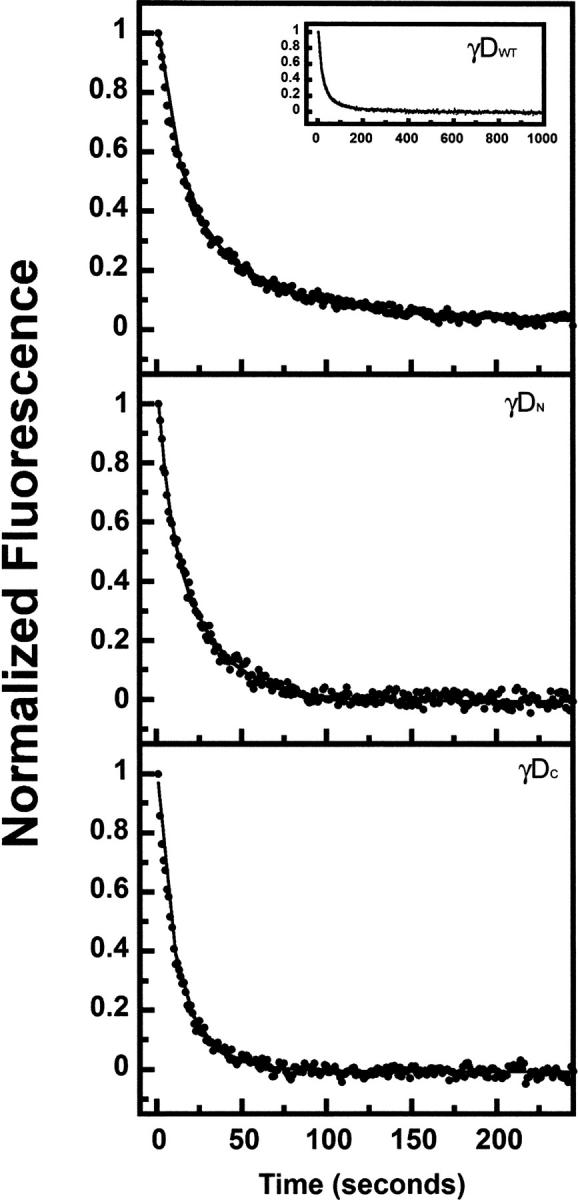
Kinetic refolding of γDWT (inset shows completion of γDWT refolding kinetics reaction), γDN, and γDC. The proteins were unfolded at high GuHCl concentration, then diluted into 100 mM sodium phosphate, 1 mM EDTA, 5 mM DTT (pH 7.0) buffer for a final protein concentration of 10 μg/mL. Protein tryptophan fluorescence emission at 350 nm was recorded every second, and data were normalized for comparison. All experiments were performed at 18°C. The final GuHCl concentrations were 1 M for γDWT and 0.55 M for γDN, and γDC (see text for details).
Figure 10.
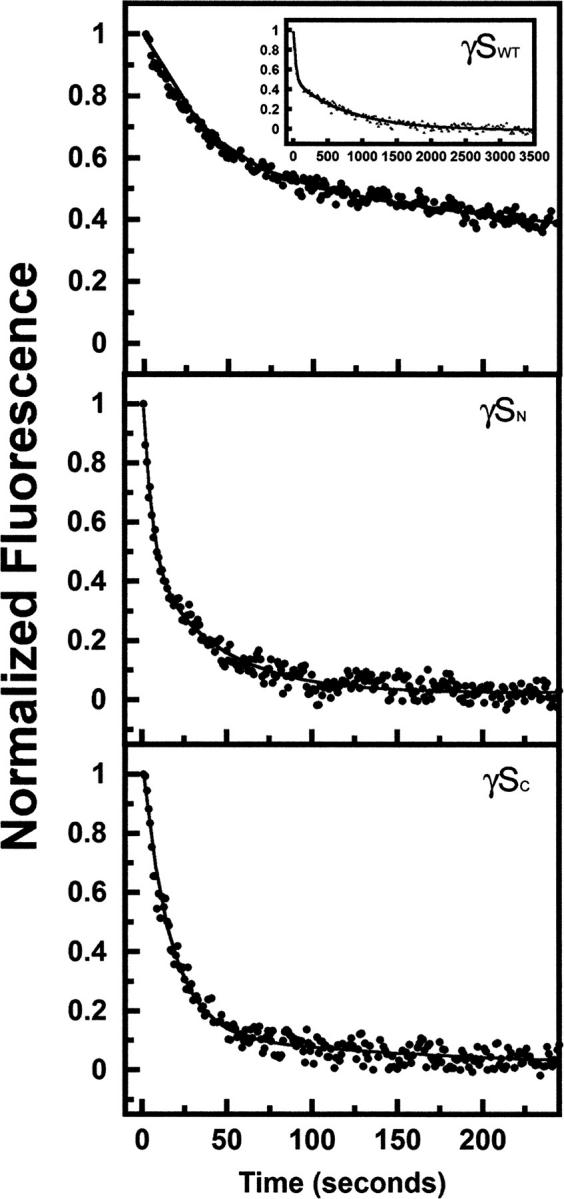
Kinetic refolding of isolated domains of γSWT (inset shows completion of γSWT refolding kinetics reaction), γSN, and γSC. Protein was unfolded at high GuHCl concentration, then diluted into 100 mM sodium phosphate, 1 mM EDTA, 5 mM DTT (pH 7.0) buffer for a final protein concentration of 10 μg/mL. Protein tryptophan fluorescence emission at 350 nm was recorded every second, and data were normalized for comparison. All experiments were performed at 18°C. The final GuHCl concentration was 0.55 M for γSWT, γSN, and γSC (see text for details).
The kinetic refolding transitions of γDWT were best fit to a four-state model in these experiments with an average calculated t 1/2 of 10 sec, 36 sec, and 252 sec for the three exponentials (k r1, k r2, k r3) (Fig. 9; Table 4) at 18°C (Equation 1).
Table 4.
Productive refolding kinetic parameters of γD and γS wild-type and isolated domain proteins at 18°C

Mutations in the interface affect the refolding rate of γDWT (Flaugh et al. 2005a,b, 2006). These mutations suggested that two refolding intermediates are populated; the first intermediate may be the C-terminal domain Greek key motif 4 (one closest to the interface) folded and motif 3 of the C-terminal domain as well as the N-terminal domain unfolded. The second intermediate is likely the C-terminal domain fully folded and the N-terminal domain unfolded.
All of the refolding reactions of the isolated domains were best fit by a three-state model indicating one observable intermediate for each refolding pathway. For the isolated domains, this kinetic refolding intermediate may be a partially folded individual Greek key domain.
The first refolding transitions for isolated γDN and γDC had t 1/2 values of 3 sec and 4.5 sec, and the second refolding transitions had t 1/2 values of 17 sec and 14 sec (Equation 2). The kinetic refolding parameters of both individual domains were within standard deviations of each other for both kinetic refolding transitions. The first kinetic refolding transition t 1/2 was 14.5 sec for γSN and ∼12 sec for γSC, and for the second kinetic refolding transition, the t 1/2 was 190 sec for γSN and 122 sec for γSC. These parameters were also within standard deviations of one another for both kinetic refolding transitions (Table 4).
γSWT refolding kinetics was best fit to a four-state model, indicating two intermediates (Fig. 10). The refolding rates were considerably slower than γDWT, with calculated average t 1/2s of 20 sec, 194 sec, and 1043 sec for the three transitions.
Discussion
All known eye lens β- and γ-crystallins have two homologous Greek key domains interacting through a tight interface. These domains presumably evolved from an ancestral single-domain protein. Given the importance of long-term solubility and stability for the crystallins, it may be likely that the evolution of a two-domain form was related to the need for very long-term stability of the lens crystallins. Previous evidence that the C terminus of γDWT served as a template for the folding of the N terminus raised the possibility that the N-terminal domain could not fold on its own, owing to the absence of the domain interface. On the contrary, the results reported here show that the N-terminal domain of γD does fold on its own in vivo within E. coli cytoplasm and remains folded through purification and storage. In addition, both equilibrium and kinetic experiments confirmed that these isolated domains were able to refold to a native-like state upon dilution out of denaturant. The isolated N- and C-terminal domains of γS-crystallin also folded in vivo and refolded in vitro, as reported previously (Wenk et al. 2000).
Nonetheless, the isolated N-terminal domain of γD was considerably less stable than the isolated C-terminal domain, in addition to being the less stable domain in the full-length protein. Comparing the stabilities of the isolated domains and the full-length protein indicated that the domain interface contributes a of ∼4.2 kcal mol−1 to the overall stability of the complete two-domain protein. Similarly, isolated domain and full-length protein analysis of bovine γB-crystallin at pH 2 determined that the interface contributed a of ∼3.8 kcal mol−1 (Mayr et al. 1997). The differential domain stability was also observed for the γS-crystallin isolated domains, although the stability differences between the two domains were not as significant as γD-crystallin isolated domains.
Native-like folded conformation of the isolated domains
The unusual fluorescence quenching of the buried tryptophans provides a very sensitive reporter of the folded state of the crystallins (Chen et al. 2006). Both fluorescence emission maxima and intensities of the isolated domains indicated that they were in the native-like fold. The two tryptophans in γSC are more fluorescent than the tryptophans in γSN. This is consistent with the fluorescence spectrum of the intact protein when using N-terminal Trp → Phe mutants to probe the C-terminal domain tryptophans in γS-crystallin (J. Chen, pers. comm.). Circular dichroism spectroscopy also confirmed the native-like structure of the isolated domains, indicating only minor structural differences between the domains themselves and their full-length counterparts. Analytical size exclusion chromatography demonstrated that all of the isolated domains were monomeric species under these conditions and did not form stable dimers or multimeric species. However, the possibility remains that these proteins may form multimers at higher protein concentrations.
Stability of the full-length γ-crystallins and their isolated domains
A comparison of both proteins demonstrated that γDWT is more stable than γSWT. This result was suggested empirically because γSWT could be unfolded in high molar urea, whereas γDWT could not (Wenk et al. 2000; Kosinski-Collins and King 2003). By comparing the γD and γS isolated domains, we observed that γDN was less stable in isolation than γSN, while γDC was more stable than the γSC (Tables 2 and 3).
The qualitative thermal denaturation data agreed with our chemical unfolding/refolding equilibrium studies, showing that the N-terminal domains (γD 64.5°C, γS 69.1°C) are less stable than the C-terminal domains (γD 76.2°C, γS 75.1°C) of both γDWT and γSWT. Also, γDWT (83.8°C) is overall more stable than γSWT (74.1°C). The γDWT T M was higher than both individual domains, emphasizing that the domain interface contributes to the overall stability of the full-length protein. The T M of γSC was similar to the T M of full-length γSWT, and the domain interface presumably makes a smaller contribution. Although the average of the T M measurements of γSC was higher than its full-length protein, it is within standard deviation of the average of the γSWT. Therefore, the intrinsic stability of the C-terminal domain contributes most of the overall stability of γSWT. Thermodynamic parameters were not calculated from these experimental results because of the irreversibility of the reaction due to protein aggregation at high temperatures.
Exposure to high heat among glass blowers and chain welders has been correlated with a high incidence of cataract (D'Onofrio and Mosci 1960; Vos and van Norren 1998; Vos and van Norren 2004). The thermal denaturation experiments measure the stability of the proteins in more physiological conditions than that of the equilibrium experiments, which use high concentrations of GuHCl to denature. During thermal denaturation, the unfolding protein molecules aggregate irreversibly, which may be relevant to the aforementioned heat-induced cataract. Thermally unfolded proteins could lead to partially unfolded species that are aggregation prone and subsequently initiate cataract formation.
Kinetic refolding of the isolated domains and their respective full-length proteins
Equilibrium unfolding/refolding experiments at 37°C demonstrated full reversibility, establishing that the isolated domain crystallins could efficiently refold in vitro in the absence of chaperones. The refolding kinetics of the isolated domain crystallins were best fit to three-state models, suggesting one detectable intermediate. The kinetic rates of γD individual domains were similar and within standard deviation of one another. The γD isolated domains exhibited higher kinetic rates compared to the γS isolated domains, an approximately threefold increase for k 1 and an ∼11-fold increase for k 2. The γS isolated domains also had similar kinetic rates to one another.
Since each crystallin domain contains two Greek key motifs, one may predict that the transitions of the isolated domains may be monitoring the sequential refolding of each Greek key motif. Previous studies with a βγ-crystallin fold protein have suggested that the interface Greek key motif and stabilizing tyrosine corner can act as a nucleation center for folding of the second outer Greek key motif (Fig. 1; Bagby et al. 1998).
γDWT kinetic refolding data were best fit to a four-state model, suggesting two detectable folding intermediates. Previous analysis of γDWT refolding kinetics suggested that the first transition monitors the refolding of the C-terminal innermost Greek key motif. The second transition is thought to monitor the refolding of the second, outermost C-terminal Greek key motif, while the third transition is complete refolding of the N terminus (Flaugh et al. 2006). This analysis was based on interdomain interface mutants, which mostly affected the third refolding transition, indicating that the interface was important for the folding of the N terminus.
The refolding of γSWT was best fit to a four-state model, suggesting two partially folded intermediates along the folding pathway. The overall refolding t 1/2 was considerably longer than γDWT, with a fourfold increase in the overall refolding half-life (Table 3). Since the folding pathway of γSWT has not been elucidated, it is not possible to compare its transitions to γDWT. As mentioned above, previous studies have suggested that the interface Greek key motif and the stabilizing tyrosine corner can act as a nucleation center for folding of the second Greek key motif. Thus, it is possible that the first and second transitions monitored the refolding of these two innermost Greek key motifs and interface interactions, while the third transition monitored the folding of the outermost Greek keys. The slower refolding step of γSWT may be due to slight arrangements in the core of the Greek keys, maximizing the quenching of the protein.
Biochemical basis of stability differences of the isolated domains
Although the chain folds in the γD and γS N- and C-terminal domains are highly homologous, the primary sequences are 39% identical and 52% similar for γD domains and 30% identical and 47% similar for the γS domains. However, upon structural and sequence comparison of the two domains, it is not obvious what causes the differences in stability. There is also variation in the stability between different β- and γ-crystallins, although structurally the double Greek key domains are homologous. In this study, we observed that γDWT and γSWT have different conformational stabilities when analyzed under the same conditions. Moreover, it was previously shown that in high concentrations of urea, γSWT completely unfolds, which is not the case for γDWT (Wenk et al. 2000; Kosinski-Collins and King 2003). A comparison of βB2- and bovine γB-crystallin indicates that βB2-crystallin can unfold in low urea concentrations, while γB-crystallin requires acidic pH (Mayr et al. 1997). There are no obvious reasons for these drastic stability differences based on comparing these two structures (Jaenicke and Seckler 1997). Furthermore, one would suspect that the oligomeric protein would be more stable than the monomeric protein (Jaenicke and Sterner 2003).
The γDWT interface consists of six hydrophobic residues, three in each domain (M43, F56, I81, V132, L145, V170), and two pairs of peripheral residues (Q54 and Q143, R79 and M147) at the top and bottom of the hydrophobic interface. The interdomain interface of γSWT is similar to γDWT, and the major difference is within the hydrophobic region, where γSWT has an Ile in place of Phe at position 56 (numbered based on γDWT). Among alignments of 35 various γ-crystallin sequences, Phe at position 56 is 80% conserved. Mutations of Phe → Ala have demonstrated destabilization in both bovine γB- and human γD-crystallin (Palme et al. 1997, 1998a; Flaugh et al. 2005b). The introduction of a smaller hydrophobic group in this position might contribute to less hydrophobic packing in the interface (Palme et al. 1998b). Additional mutations of Phe → Trp and Phe → Asp at position 56 in the bovine γB-crystallin led to destabilization of the protein, emphasizing the importance of the Phe at this position in the interface (Palme et al. 1997).
One possible contributor to the stability of β- and γ-crystallins could be the packing of hydrophobic residues within the two domains, particularly aromatic residues. Both γDWT and γSWT have high percentages of aromatic residues with four tryptophans, 14 tyrosines, six phenylalanines (γDWT), and nine phenylalanines (γSWT), consisting of ∼13%–14% of residues compared to ∼8% in other small globular proteins (McCaldon and Argos 1988). When comparing a species-diverse set of 35 γ-crystallin sequences, most of the aromatic residues are strongly conserved (>80% identity), with the exception of a few tyrosines and phenylalanines. Aromatic–aromatic interactions identified in high-resolution structures are predicted to stabilize a protein's native state (Burley and Petsko 1985). Long-range hydrophobic interactions have been computed by structure and sequence comparison analysis in the cellular retinoic acid binding protein and have been found to be important for its stability (Gunasekaran et al. 2004).
In addition to the stabilizing interdomain interface, the tyrosine corner of the γ-crystallin proteins has been proposed to be a contributor to stability. The tyrosine corner is present between strand c and strand d of the second Greek key motif (Bax et al. 1990; Hemmingsen et al. 1994). The tyrosine corner has a slightly different conformation in the isolated γS C-terminal domain when compared to other solved γ-crystallin structures such as γDWT (Purkiss et al. 2002). In the murine γS-crystallin NMR structure, γSN did not appear to have a tyrosine corner present (Wu et al. 2005). It is possible that the lack of the tyrosine corner in γSN has an effect on the stability of this domain.
Electrostatic interactions on the surface of the proteins may also be contributing to the overall intrinsic stability. Surface charge networks in the human and bovine γA-D crystallins have been observed through comparative and homology modeling (Salim and Zaidi 2003). These electrostatic interactions range from ionic pairs to interaction clusters, some of which are conserved and are located in particular regions of the Greek key motifs. For instance, one cluster is found on the loop regions in motifs 3 and 4 of γA-D crystallins' C-terminal domain. Many of these ionic interactions have been confirmed in the γDWT structure (Basak et al. 2003). We observe potential surface electrostatic interactions in the crystal structure of the γS C-terminal domain, albeit these interactions seem to be at a longer range (>4 Å) than in the γA-D crystallins.
Although surface electrostatic residues are thought to have a limited effect on stability because of water's dielectric constant, thermophilic proteins have ionic networks essential for stability (Jaenicke 1996; Perl and Schmid 2001; Wunderlich et al. 2005). We calculated the net charge of each γD- and γS-crystallin isolated domain by using the Web site http://zbio.net, which calculates net charge based on amino acid sequence. Although all proteins were close to their isoelectric points, there were slight differences in the net charge of the γDWT and the γSWT isolated domains at pH 7. The calculated net charge (Δz) was 2.2, 1.9, and 1.6 for γDWT, γDN, and γDC, respectively. The calculated net charges (Δz) for γSWT, γSN, and γSC were 0.9, 1.7, and 0.7, respectively.
As a protein increases in net charge, the stability of the protein becomes more sensitive to ionic changes in the environment (Negin and Carbeck 2002). Since there were slight differences in net charge as well as possibilities of nonmeasurable local charge interactions, we performed thermal denaturation experiments in a low ionic strength buffer to better assess intrinsic stability. In the thermal denaturation experiments, differential domain stability observed in the equilibrium studies was maintained as the C-terminal domains were more stable than the N-terminal domains. These results suggest that electrostatic interactions may have only minor effects in the intrinsic conformational stability of the isolated domains of γD- and γS-crystallins. This is contrary to bovine γB-crystallin studies, which demonstrated that at neutral pH the individual domains were similar in stability (Mayr et al. 1997). In these studies of the bovine γB N and C termini, the C-terminal domain was greatly destabilized at acidic pH and not at neutral pH. This suggested that the stabilizing interface contacts may not be absent at neutral pH but difficult to assess using equilibrium unfolding/refolding methods monitored spectroscopically (Mayr et al. 1997).
Comparisons with other crystallins
The expression of β- and γ-crystallins is specific to lens fiber cells, but their expression varies during development and in the different lens regions. In particular, γA-, γB-, γC-, and γD-crystallins (γE and F are pseudogenes in humans) and βB1-crystallins are expressed in utero and are localized to the lens nucleus (Aarts et al. 1989; Chambers and Russell 1991; Lampi et al. 2002a). In contrast, βB2- and γS-crystallin are post-natally expressed in the secondary fiber cells and are expressed throughout life (Peek et al. 1992; Wistow et al. 2000; Ueda et al. 2002). These physiological differences in the crystallins may explain the stability differences observed biophysically. For example, the observation that γSWT is less stable than γDWT may be related to the location in the lens: γSWT localized to a region that exhibits some protein synthesis due to the continual lens growth throughout life. Thus in the lens, γSWT is a “younger” protein than γDWT.
In addition to the aforementioned studies of γB-crystallin, isolated domain studies of the rat and human domain-swapped dimeric βB2-crystallin found that the N-terminal domain was marginally less stable than the C-terminal domain in isolation (Wieligmann et al. 1999; Fu and Liang 2002). The N-terminal domain in the monomeric form had a higher propensity to unfold. These results suggested that the unfolding of the N-terminal domain promotes dissociation of the N-terminal domain from the C terminus of its partner molecule, forming a monomeric intermediate in which the N terminus is unstructured and the C-terminal domain is folded. Thus, the second equilibrium transition monitors the unfolding of the C-terminal domain (Wieligmann et al. 1999; Fu and Liang 2002).
βB1-crystallin forms dimers and oligomers not by domain swapping but by intramolecular association (Bateman et al. 2001; Lampi et al. 2001; Van Montfort et al. 2003). Studies using spin-labeling of the βB1 N terminus demonstrated that the N-terminal domain of βB1 unfolded first. Therefore, the N-terminal domain was hypothesized to be less stable than the C-terminal domain (Kim et al. 2002). In addition, an extensive truncated protein analysis of βA3 crystallin has been performed, suggesting the possibility that the βA3 N terminus is more stable than the C terminus (Gupta et al. 2006). All of these studies demonstrated the importance of the domain interface in the stability of the β-crystallins.
The high thermal stability observed in our thermal denaturation experiments is also consistent with other β- and γ-crystallins analyzed previously. For example, calorimetry experiments on bovine γB, γF, γE, and γD established T Ms of 71.5°C, 70°C, 73°C, and 74°C, respectively (Sen et al. 1992). β-Crystallins have lower thermal stabilities; for instance, βB2 has a T M of 67°C. Thermal denaturation of βB1-crystallin also demonstrates lower thermal stability, with a loss of secondary structure and precipitation of the protein at temperatures >70°C (Lampi et al. 2002b). Also, α-crystallins have lower thermal stabilities with a T Ms of ∼61°C (Surewicz and Olesen 1995; Das et al. 1997; Raman and Rao 1997). The T M values determined for the crystallins are within the range of structural proteins of thermophilic organisms (Jaenicke 1996).
Gene duplication in the crystallins
The βγ-crystallins are thought to originate from a single-domain ancestor protein, suggesting a gene duplication and fusion event in the crystallin lineage (Lubsen et al. 1988; Piatigorsky 2003; Shimeld et al. 2005). It is interesting to note that most βγ-crystallins have one domain that is more stable than the other, indicating that one domain may have evolved increasing stability or the other domain accrued destabilizing properties. The significance of this observation is unclear for γSWT, since the conformational stability of γSC domain is similar to the stability of the full-length protein in both thermal denaturation and equilibrium experiments. However, in γDWT, the addition the γDN domain and the interdomain interface to the γDC contribute to the overall stability of the full-length protein. This additional stability of both domains is demonstrated in both equilibrium and thermal denaturation experiments of γDWT. The gene duplication event in the modern crystallins could be attributed to the necessity for crystallins to have higher stabilities in longer-lived organisms.
Materials and Methods
Preparation of constructs for isolated domains and γSWT
A previously subcloned vector of γDWT as described in Kosinski-Collins and King (2003) was used to prepare γDN. This vector includes an N-terminal His-tag for purification with Ni-NTA affinity chromatography (Qiagen). PCR primers (Integrated DNA Technologies) were designed to introduce a stop codon (QuikChange Site-Directed Mutagenesis; Stratagene) at position 83 to create the γDN consisting of residues G1–P82. The rest of the single domains and γSWT were prepared using PCR primers (Integrated DNA Technologies) designed at appropriate positions in the sequence. The resulting PCR products were subcloned into the pQE-1 vector using the blunt-ended PvuII (adaptable for other blunt-ended restriction enzymes) and the HindIII enzyme restriction sites. All vectors included an N-terminal His-tag, MKHHHHHHQA, to aid in purification.
γDC consisted of residues R89–S174 (based on numbering in PDB: 1HK0). The γSN consisted of residues S1–H86, and γSC consisted of residues Y93–E177. The γSWT template was a generous gift from S. Helber at Commonwealth Biotechnologies, Inc. All vectors were sequenced to confirm the correct subcloning product and to ensure that there were no erroneous mutations, additions, or deletions in the sequences (MGH DNA Core).
Expression and purification of proteins
Recombinant full-length and variant proteins were prepared as described by Kosinski-Collins et al. (2004). Briefly, all aforementioned vectors were transformed into E. coli M15[pRep4] cells (Qiagen), used for tightly regulated protein expression. The cells were lysed by conventional methods and purified by Ni-NTA resin (Qiagen) affinity chromatography using a Pharmacia FPLC apparatus. The purity and size of each protein were confirmed by SDS-PAGE gel electrophoresis. The identities of γDWT, γDN, and γDC were additionally confirmed by mass spectrometry (CCR Biopolymers Laboratory, MIT, Cambridge, MA). This purification protocol produced proteins with a purity of >90%.
Protein concentrations were determined by unfolding the proteins in 5.5 M GuHCl, measuring absorbance at 280 nm, and calculating concentrations using their respective protein extinction coefficients (γDWT, 41,040 cm−1 M−1; γDN, 20,580 cm−1 M−1; γDC, 21,555 cm−1 M−1; γSWT, 41,040 cm−1 M−1; γSN, 21,860 cm−1 M−1; and γSC, 19,180 cm−1 M−1).
Analytical size exclusion chromatography
All samples were prepared by diluting to a final protein concentration of 80 μg/mL in 10 mM ammonium acetate buffer (pH 7.0). The samples were loaded onto the Superdex 200 10/300 GL (Pharmacia Biotech) column using a FPLC apparatus (Pharmacia). The column was equilibrated with 100 mM sodium phosphate, 1 mM EDTA, 5 mM DTT (pH 7.0) buffer. Molecular weight standards were used to calibrate relative elution times for various protein sizes. Overlays of each spectrum were made using the Unicorn program (Pharmacia) and subsequent analysis in Excel (Microsoft). Peak fractions were collected, and SDS-PAGE analysis confirmed the presence of each protein.
Circular dichroism
All experiments were performed on an Aviv Model 202 CD spectrometer with an internal Peltier thermoelectric controller used to maintain a constant 37°C temperature. Protein samples were prepared at a 100 μg/mL protein concentration in a degassed 10 mM sodium phosphate buffer (no EDTA or DTT to prevent absorption at lower wavelengths) and equilibrated overnight at 37°C. Each sample was placed in a 1-mm quartz cuvette (Starna, Inc.) and allowed to equilibrate in the CD spectrometer for 1 min, and data were recorded in the 260–195 nm wavelength range, with each wavelength averaged over a 10 sec period. The CD spectrum of the buffer was subtracted from all spectra, and the molar ellipticity was subsequently calculated. Deconvolution of the CD spectrum was performed using the CDPro suite software package consisting of the CONTILL, CDSSTR, and SELCON3 programs (http://lamar.colostate.edu/∼sreeram/CDPro/) (Sreerama and Woody 2000; Sreerama et al. 2000). The IBasis 1 parameter (Johnson 1999) was chosen since it analyzed a larger range for the recorded data, had a large reference set (29 proteins), and had a lower RMSD and NRMSD for most of the data. The results from each program were averaged to obtain the overall secondary structure percentages.
Thermal denaturation
All experiments were performed on an Aviv Model 202 CD spectrometer with an internal Peltier thermoelectric controller. A quartz four-sided screwtop cuvette with a bandwidth of 4 mm was used to prevent loss of sample due to evaporation as the temperature increased. Samples were prepared at a concentration of 100 μg/mL in a degassed 10 mM phosphate buffer (pH 7.0). Samples were equilibrated for 1 min for each 1°C increase in temperature, and all data points were averaged over a 3 sec period. Specifically, decrease in the β-sheet secondary structure minimum at 218 nm versus increase in temperature was monitored. The buffer spectrum was subtracted for each data point. Owing to aggregation of the proteins at high temperatures, the fraction native of each protein was calculated by the following equation:
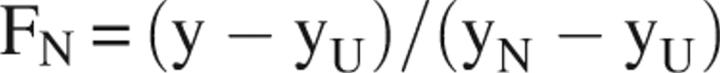 |
where FN is the fraction native, y is the ellipticity at 218 nm, y U is the unfolded/aggregation baseline, and y N is the native baseline. All experiments were repeated three times, calculating averages and standard deviations.
Fluorescence spectroscopy
Fluorescence emission spectra were taken using a Hitachi F-4500 fluorimeter equipped with a temperature control circulating water bath to maintain 37°C. The fluorimeter parameters were a bandpass of 10 nm for excitation and emission monochromators, scan speed 60 nm/min, and a 2 sec response time. All proteins were analyzed at a concentration of 10 μg/mL in 100 mM phosphate, 1 mM EDTA, and 5 mM DTT buffer (pH 7.0) for the native sample (the crystallins do not required added salt for stability) and 5.5 M GuHCl for the unfolded protein samples. The protein samples were excited at 295 nm, and the emission fluorescence was recorded over a wavelength range of 310–400 nm.
Equilibrium unfolding and refolding
Equilibrium unfolding samples were diluted to a protein concentration of 10 μg/mL with increasing concentrations of GuHCl (0–5.5 M) in 100 mM sodium phosphate, 1 mM EDTA, and 5 mM DTT (pH 7.0) buffer (guanidine hydrochloride solution, 8 M GuHCl) (Sigma). In equilibrium unfolding experiments, all samples were equilibrated for 24 h in order to reach equilibrium. For equilibrium refolding experiments, a 10× protein solution was unfolded at 5.5 M GuHCl for 5 h. The unfolded protein was then diluted 10-fold into various concentrations of GuHCl (0–5.5 M), giving a lowest GuHCl concentration of 0.55 M GuHCl. These samples were also allowed to equilibrate for 24 h. Exact GuHCl concentrations of each sample were determined by refractometer readings. Emission spectra were recorded over a wavelength range of 310–400 nm. All spectra were corrected for buffer. Equilibrium unfolding/refolding curves were fit to a two-state model according to the methods of Greene and Pace (1974), or a three-state model according to the methods of Clark et al. (1993). Calculations of thermodynamic parameters were performed from both 360 nm emission data and 360/320 nm emission ratio data using Kalediagraph software version 4.0 (Synergy Software). Both analyses were comparable and within standard error of one another. 360/320 nm is shown for visual clarity of equilibrium transitions. Single-wavelength 360 nm data were used to calculate m and ΔG values. Each experiment was repeated three times to determine averages and standard deviations.
Productive refolding kinetics
Productive refolding kinetics experiments were performed by first unfolding 10× protein in 5.5 M GuHCl for 5 h to guarantee complete unfolding of the protein. The 10× unfolded protein sample was diluted 10-fold into agitated 100 mM phosphate, 1 mM EDTA, and 5 mM DTT (pH 7.0) buffer using an injection port system with a dead time of ∼1 sec. For γDWT, the 10× protein solution was diluted to a final GuHCl concentration of 1 M. Temperature was maintained at 18°C using a circulating water bath. The samples were excited at 295 nm with 10 nm bandpass. Emission at 350 nm with 10 nm bandpass was recorded over time. Unfolded and native control spectra were recorded at the beginning and at the end of the experiment. The refolding kinetic data were analyzed using different exponential model equations to fit by Fersht (1999) and residual distribution to determine the best fit. The data were analyzed by fitting it to equations depicting two-, three-, or four-state models by Kaleidagraph 4.0 software. All experiments were performed three times to calculate average kinetic rates and standard deviations. All refolding data are depicted as normalized fluorescence data for comparison.
Acknowledgments
We thank Yongting Wang, Jiejin Chen, Ligia Acosta, Kate Drahos, Xionan Lin, and Robin Nance for helpful discussions and/or technical assistance. The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-0070319 and NIH GM68762) is gratefully acknowledged. This work was supported by an NIH grant GM17980 awarded to J.A.K. I.A.M. was supported by a UNCF·Merck Dissertation Graduate Fellowship and S.L.F. by a Cleo and Paul Schimmel Graduate Fellowship.
Footnotes
Reprint requests to: Jonathan A. King, Department of Biology, Massachusetts Institute of Technology, Building 68, Room 330, 31 Ames Street, Cambridge, MA 02139, USA; e-mail: jaking@mit.edu; fax: (617) 252-1843.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072970207.
References
- Aarts H.J., Lubsen, N.H., and Schoenmakers, J.G. 1989. Crystallin gene expression during rat lens development. Eur. J. Biochem. 183: 31–36. [DOI] [PubMed] [Google Scholar]
- Bagby S., Go, S., Inouye, S., Ikura, M., and Chakrabartty, A. 1998. Equilibrium folding intermediates of a Greek key β-barrel protein. J. Mol. Biol. 276: 669–681. [DOI] [PubMed] [Google Scholar]
- Basak A., Bateman, O., Slingsby, C., Pande, A., Asherie, N., Ogun, O., Benedek, G.B., and Pande, J. 2003. High-resolution X-ray crystal structures of human γD crystallin (1.25 Å) and the R58H mutant (1.15 Å) associated with aculeiform cataract. J. Mol. Biol. 328: 1137–1147. [DOI] [PubMed] [Google Scholar]
- Bateman O.A., Lubsen, N.H., and Slingsby, C. 2001. Association behaviour of human βB1-crystallin and its truncated forms. Exp. Eye Res. 73: 321–331. [DOI] [PubMed] [Google Scholar]
- Bateman O.A., Sarra, R., van Genesen, S.T., Kappe, G., Lubsen, N.H., and Slingsby, C. 2003. The stability of human acidic β-crystallin oligomers and hetero-oligomers. Exp. Eye Res. 77: 409–422. [DOI] [PubMed] [Google Scholar]
- Bax B., Lapatto, R., Nalini, V., Driessen, H., Lindley, P.F., Mahadevan, D., Blundell, T.L., and Slingsby, C. 1990. X-ray analysis of β B2-crystallin and evolution of oligomeric lens proteins. Nature 347: 776–780. [DOI] [PubMed] [Google Scholar]
- Bloemendal H., de Jong, W., Jaenicke, R., Lubsen, N.H., Slingsby, C., and Tardieu, A. 2004. Ageing and vision: Structure, stability and function of lens crystallins. Prog. Biophys. Mol. Biol. 86: 407–485. [DOI] [PubMed] [Google Scholar]
- Blundell T., Lindley, P., Miller, L., Moss, D., Slingsby, C., Tickle, I., Turnell, B., and Wistow, G. 1981. The molecular structure and stability of the eye lens: X-ray analysis of γ-crystallin II. Nature 289: 771–777. [DOI] [PubMed] [Google Scholar]
- Bron A.J., Vrensen, G.F., Koretz, J., Maraini, G., and Harding, J.J. 2000. The ageing lens. Ophthalmologica 214: 86–104. [DOI] [PubMed] [Google Scholar]
- Burley S.K. and Petsko, G.A. 1985. Aromatic–aromatic interaction: A mechanism of protein structure stabilization. Science 229: 23–28. [DOI] [PubMed] [Google Scholar]
- Chambers C. and Russell, P. 1991. Deletion mutation in an eye lens β-crystallin. An animal model for inherited cataracts. J. Biol. Chem. 266: 6742–6746. [PubMed] [Google Scholar]
- Chen J., Flaugh, S.L., Callis, P.R., and King, J. 2006. Mechanism of the highly efficient quenching of tryptophan fluorescence in human γD-crystallin. Biochemistry 45: 11552–11563. [DOI] [PubMed] [Google Scholar]
- Clark A.C., Sinclair, J.F., and Baldwin, T.O. 1993. Folding of bacterial luciferase involves a non-native heterodimeric intermediate in equilibrium with the native enzyme and the unfolded subunits. J. Biol. Chem. 268: 10773–10779. [PubMed] [Google Scholar]
- Cook C.A., Koretz, J.F., Pfahnl, A., Hyun, J., and Kaufman, P.L. 1994. Aging of the human crystalline lens and anterior segment. Vision Res. 34: 2945–2954. [DOI] [PubMed] [Google Scholar]
- Das B.K., Liang, J.J., and Chakrabarti, B. 1997. Heat-induced conformational change and increased chaperone activity of lens α-crystallin. Curr. Eye Res. 16: 303–309. [DOI] [PubMed] [Google Scholar]
- Delaye M. and Tardieu, A. 1983. Short-range order of crystallin proteins accounts for eye lens transparency. Nature 302: 415–417. [DOI] [PubMed] [Google Scholar]
- D'Onofrio V. and Mosci, L. 1960. Cataract caused by heat: Observations in metallurgic workers and glass blowers. Rass. Med. Ind. Ig. Lav. 29: 253–267. [PubMed] [Google Scholar]
- Evans P., Wyatt, K., Wistow, G.J., Bateman, O.A., Wallace, B.A., and Slingsby, C. 2004. The P23T cataract mutation causes loss of solubility of folded γD-crystallin. J. Mol. Biol. 343: 435–444. [DOI] [PubMed] [Google Scholar]
- Fagerholm P., Philipson, B., and Carlstrom, D. 1981. Calcification in the human lens. Curr. Eye Res. 1: 629–633. [DOI] [PubMed] [Google Scholar]
- Fernald R.D. and Wright, S.E. 1983. Maintenance of optical quality during crystalline lens growth. Nature 301: 618–620. [DOI] [PubMed] [Google Scholar]
- Fersht A. 1999. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. W.H. Freeman and Company, New York.
- Flaugh S.L., Kosinski-Collins, M.S., and King, J. 2005a. Contributions of hydrophobic domain interface interactions to the folding and stability of human γD-crystallin. Protein Sci. 14: 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaugh S.L., Kosinski-Collins, M.S., and King, J. 2005b. Interdomain side-chain interactions in human γD crystallin influencing folding and stability. Protein Sci. 14: 2030–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaugh S.L., Mills, I.A., and King, J. 2006. Glutamine deamidation destabilizes human γD-crystallin and lowers the kinetic barrier to unfolding. J. Biol. Chem. 281: 30782–30793. [DOI] [PubMed] [Google Scholar]
- Fu L. and Liang, J.J. 2002. Unfolding of human lens recombinant βB2- and γC-crystallins. J. Struct. Biol. 139: 191–198. [DOI] [PubMed] [Google Scholar]
- Greene R.F. and Pace, C.N. 1974. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, α-chymotrypsin, and β-lactoglobulin. J. Biol. Chem. 249: 5388–5393. [PubMed] [Google Scholar]
- Gunasekaran K., Hagler, A.T., and Gierasch, L.M. 2004. Sequence and structural analysis of cellular retinoic acid-binding proteins reveals a network of conserved hydrophobic interactions. Proteins 54: 179–194. [DOI] [PubMed] [Google Scholar]
- Gupta R., Srivastava, K., and Srivastava, O.P. 2006. Truncation of motifs III and IV in human lens βA3-crystallin destabilizes the structure. Biochemistry 45: 9964–9978. [DOI] [PubMed] [Google Scholar]
- Hanson S.R., Hasan, A., Smith, D.L., and Smith, J.B. 2000. The major in vivo modifications of the human water-insoluble lens crystallins are disulfide bonds, deamidation, methionine oxidation and backbone cleavage. Exp. Eye Res. 71: 195–207. [DOI] [PubMed] [Google Scholar]
- Harding J.J. and Crabbe, M.J.C. 1984. The lens: Development, proteins, metabolism and cataract. In The eye (ed. H. Davson), pp. 207–492. Academic Press, Orlando, FL.
- Hejtmancik J.F., Wingfield, P.T., and Sergeev, Y.V. 2004. β-Crystallin association. Exp. Eye Res. 79: 377–383. [DOI] [PubMed] [Google Scholar]
- Hemmingsen J.M., Gernert, K.M., Richardson, J.S., and Richardson, D.C. 1994. The tyrosine corner: A feature of most Greek key β-barrel proteins. Protein Sci. 3: 1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heon E., Priston, M., Schorderet, D.F., Billingsley, G.D., Girard, P.O., Lubsen, N., and Munier, F.L. 1999. The γ-crystallins and human cataracts: A puzzle made clearer. Am. J. Hum. Genet. 65: 1261–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenicke R. 1996. Stability and folding of ultrastable proteins: Eye lens crystallins and enzymes from thermophiles. FASEB J. 10: 84–92. [DOI] [PubMed] [Google Scholar]
- Jaenicke R. and Seckler, R. 1997. Protein misassembly in vitro. Adv. Protein Chem. 50: 1–59. [DOI] [PubMed] [Google Scholar]
- Jaenicke R. and Sterner, R. 2003. Protein design at the crossroads of biotechnology, chemistry, theory, and evolution. Angew. Chem. Int. Ed. Engl. 42: 140–142. [DOI] [PubMed] [Google Scholar]
- Johnson W.C. 1999. Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 35: 307–312. [PubMed] [Google Scholar]
- Kim Y.H., Kapfer, D.M., Boekhorst, J., Lubsen, N.H., Bachinger, H.P., Shearer, T.R., David, L.L., Feix, J.B., and Lampi, K.J. 2002. Deamidation, but not truncation, decreases the urea stability of a lens structural protein, βB1-crystallin. Biochemistry 41: 14076–14084. [DOI] [PubMed] [Google Scholar]
- Kosinski-Collins M.S. and King, J. 2003. In vitro unfolding, refolding, and polymerization of human γD crystallin, a protein involved in cataract formation. Protein Sci. 12: 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski-Collins M.S., Flaugh, S.L., and King, J. 2004. Probing folding and fluorescence quenching in human γD-crystallin Greek key domains using triple tryptophan mutant proteins. Protein Sci. 13: 2223–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmar M., Mayr, E.M., and Jaenicke, R. 1999. Homo-dimeric spherulin 3a: A single-domain member of the β γ-crystallin superfamily. Biol. Chem. 380: 89–94. [DOI] [PubMed] [Google Scholar]
- Lampi K.J., Ma, Z., Hanson, S.R., Azuma, M., Shih, M., Shearer, T.R., Smith, D.L., Smith, J.B., and David, L.L. 1998. Age-related changes in human lens crystallins identified by two-dimensional electrophoresis and mass spectrometry. Exp. Eye Res. 67: 31–43. [DOI] [PubMed] [Google Scholar]
- Lampi K.J., Oxford, J.T., Bächinger, H.P., Shearer, T.R., David, L.L., and Kapfer, D.M. 2001. Deamidation of human β B1 alters the elongated structure of the dimer. Exp. Eye Res. 72: 279–288. [DOI] [PubMed] [Google Scholar]
- Lampi K.J., Shih, M., Ueda, Y., Shearer, T.R., and David, L.L. 2002a. Lens proteomics: Analysis of rat crystallin sequences and two-dimensional electrophoresis map. Invest. Ophthalmol. Vis. Sci. 43: 216–224. [PubMed] [Google Scholar]
- Lampi K.J., Kim, Y.H., Bächinger, H.P., Boswell, W.A., Lindner, R.A., Carver, J.A., Shearer, T.R., David, L.L., and Kapfer, D.M. 2002b. Decreased heat stability and increased chaperone requirement of modified human βB1-crystallins. Mol. Vis. 8: 359–366. [PubMed] [Google Scholar]
- Lampi K.J., Amyx, K.K., Ahmann, P., and Steel, E.A. 2006. Deamidation in human lens βB2-crystallin destabilizes the dimer. Biochemistry 45: 3146–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapko V.N., Cerny, R.L., Smith, D.L., and Smith, J.B. 2005. Modifications of human βA1/βA3-crystallins include S-methylation, glutathiolation, and truncation. Protein Sci. 14: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.F. and Liang, J.J. 2006. Domain interaction sites of human lens βB2-crystallin. J. Biol. Chem. 281: 2624–2630. [DOI] [PubMed] [Google Scholar]
- Lubsen N.H., Aarts, H.J., and Schoenmakers, J.G. 1988. The evolution of lenticular proteins: The β- and γ-crystallin super gene family. Prog. Biophys. Mol. Biol. 51: 47–76. [DOI] [PubMed] [Google Scholar]
- MacDonald J.T., Purkiss, A.G., Smith, M.A., Evans, P., Goodfellow, J.M., and Slingsby, C. 2005. Unfolding crystallins: The destabilizing role of a β-hairpin cysteine in βB2-crystallin by simulation and experiment. Protein Sci. 14: 1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr E.M., Jaenicke, R., and Glockshuber, R. 1997. The domains in γB-crystallin: Identical fold-different stabilities. J. Mol. Biol. 269: 260–269. [DOI] [PubMed] [Google Scholar]
- McCaldon P. and Argos, P. 1988. Oligopeptide biases in protein sequences and their use in predicting protein coding regions in nucleotide sequences. Proteins 4: 99–122. [DOI] [PubMed] [Google Scholar]
- Negin R.S. and Carbeck, J.D. 2002. Measurement of electrostatic interactions in protein folding with the use of protein charge ladders. J. Am. Chem. Soc. 124: 2911–2916. [DOI] [PubMed] [Google Scholar]
- Norledge B.V., Trinkl, S., Jaenicke, R., and Slingsby, C. 1997. The X-ray structure of a mutant eye lens β B2-crystallin with truncated sequence extensions. Protein Sci. 6: 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyster C. 1999. The lens and the vitreous. In The human eye: Structure and function, pp. 492–510. Sinauer Associates, Inc, Sunderland, MA.
- Palme S., Slingsby, C., and Jaenicke, R. 1997. Mutational analysis of hydrophobic domain interactions in γ B-crystallin from bovine eye lens. Protein Sci. 6: 1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palme S., Jaenicke, R., and Slingsby, C. 1998a. Unusual domain pairing in a mutant of bovine lens γB-crystallin. J. Mol. Biol. 279: 1053–1059. [DOI] [PubMed] [Google Scholar]
- Palme S., Jaenicke, R., and Slingsby, C. 1998b. X-ray structures of three interface mutants of γB-crystallin from bovine eye lens. Protein Sci. 7: 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande A., Pande, J., Asherie, N., Lomakin, A., Ogun, O., King, J.A., Lubsen, N.H., Walton, D., and Benedek, G.B. 2000. Molecular basis of a progressive juvenile-onset hereditary cataract. Proc. Natl. Acad. Sci. 97: 1993–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande A., Annunziata, O., Asherie, N., Ogun, O., Benedek, G.B., and Pande, J. 2005. Decrease in protein solubility and cataract formation caused by the Pro23 to thr mutation in human γ D-crystallin. Biochemistry 44: 2491–2500. [DOI] [PubMed] [Google Scholar]
- Peek R., McAvoy, J.W., Lubsen, N.H., and Schoenmakers, J.G. 1992. Rise and fall of crystallin gene messenger levels during fibroblast growth factor induced terminal differentiation of lens cells. Dev. Biol. 152: 152–160. [DOI] [PubMed] [Google Scholar]
- Perl D. and Schmid, F.X. 2001. Electrostatic stabilization of a thermophilic cold shock protein. J. Mol. Biol. 313: 343–357. [DOI] [PubMed] [Google Scholar]
- Piatigorsky J. 2003. Crystallin genes: Specialization by changes in gene regulation may precede gene duplication. J. Struct. Funct. Genomics 3: 131–137. [PubMed] [Google Scholar]
- Purkiss A.G., Bateman, O.A., Goodfellow, J.M., Lubsen, N.H., and Slingsby, C. 2002. The X-ray crystal structure of human γ S-crystallin C-terminal domain. J. Biol. Chem. 277: 4199–4205. [DOI] [PubMed] [Google Scholar]
- Raman B. and Rao, C.M. 1997. Chaperone-like activity and temperature-induced structural changes of α-crystallin. J. Biol. Chem. 272: 23559–23564. [DOI] [PubMed] [Google Scholar]
- Rudolph R., Siebendritt, R., Neßlauer, G., Sharma, A.K., and Jaenicke, R. 1990. Folding of an all-β protein: Independent of domain folding in γII-crystallin from calf lens. Proc. Natl. Acad. Sci. 87: 4625–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim A. and Zaidi, Z.H. 2003. Homology models of human γ-crystallins: Structural study of the extensive charge network in γ-crystallins. Biochem. Biophys. Res. Commun. 300: 624–630. [DOI] [PubMed] [Google Scholar]
- Santhiya S.T., Shyam Manohar, M., Rawlley, D., Vijayalakshmi, P., Namperumalsamy, P., Gopinath, P.M., Loster, J., and Graw, J. 2002. Novel mutations in the γ-crystallin genes cause autosomal dominant congenital cataracts. J. Med. Genet. 39: 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle B.C., Dasari, S., Wilmarth, P.A., Turner, M., Reddy, A.P., David, L.L., and Nagalla, S.R. 2005. Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J. Proteome Res. 4: 546–554. [DOI] [PubMed] [Google Scholar]
- Sen A.C., Walsh, M.T., and Chakrabarti, B. 1992. An insight into domain structures and thermal stability of γ-crystallins. J. Biol. Chem. 267: 11898–11907. [PubMed] [Google Scholar]
- Shimeld S.M., Purkiss, A.G., Dirks, R.P., Bateman, O.A., Slingsby, C., and Lubsen, N.H. 2005. Urochordate βγ-crystallin and the evolutionary origin of the vertebrate eye lens. Curr. Biol. 15: 1684–1689. [DOI] [PubMed] [Google Scholar]
- Siezen R.J., Wu, E., Kaplan, E.D., Thomson, J.A., and Benedek, G.B. 1988. Rat lens γ-crystallins. Characterization of the six gene products and their spatial and temporal distribution resulting from differential synthesis. J. Mol. Biol. 199: 475–490. [DOI] [PubMed] [Google Scholar]
- Slingsby C. and Clout, N.J. 1999. Structure of the crystallins. Eye 13: 395–402. [DOI] [PubMed] [Google Scholar]
- Smith R.S., Hawes, N.L., Chang, B., Roderick, T.H., Akeson, E.C., Heckenlively, J.R., Gong, X., Wang, X., and Davisson, M.T. 2000. Lop12, a mutation in mouse Crygd causing lens opacity similar to human Coppock cataract. Genomics 63: 314–320. [DOI] [PubMed] [Google Scholar]
- Smith M.A., Bateman, O.A., Jaenicke, R., and Slingsby, C. 2007. Mutations of interfaces in domain-swapped human βB2-crystallin. Protein Sci. 16: 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N. and Woody, R.W. 2000. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287: 252–260. [DOI] [PubMed] [Google Scholar]
- Sreerama N. and Woody, R.W. 2003. Structural composition of βI- and βII-proteins. Protein Sci. 12: 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N., Venyaminov, S.Y., and Woody, R.W. 2000. Estimation of protein secondary structure from circular dichroism spectra: Inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem. 287: 243–251. [DOI] [PubMed] [Google Scholar]
- Stephan D.A., Gillanders, E., Vanderveen, D., Freas-Lutz, D., Wistow, G., Baxevanis, A.D., Robbins, C.M., VanAuken, A., Quesenberry, M.I., Bailey-Wilson, J., et al. 1999. Progressive juvenile-onset punctate cataracts caused by mutation of the γD-crystallin gene. Proc. Natl. Acad. Sci. 96: 1008–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surewicz W.K. and Olesen, P.R. 1995. On the thermal stability of α-crystallin: A new insight from infrared spectroscopy. Biochemistry 34: 9655–9660. [DOI] [PubMed] [Google Scholar]
- Takata T., Oxford, J.T., Brandon, T.R., and Lampi, K. 2007. Deamidation alters the structure and decreases the stability of human lens βA3-crystallin. Biochemistry 46: 8861–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y., Duncan, M.K., and David, L.L. 2002. Lens proteomics: The accumulation of crystallin modifications in the mouse lens with age. Invest. Ophthalmol. Vis. Sci. 43: 205–215. [PubMed] [Google Scholar]
- Van Montfort R.L., Bateman, O.A., Lubsen, N.H., and Slingsby, C. 2003. Crystal structure of truncated human βB1-crystallin. Protein Sci. 12: 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos J.J. and van Norren, D. 1998. On the relevant spectral parts for glassblowers' cataract. Ophthalmic Physiol. Opt. 18: 311–313. [DOI] [PubMed] [Google Scholar]
- Vos J.J. and van Norren, D. 2004. Thermal cataract, from furnaces to lasers. Clin. Exp. Optom. 87: 372–376. [DOI] [PubMed] [Google Scholar]
- Wenk M., Herbst, R., Hoeger, D., Kretschmar, M., Lubsen, N.H., and Jaenicke, R. 2000. γS-Crystallin of bovine and human eye lens: Solution structure, stability and folding of the intact two-domain protein and its separate domains. Biophys. Chem. 86: 95–108. [DOI] [PubMed] [Google Scholar]
- Werten P.J., Lindner, R.A., Carver, J.A., and de Jong, W.W. 1999. Formation of βA3/βB2-crystallin mixed complexes: Involvement of N- and C-terminal extensions. Biochim. Biophys. Acta 1432: 286–292. [DOI] [PubMed] [Google Scholar]
- Wieligmann K., Mayr, E.M., and Jaenicke, R. 1999. Folding and self-assembly of the domains of βB2-crystallin from rat eye lens. J. Mol. Biol. 286: 989–994. [DOI] [PubMed] [Google Scholar]
- Wilmarth P.A., Tanner, S., Dasari, S., Nagalla, S.R., Riviere, M.A., Bafna, V., Pevzner, P.A., and David, L.L. 2006. Age-related changes in human crystallins determined from comparative analysis of post-translational modifications in young and aged lens: Does deamidation contribute to crystallin insolubility? J. Proteome Res. 5: 2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistow G., Sardarian, L., Gan, W., and Wyatt, M.K. 2000. The human gene for γS-crystallin: Alternative transcripts and expressed sequences from the first intron. Mol. Vis. 6: 79–84. [PubMed] [Google Scholar]
- Wistow G., Bernstein, S.L., Wyatt, M.K., Behal, A., Touchman, J.W., Bouffard, G., Smith, D., and Peterson, K. 2002. Expressed sequence tag analysis of adult human lens for the NEIBank Project: Over 2000 non-redundant transcripts, novel genes and splice variants. Mol. Vis. 8: 171–184. [PubMed] [Google Scholar]
- Wu Z., Delaglio, F., Wyatt, K., Wistow, G., and Bax, A. 2005. Solution structure of γS-crystallin by molecular fragment replacement NMR. Protein Sci. 14: 3101–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M., Martin, A., and Schmid, F.X. 2005. Stabilization of the cold shock protein CspB from Bacillus subtilis by evolutionary optimization of Coulombic interactions. J. Mol. Biol. 347: 1063–1076. [DOI] [PubMed] [Google Scholar]
- Zarina S., Slingsby, C., Jaenicke, R., Zaidi, Z.H., Driessen, H., and Srinivasan, N. 1994. Three-dimensional model and quaternary structure of the human eye lens protein γ S-crystallin based on β- and γ-crystallin X-ray coordinates and ultracentrifugation. Protein Sci. 3: 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]