

Abstract
The limited repertoire of drug-resistance markers imposes a serious obstacle to genetic manipulation of Trypanosoma brucei. Here we describe experiments with a fusion protein that allows positive selection for genome integration followed by CRE recombinase-mediated excision of the marker cassette that can be selected by ganciclovir, although the excision event is so efficient that selection is not strictly necessary. We describe two variants of the tetracycline-inducible pLEW100-based CRE-expression vector that reduced its toxicity when stably integrated into the genome, and we demonstrate that transient transfection of circular pLEW100-CRE is highly efficient at catalyzing marker excision. We used this approach to delete the last two enzymes of the pyrimidine synthesis pathway, creating a cell line that is resistant to fluoroorotic acid, which would allow the same enzymes (PYR6-5) to be used as an alternative negative selectable marker.
Keywords: Trypanosoma brucei, Genetic tools, Negative selection, CRE recombinase, Ganciclovir, Fluoroorotic acid, Pyrimidine synthesis, Pyrimidine auxotroph, Culture, Thymidine, ura3
1. Introduction
Bacteriophage P1 encodes CRE, a recombinase of the integrase family [1] that helps to maintain and segregate the viral genome as a single-copy plasmid during lysogeny [2,3]. In its most common use, CRE catalyzes the excision of a ‘floxed’ DNA fragment—one flanked by 34-bp loxP elements in direct orientation—to an episome, leaving one residual loxP sequence in the genome. CRE also catalyzes the reverse reaction—integration of a plasmid containing one loxP site to a genomic locus containing another loxP site—but equilibrium favors excision. If a DNA fragment is flanked by loxP elements in opposite orientation, CRE will invert the intervening sequence [4–6]. As the enzyme requires no additional co-factors, it has been adapted for genetic manipulation in myriad contexts, including yeast and human cells, in vitro and in vivo [7] (reviewed in [8–10]). Most notably, the CRE/loxP system has been used for removing selectable markers [11], excising large tracts of genomic DNA [12], triggering chromosomal rearrangements [13] and integrating exogenous DNA to a specific locus [14], none of which has been achieved in Trypanosoma brucei.
As the limited repertoire of selectable drug markers presents a major barrier to research with T. brucei, we decided first to evaluate the potential of the CRE/loxP system for marker removal. In Leishmania, markers have been removed by targeted replacement of a negative-selectable marker via ‘null cassette’ transfection [15] and by selection for marker loss via intrinsic recombination [16]. In both cases, the methods are labor intensive and inefficient. We hypothesized that CRE/loxP could present a much simpler and more versatile solution. To this end, we designed a cassette encoding a fusion protein consisting of positive (hygromycin phosphotransferase: HYG) and negative (Herpes simplex virus thymidine kinase: HSVTK) markers flanked by loxP sites in direct orientation. The action of CRE would excise the HYG-HSVTK coding region, leaving one residual loxP sequence. Wild-type T. brucei are indifferent to the nucleoside analog ganciclovir (GCV), which is toxic to cells expressing HSVTK. We could therefore select cells that reverted to hygromycin sensitivity and GCV resistance as markers for CRE action.
Given previous results [17], we were not surprised to find CRE-mediated recombination occurred very efficiently but that CRE expression slowed cell growth. Similar effects have been observed in mammalian systems both in vitro [18,19] and in vivo [20,21]. We explored two approaches to tighten regulation of expression of stably integrated CRE, but—considering the perpetual problem of read-through transcription [22]—our preferred solution is transient transfection of a CRE-expressing plasmid, which proved to be very effective without observable toxicity.
We tested CRE-mediated excision in two loci: first in β-TUBULIN (β-TUB) and then in PYR6-5 (Tb 927.5.3810). PYR6-5 is a fusion of the final two enzymes of the pyrimidine synthesis pathway, orotate phosphoribosyltransferase (IUBMB EC 2.4.10) or PYR5 and orotidine-5′-phosphate decarboxylase (EC 4.1.1.23) or PYR6 [23]. PYR6, known as ura3 in Saccharomyces cerevisiae, is universally used as a positive/negative marker in yeast [24]. Wild-type cells that express the enzyme die in the presence of 5-fluoroorotic acid (FOA). PYR6−/− cells can therefore be selected for resistance to FOA and pyrimidine auxotrophy. As wild-type bloodstream-form T. brucei are highly sensitive to FOA (IC50 ~ 1 μg/ml in HMI-9: data not shown), we generated PYR6-5−/− trypanosomes by knocking out the first allele with the loxP-HYG-TK-loxP cassette and then selecting for gene-conversion-mediated loss of heterozygosity (LOH) with FOA. We then simultaneously excised both copies of the loxP-HYG-TK-loxP cassette, creating a PYR6-5−/− cell line lacking any drug markers and making PYR6-5 available as an additional negative selectable marker in this cell line.
2. Materials and methods
2.1 Plasmids
pLHTL, a cassette of loxP-HYG-Ty1-HSVTK-loxP flanked by HindIII and BamHI sites, was assembled in pGEM4 from PCR products. HSVTK was amplified from pORF-HSVTK (Invitrogen) with a 5′ primer incorporating an XbaI site and the Ty1 epitope tag [25] and a 3′ primer containing BamHI and loxP sites. HYG was amplified from pHD309 [26] with a 5′ primer incorporating HindIII and loxP sites, and a 3′ primer containing an XbaI site. The fragments were cloned sequentially into pGEM4 (Promega). In the assembled chimera, the two loxP sites are in direct orientation and the open reading frame is continuous. The chimeric HindIII and BamHI fragment was excised from pGEM4 and cloned into pHD309, from which the region between the SmaI and StuI sites had been removed.
To target pHD309-LHTL to the PYR6-5 locus (Tb 927.5.3810), 496 bp immediately downstream of the coding region were amplified by a PCR primer pair that introduced BamHI and SbfI terminal restriction sites and cloned into the respective sites in pHD309-LHTL. To the resulting plasmid, a 365 bp fragment, 134 bp upstream of the PYR6-5 coding region, amplified with PvuII and HindIII terminal sites was cloned, resulting in pLHTL-PYR6-5 (Fig. 1A).
Fig. 1.

(A) pLHTL-PYR6-5 incorporates HYG (positive selectable marker) and Herpes simplex virus thymidine kinase (HSVTK, negative selectable marker) fused via a Ty1 epitope tag. The markers are flanked by loxP recognition elements in direct orientation, to allow its excision by CRE. The illustrated vector is targeted for homologous recombination into the PYR6-5 locus. Its parent, pHD309-LHTL, targets β-TUB. The open arrows denote primers used for diagnostic PCR in Fig. 3B and C. (B) pLEW100Cre (gift of J.E. Donelson [17]) replaces LUC [27] with CRE, whose expression is driven by the GPEET promoter under the control of two Tet operators. The sequence of the polypyrimidine tract of the SAS in the GPEET upstream region is shown. In pLEW100CreTS, the ALD 3′ UTR between PstI sites is replaced by the temperature regulatory element of the EP1 3′ UTR (gift of I. Roditi [28]). In pLEW100CreTS-SAS, the SAS is mutated to contain six purines. (C) Western blot with anti-Ty1 monoclonal antibody BB2 verifies that the HYG-Ty1-HSVTK product is expressed in three independent clones transfected with pHD309-pLHTL, with the parental SM line as a negative control. The lower band is consistent with a Ty1-HSVTK cleavage product.
pLEW100Cre [17] (generous gift of J. E. Donelson) replaces the Luciferase coding sequence (LUC) of pLEW100 [27] with CRE (GenBank X03453). To make pLEW100CreTS, the final 612 bp of the ALD 3′ UTR of pLEW100Cre were excised by digestion with PstI and replaced with the temperature-sensitive region (bp 174–298) of the EP1 3′ UTR [28], which was amplified from pGAPRONE [29] (generous gift of I. Roditi) with terminal PstI sites. Orientation was confirmed by sequencing and conservation of the essential mRNA structure was confirmed by the method of Drozdz and Clayton [30]. Mutations in the GPEET splice-acceptor site (SAS) of pLEW100CreTS (Fig. 1B) were introduced by QuikChange (Stratagene), following the predictions of Seigel et al. [31], and confirmed by sequencing.
2.2 Strains, growth and transfection
T. brucei bloodstream forms (strain Lister 427 antigenic type MITat 1.2 clone 221a [32] were cultured in HMI-9 medium [33] without thymidine at 37 °C. All experiments used the ‘single-marker’ derivative, which constitutively expresses T7 RNA polymerase and the Tet repressor [27], maintained in the presence of 2 μg/ml G418 (Sigma). For stable transfection of pHD309-LHTL and pLEW100 or its CRE derivatives, the plasmids were linearized with NotI. The LHTL-PYR6-5 cassette was liberated by cleavage with PvuII. Circular pLEW100 and its CRE derivatives were used for transient transfection. All transfections were performed with an Amaxa Nucleofactor electroporator using program X-001 and Human T Cell solution, using 2.5 × 107 cells and 10 μg of DNA. pLEW100-derived clones were selected in 2.5 μg/ml phleomycin (Sigma) and maintained in 1.2 μg/ml. LHTL derived clones were selected and maintained in 4.5 μg/ml hygromycin (Sigma). To select for LOH, PYR6-5::LHTL cells were distributed into 96-well plates in 10-fold serial dilutions, from 106 to 10 cells, in 6 μg/ml FOA (Sigma).
2.3 CRE expression
Cells that had been stably transfected with LHTL cassettes and pLEW100 or its CRE derivatives were washed and resuspended in HMI-9 containing G418 and phleomycin, to remove hygromycin. CRE or LUC expression was induced by adding 100 ng/ml doxycycline (Sigma) for 12 h. Cells were washed again and plated in serial 10-fold dilutions from 5 × 105 to 50 cells per 96-well plate. GCV (Sigma) was added to 10 μg/ml (β-TUB::LHTL) or 25 μg/ml (PYR6-5::LHTL), leaving one additional plate of 50 cells without GCV as a control for viability. For transient transfection, the same procedure was used except that phleomycin was omitted and CRE or LUC was induced by adding 100 ng/ml doxycycline for 7 h immediately after transfecting the cells with pLEW100 or its pLEW100CRE.
2.4 DNA isolation and analysis
Genomic DNA was isolated as previously described [34] and digested with BglII and SalI (New England Biolabs) according to the manufacturer’s recommendations. Fragments were detected by blotting, hybridization, and phosphorimaging [35]. HYG probe was prepared by digestion of pHD309 with XbaI and MscI and gel purification. PYR6 was PCR amplified from T. brucei DNA, and the desired fragment was gel purified after FspI, XhoI and NcoI digestion. PCR was performed on 200 ng DNA, isolated with DNAzol (Invitrogen), using primers that would reveal the 34-bp loxP ‘scar’ that remains after excision of the loxP-flanked marker cassette. Primers annealed at 53 °C. In the β-TUB locus, targeted by pHD309-LHTL, primers to β-TUB (5′-GTTTGCGATATCCCACCCAAGGGACTC) and the pHD309 backbone (5′-GCTAGCTTGCATGCCTGCAAGG) were added to 0.9 pmol/μl. Primers flanking the endogenous GAPDH locus (Tb927.6.4280 & 4300; 5′-AGGCACTGTGCGACGAC and 5′-GACCGTGCAGCAAAGCTATAA) were used at 0.2 pmol/μl, as GAPDH is present in four copies. Extension was done for 30 cycles at 72 °C for 65 s. In the PYR6-5 locus, primers to the upstream (5′-TCTCTCACTTGGCGCCATCTCTGTGTTACAA) and downstream regions (5′-CGTCCGCAATAGCTTAAAGATAGAGCTCCG) were added to 0.9 pmol/μl, with annealing at 53 °C and 30 cycles of extension at 72 °C for 60 s.
2.5 Statistics
Frequency values in CRE-mediated excision assays were calculated from GCV-resistant clones relative to the number of clones without selection, multiplied by the dilution factor where applicable. Only plates considered at limiting dilution, namely those in which fewer than forty-eight wells showed growth, were counted. Error bars in all figures are standard deviations calculated from at least three independent trials (n values >3 are noted). Probabilities were calculated by Student’s T test – one-tailed, when comparing background (LUC) to experimental samples, and two-tailed when comparing two experimental conditions.
3. Results
In 2004, Barrett and colleagues [17] adapted pLEW100 to express CRE in procyclic T. brucei, under the control of the Tet operator (Fig. 1B). They observed very efficient CRE-mediated recombination from stably integrated pLEW100Cre, but found that the expression of CRE driven by pLEW100’s GPEET promoter (PGPEET) was toxic, even in the absence of tetracycline. Knowing PGPEET to be at least ten times less active in bloodstream cells, we cloned bloodstream-form ‘single marker’ (SM) Lister 427 cells transfected with either pLEW100 (expressing LUC) or pLEW100Cre. CRE integration was confirmed by Southern blot (data not shown).
3.1 Validating HSVTK as a negative marker selectable with GCV
We chose to employ the well-studied Herpes simplex virus thymidine kinase (HSVTK) (EC 2.7.1.21) [36] as a negative selectable marker that had been used previously in bloodstream forms of T. brucei [37]. HSVTK phosphorylates nucleosides more promiscuously than its cellular homologues, rendering cells that express it sensitive to nucleoside analog prodrugs such as GCV. As such, GCV would select cells that had lost HSVTK function. We adapted pHD309 [26], which targets the T. brucei β-TUB coding sequence, to express a fusion of HYG for positive selection and HSVTK for negative selection. We flanked the fusion with loxP sites in direct orientation, so that CRE would excise only the coding region and one loxP element [4]. A second version of pHD309, called pLHTL-PYR6-5, was constructed so that homologous recombination would cause the LHTL cassette to replace the endogenous PYR6-5 coding region (Fig. 1A).
As we had inserted a Ty1 epitope between HYG and HSVTK, we performed Western blots using an anti-Ty1 antibody [25] on bloodstream trypanosome clones transfected with pHD309-LHTL. Three independent clones showed a prominent band of the expected size for the Hyg-Ty1-HSVTK fusion protein that is absent from the parental SM cells (Fig. 1C). The smaller band is consistent with the expected size of either a Hyg-Ty1 or Ty1-HSVTK product. In an analogous construct where HYG was replaced by PUR, the only visible band on a western blot corresponded to Ty1-HSVTK (data not shown). Regardless, the cells displayed the expected phenotype of resistance to hygromycin or puromycin and sensitivity to GCV.
Preliminary experiments required high concentrations of GCV for selection against trypanosomes expressing HSVTK. We suspected the unusually high amount of thymidine (39 mg/l) included in the formulation of HMI-9 [33], in addition to any contribution from the serum or SerumPlus™, might be competing with the drug. As trypanosomes are pyrimidine prototrophs, an external source of thymidine should be unnecessary [38–40]. When thymidine was omitted, cells grew normally and HSVTK-expressing clones were about ten times more sensitive to GCV (IC50 ~ 0.5 μg/ml: data not shown), so all experiments were conducted in HMI-9 without thymidine.
3.2 Marker excision via tetracycline-inducible stably integrated CRE
With a sensitive negative-selectable marker in hand, we serially transfected and cloned pLEW100 or pLEW100Cre and then pLHTL into SM cells [27]. After selecting clones, hygromycin was removed from the medium and LUC or CRE expression was immediately induced for 12 h [27]. After removing doxycycline, cells were distributed in a dilution series ranging from 5,000 to 50 cells per 96-well plate. GCV was added to all wells except in one additional plate with 50 cells, which served as a viability control. We observed reversion to GCV resistance in ~50% of the cells in which CRE was induced, compared to 1 in ~5 × 105 cells in which LUC was induced. However, even uninduced CRE cells reverted to GCV resistance ~100 times more frequently than the control cells expressing LUC, implying leakage of CRE expression in uninduced cells.
This protocol was repeated with LHTL inserted into the endogenous PYR6-5 locus (Fig. 1A). Cells in which CRE was expressed reverted to GCV resistance at a very high rate (Fig. 2A), while those expressing LUC from the same vector rarely reverted to the parental phenotype, presumably due to gene conversion from the second PYR6-5 allele. Thus it seems that CRE can excise targets equally well from the β-TUB locus on chromosome 1 or the PYR6-5 locus on chromosome 5.
Fig. 2.

Graphs show the frequency at which cells reverted to GCV resistance, which implies loss of HSVTK. (A) Clones bore stably integrated copies of pHD309-LHTL and pLEW100 (Luciferase, n=5) or pLEW100Cre (Cre, n=4), whose expression was induced with doxycycline. (B) Clones with stably integrated pHD309-LHTL and pLEW100 versions bearing Luciferase (n=4), Cre (n=9), CreTS (n=6) or CreTS-SAS (n=4) were selected in GCV without induction, to assay leakiness of the constructs. (C) On the left, a clone with LHTL in one allele of the pyFE locus (pyFE+/LHTL) was transiently transfected with pLEW100 (Luciferase, n=4) or pLEW100Cre (Cre, n=6) and induced with doxycycline. On the right, the pLHTL-pyFE clone had been with FOA to select a PYR6-5LHTL/LHTL clone, which was then transiently transfected as before (Luciferase n=6, Cre n=6).
Assuming GCV resistance arose by loss of the LHTL cassette from the β-TUB locus, these cells should have reverted to hygromycin sensitivity. Indeed, of the ten randomly selected independent clones believed to have undergone CRE-mediated reversion to GCV resistance, none survived when hygromycin was added (data not shown).
To confirm that these cells had been converted by CRE-mediated excision of the LHTL cassette, DNA was isolated from randomly selected clones and analyzed by PCR. We designed primers to the pLHTL vector region just outside the loxP sites. Excision of the 2.2-kb HYG-HSVTK-loxP region should leave a ‘scar’ consisting of the upstream and downstream targeting regions fused via a single loxP site. PCR should therefore yield a product 2.2 kb shorter than that from the parental construct. Fig. 3A shows the results of this PCR on the β-TUB version of LHTL. The 3-kb parental product was not amplified by this short-cycle PCR, but the predicted 730-bp scar is clearly visible. Primers for GAPDH (Tb927.6.4300) were included as an internal control. The first three clones in Fig. 3A show the CRE scar as expected. Two of the second set of three clones also show a scar, which indicates that, even in the absence of GCV selection, CRE-mediated excision occurred in the majority of cells in which CRE was induced. The third set of clones are from CRE cells that were not induced but were selected in GCV. The two observed scars confirm that CRE expression is leaky in these cells. As expected, none of the parental cell lines or cells bearing LUC instead of CRE show a scar.
Fig. 3.
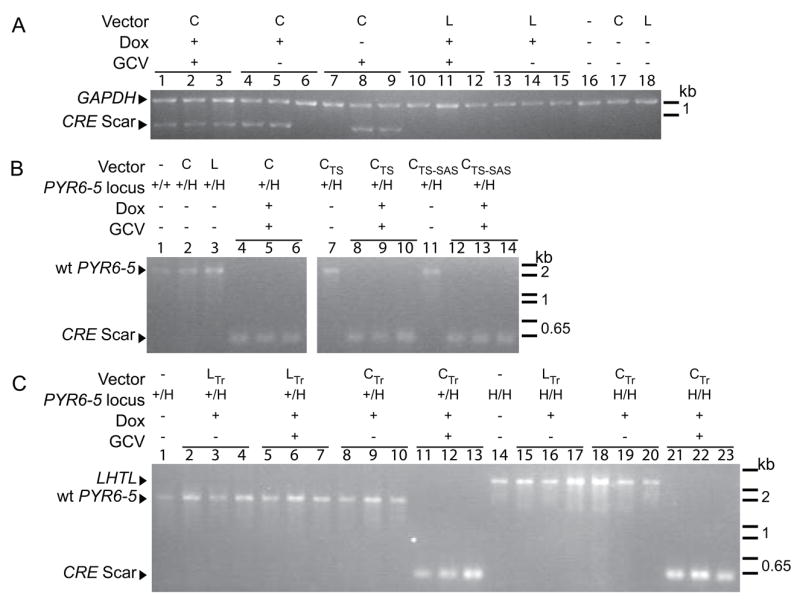
PCR confirms that reversion to GCV resistance is attributable to CRE. Horizontal bars above the lane numbers indicate triplet independent clones. (A) Primers flanking the LHTL cassette in the β-TUB locus show either the diagnostic scar of CRE-mediated excision or no product, under the conditions used. Primers to the GAPDH coding sequence were included as an internal control. Cells had stably integrated pLEW100 (L) or pLEW100Cre (C), and were treated with or without doxycycline and GCV. (B) CRE-mediated excision from the PYR6-5 locus. The PYR6-5 locus is either wild type (+/+) or heterozygous with LHTL (+/H). In the absence of a CRE scar, primers amplify the endogenous PYR6-5. Cells had stably integrated pLEW100 (L) or pLEW100Cre and its variants (C, CTS and CTS-SAS), and were treated with or without doxycycline and GCV. (C) pLEW100 (LTr) or pLEW100Cre (CTr) was transiently transfected in the presence of doxycycline prior to GCV selection. CRE was also effective in cells homozygous for LHTL at PYR6-5 (H/H). In these cells, without an endogenous PYR6-5 and without CRE-mediated excision creating a CRE scar that is preferentially amplified under the conditions used, the primers amplify the larger LHTL cassette.
The PCR protocol was repeated for PYR6-5::LHTL, using primers (Fig. 1A) to sequences flanking PYR6-5. Three products were possible: the endogenous PYR6-5 (1.9 kb), the LHTL cassette inserted into PYR6-5 (2.6 kb), and the CRE scar (400 bp). With a short PCR extension step, competition favored the shortest product where multiple products would be expected. Lane 1 of Fig. 3B shows the parental SM cells. The only detected product arises from the wild-type 1.9-kb allele. The same result is apparent in lanes 2 and 3, cells transfected with pLEW100Cre or pLEW100 without tetracycline induction, in which one PYR6-5 allele was replaced by LHTL, which is out-competed by the smaller wild-type allele. Lanes 4–6 show clones in which CRE has been induced and cells have been selected in GCV. The favored 400-bp CRE scar verifies CRE-mediated excision.
Southern blots on representative clones confirmed the PCR results. In lane 1 of Fig. 4, SM cells displayed the expected band for the endogenous PYR6 without a HYG band corresponding to LHTL. Because of the cloning strategy used to make them [27], SM cells contain a 250-bp fragment of HYG in the TUB locus. As all cell lines used in this project were derived from SM cells, this band serves as a loading control. In lanes 2 and 3, parental cells transfected with pLHTL-PYR6-5 show an LHTL band in addition to PYR6 from the remaining wild-type allele. The cells in lane 6 (the same cells analyzed in Fig. 3B, lane 6) retain an endogenous PYR6 but have lost LHTL by the action of CRE.
Fig. 4.
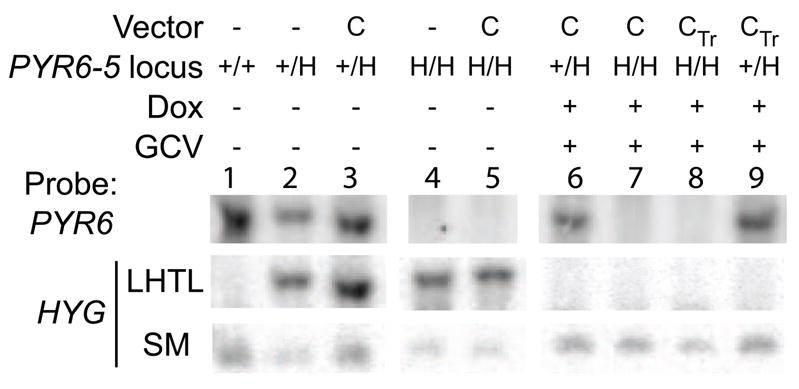
Southern blot of genomic DNA confirms spontaneous LOH and CRE-mediated loss of LHTL cassette at the PYR6-5 locus. C and CTr denote cells transfected with pLEW100Cre stably or transiently, respectively. In the PYR6-5 locus, clones are wild type (+/+), heterozygous (+/H) or homozygous (H/H) for LHTL, prior to CRE induction. In lanes 6–9, cells were induced with doxycycline and selected in GCV. Blot was probed for HYG, which detects LHTL and a smaller band from a fragment of HYG, which is present in all SM-derived cells. The blot was reprobed for PYR6.
For the final proof of CRE excision, the scar PCR product from the three independent clones in Fig. 3A lanes 1–3 was purified and sequenced. The sequences were exactly as expected, encompassing the upstream UTR, one loxP element and the downstream UTR (data not shown). Thus it seems that the 10,000-fold increase in frequency of reversion to GCV resistance shown in Fig. 2A is truly attributable to specific excision by CRE.
3.3 CRE toxicity concerns and solutions
Considering previous experience with pLEW100Cre in procyclic forms [17], which did not tolerate CRE induction, we were concerned that CRE expression might be toxic, despite the reduced expression level of pLEW100 in bloodstream forms. Without induction, populations of pLEW100Cre-transfected cells exhibited no significant growth defect. Under doxycycline induction, however, the doubling time increased by ~ 15%. We felt, therefore, that certain applications would require a system that demonstrated no toxicity.
Our first approach was to tighten the regulation of CRE expression by using a temperature sensitive motif from the 3′ UTR of the EP1 mRNA [29,30,28]. This 124-bp element confers a 10- to 20-fold decrease in expression at 37 ºC relative to 20 ºC, in bloodstream forms. By inserting this motif into the 3′ UTR of CRE in pLEW100, creating pLEW100CreTS (Fig. 1B), we expected to decrease the leaky expression of CRE at 37 °C. At the same time, if tetracycline induction alone generated insufficient amount of CRE at 37 °C, we would have the option of combining tetracycline induction with a short exposure to 20 °C. We amplified the essential motif, bases 173–298 from the stop codon, plus the next 63 bases, thought to be necessary for polyadenylation, from pGAPRONE (generous gift of Isabel Roditi). Due to the paucity of unique restrictions sites in the region, we were forced to insert between two PstI sites in the ALD 3′ UTR of pLEW100. This created a hybrid UTR in which the first 93 bases of the ALD 3′ UTR were fused to the EP1 motif. Structural predictions with M-fold (http://www.bioinfo.rpi.edu/applications/mfold, [41]) using the previously described constraints [30], suggested that this fusion would have no impact on the stability of the temperature-sensitive motif (data not shown).
As CRE expression from pLEW100CreTS appeared to be adequate at 37 °C (see below), our second approach was to further decrease CRE expression from pLEW100CreTS, by replacing six pyrimidines in the SAS with purines [31]. We anticipated that CRE expression from the resulting construct, pLEW100CreSAS-TS (Fig. 1B), would decrease by at least 100-fold, due to reduced trans-splicing of the mRNA. We created SM cell lines with stably integrated copies of pLEW100CreTS or pLEW100CreTS-SAS plus pLHTL-PYR6-5. Surprisingly and inexplicably, the pLEW100CreTS-SAS cells appeared to have a slight growth defect, whereas pLEW100CreTS cells did not (data not shown). The frequency of CRE-induced reversion to GCV resistance (data not shown) with either of the modified constructs was indistinguishable from the previously measured frequency with the unmodified pLEW100Cre (Fig. 2A). In uninduced cells (Fig. 2B), however, whereas ‘leaky’ reversion from the original pLEW100Cre was well above the LUC control (p = 0.0030), the background reversion level was significantly lower for both pLEW100CreTS (p=0.0018) and pLEW100CreTS-SAS (p=0.050). The difference between pLEW100CreTS and pLEW100CreTS-SAS was surprising (p=0.014), as we had expected the SAS mutant would have a very low expression level. PCR (Fig. 3B, lanes 7–14) and Southern blot (data not shown) confirmed that CRE-mediated excision of the LHTL cassette was responsible for the high frequency of GCV resistance from the modified CRE constructs.
Though pleased with the increased regulation provided by the modified CRE constructs, we wanted to test a transient transfection protocol for two reasons: even leaky CRE-excision frequencies of 10−4 may be too high for certain applications, and the use of a stably-integrated version of pLEW100Cre would preclude the concurrent presence of other Tet-regulated constructs. An earlier study had suggested that transient transfection of CRE constructs could be an effective delivery system [17]. Because CRE-mediated excision was very efficient, transient transfection of a CRE expression vector seemed likely to be an ideal means to avoid toxicity or inappropriate expression. We therefore cloned SM cells in which one or both alleles of PYR6-5 had been replaced with LHTL and transiently transfected them with circular pLEW100Cre. The results (Fig. 2C) proved that transient transfection is very effective: nearly 3% of the cells, relative to transfection survival controls, reverted to GCV resistance.
As before, random clones were selected for analysis by PCR and Southern blot. In Fig. 3C, lane 1 shows the endogenous PYR6-5 product in the parental SM PYR6-5::LHTL heterozygous line. Again, because of competition for PCR amplification between the short wild-type and longer PYR6-5::LHTL alleles, only the former is amplified in the heterozygote. Lanes 2–4 show that transfection of pLEW100 has no effect on the locus. Lanes 5–7 represent the rare clones that reverted to GCV resistance without being exposed to CRE. Lanes 8–10 are clones from the pLEW100Cre transfection that were not selected with GCV. Not surprisingly, with a 3% excision frequency, none of the three clones showed a CRE excision scar. Clones 11–13 were transfected with pLEW100Cre and selected in GCV, and show the characteristic CRE excision scar. In Southern blots (Fig. 4), the PYR6-5::LHTL heterozygote in lane 2 clearly shows the wild-type PYR6-5 allele and the LHTL cassette in the other allele. In lane 9 (same clone as Fig. 3C lane 11), the LHTL cassette is clearly lost after pLEW100Cre transfection and GCV selection. As a large-scale proof, clones were replica plated to medium with hygromycin to screen for loss of LHTL. Of 63 clones transfected with pLEW100 and unexposed to GCV, all survived in hygromycin. Of 112 clones transfected with pLEW100Cre and selected in GCV, none survived in hygromycin.
Growth curves confirmed that pLEW100Cre transfection and GCV selection had no detectable impact on cell viability, and none of the transiently transfected clones were resistant to phleomycin one week after transfection, implying no retention of the circular plasmid (data not shown). Thus, our data confirm that transient transfection of a CRE expression vector provides a very effective delivery protocol without detectable side effects.
3.4 Gene conversion at the PYR6-5 locus
Gene conversion, the genetic mechanism underlying most VSG switching, is much pondered but little studied in T. brucei. Studies of gene-conversion-mediated LOH in trypanosomatid protozoa are scant and show a wide range of frequencies [42,43]. We targeted the LHTL cassette to the PYR6-5 locus in part to assay LOH, as half the gene conversion events at that locus would produce cells with two copies of LHTL and no PYR6-5, which would render them resistant to FOA. After adding 6 μg/ml of FOA, which rapidly kills wild-type cells, to serial dilutions of PYR6-5::LHTL heterozygous cells, we found resistance arose at a frequency ~ 10−5, which is consistent with results from L. major [42].
We confirmed, by PCR and Southern analysis, that the FOA-resistant cells had arisen by LOH. Comparing, for example, lanes 1 and 14 in Figure 3C, it is clear that the wild-type PYR6-5 is no longer present to compete out the considerably larger LHTL product from the same primer pair. In Figure 4, the PYR6-5 band in lanes 1–3 is clearly lost in lanes 4 and 5. While these data do not exclude the remote possibility that the second PYR6-5 allele was lost by some other means, previous work from our lab on the ALG12 locus suggests that LOH by duplicative gene conversion is by far the most likely mechanism [43].
Manipulating the pyrimidine synthesis pathway is particularly useful in yeast, where ura3−/− (PYR6−/−) can be both a positive (allowing growth in the absence of exogenous pyrimidine) or a negative (rendering cells sensitive to FOA) marker. We were disappointed though not surprised to find that PYR6-5−/− T. brucei did not require uracil supplementation for growth. Considering the low yields of bloodstream-form T. brucei in culture, it seems likely that the fetal bovine serum and/or Serum Plus™, whose content is proprietary, provide ample pyrimidines.
With PYR6-5LHTL/LHTL cells in hand, we wondered if CRE could simultaneously and efficiently excise both copies of the LHTL cassette. The results when CRE was expressed from a stably integrated copy of pLEW100Cre were indistinguishable from those shown in Fig. 2A (data not shown). Excision from a transiently transfected copy of pLEW100Cre was similarly successful (Fig. 2C). The excision frequency was not significantly different between the homozygous and heterozygous cell lines.
Again, randomly selected clones were selected for analysis by PCR and Southern blot and were confirmed to be sensitive to hygromycin. In Fig. 3C lanes 14–20, the parental homozygous PYR6-5::LHTL cells and control clones transfected with pLEW100 and/or not selected in GCV all show the LHTL cassette amplified by PCR whereas, in lanes 21–23, clones transfected with pLEW100Cre and selected in GCV show the characteristic scar of CRE-mediated excision. In Fig. 4, lanes 7 and 8 show the loss of the LHTL band from the parental cell lines of lanes 4 and 5 in both stable and transient CRE expression protocols.
In both stable and transient experiments in the absence CRE, LHTL insertions into β-TUB or PYR6-5 loci revert to GCV resistance at frequencies of ~ 4 × 10−5 (LUC data in Fig. 2). In PYR6-5::LHTL homozygotes, however, not a single GCV-resistant clone was recovered in all experiments, which represent a total of > 2 × 107 cells. This supports the hypothesis that the primary mechanism for spontaneous elimination of HSVTK is LOH by gene conversion. When both alleles have already been converted to LHTL, there is no efficient way to eliminate HSVTK.
4. Discussion
4.1 CRE/loxP provides a solution to the marker problem in T. brucei
At present, the repertoire of positive selectable drug markers imposes a serious obstacle to research in T. brucei [44–47]. We sought a system whereby genes could be knocked out using positive-selectable markers that could then be removed, allowing positive markers to be reused indefinitely to delete any number of genes anywhere in the genome. Such strategies have been employed before but have drawbacks that will be addressed below [15,16].
We sought to adapt the widely used CRE/loxP system to solve this marker problem. An earlier study showed that CRE was very effective in T. brucei but could not overcome its toxicity [17]. Indeed, toxicity from excess CRE has been reported recently in many systems [18–21,48], and, considering the potency of the GPEET promoter driving CRE expression in procyclic T. brucei, some degree of toxicity would not be unexpected. Our goal was to implement a system in which CRE would excise from the genome a combined positive and negative marker by recognizing the flanking loxP elements at a safe level of CRE expression.
Our data confirmed that, when expressed from a stably integrated copy, CRE is so efficient in bloodstream-form T. brucei that negative selection for excision of the marker is unnecessary: merely screening a few clones after CRE induction yielded the desired cell line. Furthermore, the data indicate CRE can excise two floxed targets almost as easily as one, so we see no theoretical limit on the number of targets that can be excised at one time. It was recently reported that, after using a different approach to achieve marker removal in Leishmania, a marker-less expression cassette was stable in the genome for as many as 200 generations [16], so the marker recycling approach can be extended beyond simple knockouts, especially in non-repetitive parts of the genome. As the GPEET promoter is at least ten times less potent in bloodstream forms than in procyclics, we expected and observed considerably less toxicity than in the previous study [17], but we were still concerned about undesirable expression.
To address this, we made two modifications to pLEW100Cre. First, we inserted a temperature-sensitive element into the 3′ UTR of CRE to confer specific and reversible post-transcriptional down regulation [28]. Second, we mutated the SAS to decrease the overall expression level by inhibiting mRNA maturation [31]. While both of these approaches reduced leaky expression of CRE, we had expected the combination of the two (pLEW100CreTS-SAS) to show less leakiness than the temperature-sensitive element alone (pLEW100CreTS). The seemingly greater leakiness and growth defect from pLEW100CreTS-SAS was unexpected and inexplicable. Based on reporter assays in procyclic forms [31], we had expected at least a 100-fold drop in CRE expression.
In cells that were homozygous for LHTL and contained a stable genomic copy of pLEW100Cre, we observed an interesting phenomenon. While the cells were in continuous culture, CRE could excise one copy of LHTL in the absence of induction or GCV selection. As they were maintained hygromycin, there was selection against loss of both copies, but a single copy was readily excised by the leaky CRE expression. Although we did not test the modified CRE cell lines, even a miniscule CRE leak would probably have the same effect over time. While this is unlikely to present a problem in practical applications of CRE/loxP, it highlights the need for an alternative CRE delivery system, probably transient transfection. Having implemented a new protocol for bloodstream-form transfection in July 2006, we found that CRE expression from a circular vector was very effective at targeted excision and produced no detectable growth defect. Our data are consistent with previous work showing that circular plasmids are quickly lost as cells divide [49] and so the method minimizes the risk of unwanted CRE expression.
As transient transfection of a CRE-expressing vector is our preferred delivery method, we plan to adapt the pLEW100 expression system, designed for stable integration [27], to this purpose. The region for homologous recombination (rRNA spacer) and drug marker (BLE) are unnecessary for transient use, and removing the Tet operators would eliminate the need for tetracycline induction cells after transient transfection into SM cells and prevent interference with any other Tet-regulated constructs that may be present in a particular SM cell line. Transient transfection also allows pLEW100Cre to be used in any wild-type T. brucei cell.
4.2 Extensions of CRE/loxP
This CRE/loxP system has several advantages over marker removal systems previously implemented in Leishmania. One approach [16] relies on random recombination events to excise a positive-negative marker, which is effective but very inefficient, but these investigators suggested CRE/loxP as a better alternative. The clever approach of Denise et al. [15] is to replace a positive-negative marker with a null fragment introduced by transfection. Although marker removal and selection are simple, it is labor intensive to tailor a null vector and repeat the transfection and cloning for each targeted locus. It is possible, however, that this method will find a use in situations where it is not permissible to leave a loxP element at the site of marker excision, or where the presence of several dispersed loxP elements, resulting from multiple CRE-mediated gene manipulations, might be undesirable.
Furthermore, the unique properties of CRE make a wide range of manipulations possible. For one, work in other organisms has shown that CRE is capable of excision and recombination over huge distances [50,12] or even between different chromosomes [13]. In T. brucei, this approach could be used, for example, to delete large tandem gene arrays.
Another property unique to CRE is that it can catalyze the inversion of a DNA fragment. Mutant loxP elements have been developed to tip the equilibrium and trap the desired inversion [51,52]. It makes one wonder how the active expression site would behave if it were suddenly centromeric [53], or what would happen if an entire cistron between two strand-switch regions were reversed [22]. Moreover, CRE can catalyze the inverse of excision, albeit at a lower frequency, and integrate an episomal or chromosomal target to a loxP element in trans [54–56]. In trypanosomes, this could be an invaluable means of targeting constructs consistently to the same locus in repetitive or scattered arrays, including in the commonly used TUB and rRNA loci.
A site-specific recombinase also opens the possibility of conditional genetic manipulation. This technique is used extensively in mammalian systems, where tissue specific promoters allow spatial and temporal regulation of CRE-mediated recombination [9]. An analogous approach has been employed in Plasmodium berghei, whereby the Flp/FRT site-specific recombination system functions only in the sporozoite life cycle stage [57]. Given the recent work on genetic exchange in T. brucei insect stages [58], such a system could extend our ability to study the trypanosome life cycle, except that few stage-specific promoters or transcription factors have been identified in trypanosomes. Finally, the use of an excisable marker cassette would be highly desirable when epitope-tagging endogenous loci, the main purpose of which is to maintain the native upstream and downstream regulatory regions, which is not possible with existing tagging approaches [59,60].
4.3 Utility of the PYR6-5−/− cell line
For over twenty years, yeast geneticists have used the unique properties of URA3 (orotidine-5′-phosphate decarboxylase; URA4 in Schizosaccharomyces pombe) [24,61,62], which can function as both a positive and negative marker. By targeting one PYR6-5 allele in SM cells, selecting with FOA for cells that had undergone LOH, then eliminating the marker cassette by transient expression of CRE, we were able to remove the drug markers from the PYR6-5−/− cell line. These cells are amenable to a wide range of manipulations, including reintroduction of PYR6-5 as an alternative negative-selectable marker trypanosomes. Indeed, PYR6-5 may be preferable to HSVTK, as the cells are more sensitive to FOA than GCV, when their respective markers are present in the same locus.
LOH remains an understudied but potentially important phenomenon in the Kinetoplastida. A study of gene conversion mechanisms in regard to antigenic variation left many questions unanswered [63]. The two previous studies assaying LOH differ in their findings. LOH occurred in the T. brucei ALG12 locus at a frequency of 0.25% [43]. At the DHFR-TS locus in L. major, LOH ranged widely, from 10−4 to 10−6 [42]. Here, we observed LOH at a frequency of ~10−5. Knowing little about the underlying mechanism, it is difficult to speculate on why the data should be so imprecise. Sequence complexity analysis of the three loci by the method of Troyanskaya et al. [64] hints that the ALG12 locus is slightly more complex, especially in the upstream region, but the differences seem far to small to explain such a difference in LOH frequency. A more systematic look at many loci is required.
4.4 Additional considerations
In addition to the specific constructs described in this paper, we have made a parallel construct (pHD309-LPTL) in which PUR replaces HYG. The current versions of both LHTL and LPTL constructs use a mutated HSVTK that was selected for increased sensitivity to GCV in Escherichia coli and rendered mammalian cells 40-fold more sensitive to GCV [65]. In T. brucei, however, this mutation had no effect on GCV sensitivity when LHTL was expressed from the β-TUB locus in HMI-9 medium from which thymidine was omitted (unpublished data). We have not tested either the wild-type or mutant HSVTK in the ΔPYR6-5 cell line, which could be more sensitive due to the lack of endogenous thymidine synthesis. Finally, these cassettes are being modified to incorporate the most efficient SAS that we know of [31], downstream of the upstream loxP site, to ensure high expression of the fusion protein at any targeted locus. The most current versions of these constructs are available upon request. As succinctly noted elsewhere [66], there are several outstanding issues to be addressed in order to develop an optimized genetic toolkit for T. brucei. We hope the approaches described in this paper will find wide application.
Acknowledgments
This work was financially supported by the National Institutes of Health (AI 64449 and AI 21729) and The Rockefeller University. Irena Pastar was supported by a Charles Revson fellowship. We thank Jenny Li and Magda Kartvelishvili for technical support, Keith Gull, John Donelson and Isabel Roditi for reagents, and our colleagues for helpful suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kwon HJ, Tirumalai R, Landy A, Ellenberger T. Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science. 1997;276:126–31. doi: 10.1126/science.276.5309.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austin S, Ziese M, Sternberg N. A novel role for site-specific recombination in maintenance of bacterial replicons. Cell. 1981;25:729–36. doi: 10.1016/0092-8674(81)90180-x. [DOI] [PubMed] [Google Scholar]
- 3.Sternberg N, Hamilton D, Austin S, Yarmolinsky M, Hoess R. Site-specific recombination and its role in the life cycle of bacteriophage P1. Cold Spring Harb Symp Quant Biol. 1981;45:297–309. doi: 10.1101/sqb.1981.045.01.042. [DOI] [PubMed] [Google Scholar]
- 4.Sternberg N. Bacteriophage P1 site-specific recombination. III. Strand exchange during recombination at lox sites. J Mol Biol. 1981;150:603–8. doi: 10.1016/0022-2836(81)90384-3. [DOI] [PubMed] [Google Scholar]
- 5.Sternberg N, Hamilton D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J Mol Biol. 1981;150:467–86. doi: 10.1016/0022-2836(81)90375-2. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg N, Hamilton D, Hoess R. Bacteriophage P1 site-specific recombination. II. Recombination between loxP and the bacterial chromosome. J Mol Biol. 1981;150:487–507. doi: 10.1016/0022-2836(81)90376-4. [DOI] [PubMed] [Google Scholar]
- 7.Sauer B. Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:2087–96. doi: 10.1128/mcb.7.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods. 1998;14:381–92. doi: 10.1006/meth.1998.0593. [DOI] [PubMed] [Google Scholar]
- 9.Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- 10.Branda CS, Dymecki SM. Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6:7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- 11.Iwaki T, Takegawa K. A set of loxP marker cassettes for Cre-mediated multiple gene disruption in Schizosaccharomyces pombe. Biosci Biotechnol Biochem. 2004;68:545–50. doi: 10.1271/bbb.68.545. [DOI] [PubMed] [Google Scholar]
- 12.Smith AJ, Xian J, Richardson M, Johnstone KA, Rabbitts PH. Cre-loxP chromosome engineering of a targeted deletion in the mouse corresponding to the 3p21.3 region of homozygous loss in human tumours. Oncogene. 2002;21:4521–9. doi: 10.1038/sj.onc.1205530. [DOI] [PubMed] [Google Scholar]
- 13.Smith AJ, De Sousa MA, Kwabi-Addo B, Heppell-Parton A, Impey H, Rabbitts P. A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination. Nat Genet. 1995;9:376–85. doi: 10.1038/ng0495-376. [DOI] [PubMed] [Google Scholar]
- 14.Sauer B, Henderson N. Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol. 1990;2:441–9. [PubMed] [Google Scholar]
- 15.Denise H, Coombs GH, Mottram JC. Generation of Leishmania mutants lacking antibiotic resistance genes using a versatile hit-and-run targeting strategy. FEMS Microbiol Lett. 2004;235:89–94. doi: 10.1016/j.femsle.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Mureev S, Kushnir S, Kolesnikov AA, Breitling R, Alexandrov K. Construction and analysis of Leishmania tarentolae transgenic strains free of selection markers. Mol Biochem Parasitol. 2007;155:71–83. doi: 10.1016/j.molbiopara.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Barrett B, LaCount DJ, Donelson JE. Trypanosoma brucei: a first-generation CRE-loxP site-specific recombination system. Exp Parasitol. 2004;106:37–44. doi: 10.1016/j.exppara.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Loonstra A, Vooijs M, Beverloo HB, Allak BA, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci USA. 2001;98:9209–14. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silver DP, Livingston DM. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol Cell. 2001;8:233–43. doi: 10.1016/s1097-2765(01)00295-7. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt EE, Taylor DS, Prigge JR, Barnett S, Capecchi MR. Illegitimate Cre-dependent chromosome rearrangements in transgenic mouse spermatids. Proc Natl Acad Sci USA. 2000;97:13702–7. doi: 10.1073/pnas.240471297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heidmann D, Lehner CF. Reduction of Cre recombinase toxicity in proliferating Drosophila cells by estrogen-dependent activity regulation. Dev Genes Evol. 2001;211:458–65. doi: 10.1007/s004270100167. [DOI] [PubMed] [Google Scholar]
- 22.Palenchar JB, Bellofatto V. Gene transcription in trypanosomes. Mol Biochem Parasitol. 2006;146:135–41. doi: 10.1016/j.molbiopara.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 23.Hammond DJ, Gutteridge WE. Enzymes of pyrimidine biosynthesis in Trypanosoma cruzi. FEBS Lett. 1980;118:259–62. doi: 10.1016/0014-5793(80)80233-x. [DOI] [PubMed] [Google Scholar]
- 24.Boeke JD, LaCroute F, Fink GR. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet. 1984;197:345–6. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- 25.Bastin P, Bagherzadeh Z, Matthews KR, Gull K. A novel epitope tag system to study protein targeting and organelle biogenesis in Trypanosoma brucei. Mol Biochem Parasitol. 1996;77:235–9. doi: 10.1016/0166-6851(96)02598-4. [DOI] [PubMed] [Google Scholar]
- 26.Wirtz E, Hartmann C, Clayton C. Gene expression mediated by bacteriophage T3 and T7 RNA polymerases in transgenic trypanosomes. Nucleic Acids Res. 1994;22:3887–94. doi: 10.1093/nar/22.19.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirtz E, Leal S, Ochatt C, Cross GAM. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 28.Engstler M, Boshart M. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev. 2004;18:2798–811. doi: 10.1101/gad.323404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furger A, Schurch N, Kurath U, Roditi I. Elements in the 3′ untranslated region of procyclin mRNA regulate expression in insect forms of Trypanosoma brucei by modulating RNA stability and translation. Mol Cell Biol. 1997;17:4372–80. doi: 10.1128/mcb.17.8.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drozdz M, Clayton C. Structure of a regulatory 3′ untranslated region from Trypanosoma brucei. RNA. 1999;5:1632–44. doi: 10.1017/s1355838299990623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siegel TN, Tan KS, Cross GAM. Systematic study of sequence motifs for RNA trans splicing in Trypanosoma brucei. Mol Cell Biol. 2005;25:9586–94. doi: 10.1128/MCB.25.21.9586-9594.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doyle JJ, Hirumi H, Hirumi K, Lupton EN, Cross GAM. Antigenic variation in clones of animal-infective Trypanosoma brucei derived and maintained in vitro. Parasitology. 1980;80:359–69. doi: 10.1017/s0031182000000810. [DOI] [PubMed] [Google Scholar]
- 33.Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989;75:985–9. [PubMed] [Google Scholar]
- 34.Munoz-Jordan JL, Cross GAM, de Lange T, Griffith JD. t-loops at trypanosome telomeres. EMBO J. 2001;20:579–88. doi: 10.1093/emboj/20.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horn D, Cross GAM. Position-dependent and promoter-specific regulation of gene expression in Trypanosoma brucei. EMBO J. 1997;16:7422–31. doi: 10.1093/emboj/16.24.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffiths PD. Progress in the clinical management of herpesvirus infections. Antiviral chemistry & chemotherapy. 1995;6:191–209. [Google Scholar]
- 37.Valdes J, Taylor MC, Cross MA, Ligtenberg MJ, Rudenko G, Borst P. The viral thymidine kinase gene as a tool for the study of mutagenesis in Trypanosoma brucei. Nucleic Acids Res. 1996;24:1809–15. doi: 10.1093/nar/24.10.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hammond DJ, Gutteridge WE. Purine and pyrimidine metabolism in the Trypanosomatidae. Mol Biochem Parasitol. 1984;13:243–61. doi: 10.1016/0166-6851(84)90117-8. [DOI] [PubMed] [Google Scholar]
- 39.Gao G, Nara T, Nakajima-Shimada J, Aoki T. Novel organization and sequences of five genes encoding all six enzymes for de novo pyrimidine biosynthesis in Trypanosoma cruzi. J Mol Biol. 1999;285:149–61. doi: 10.1006/jmbi.1998.2293. [DOI] [PubMed] [Google Scholar]
- 40.Carter NS, Rager N, Ullman B. Purine and pyrimidine transport and metabolism. In: Marr JJ, Nilsen TW, Komuniecki RW, editors. Molecular Medical Parasitology. Academic Press; Amsterdam: 2003. pp. 197–223. [Google Scholar]
- 41.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–48. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gueiros-Filho FJ, Beverley SM. Selection against the dihydrofolate reductase-thymidylate synthase (DHFR-TS) locus as a probe of genetic alterations in Leishmania major. Mol Cell Biol. 1996;16:5655–63. doi: 10.1128/mcb.16.10.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leal S, Acosta-Serrano A, Morris J, Cross GAM. Transposon mutagenesis of Trypanosoma brucei identifies glycosylation mutants resistant to concanavalin A. J Biol Chem. 2004;279:28979–88. doi: 10.1074/jbc.M403479200. [DOI] [PubMed] [Google Scholar]
- 44.ten Asbroek AL, Ouellette M, Borst P. Targeted insertion of the neomycin phosphotransferase gene into the tubulin gene cluster of Trypanosoma brucei. Nature. 1990;348:174–5. doi: 10.1038/348174a0. [DOI] [PubMed] [Google Scholar]
- 45.Lee MG, van der Ploeg LH. The hygromycin B-resistance-encoding gene as a selectable marker for stable transformation of Trypanosoma brucei. Gene. 1991;105:255–7. doi: 10.1016/0378-1119(91)90159-9. [DOI] [PubMed] [Google Scholar]
- 46.Jefferies D, Tebabi P, Le Ray D, Pays E. The ble resistance gene as a new selectable marker for Trypanosoma brucei: fly transmission of stable procyclic transformants to produce antibiotic resistant bloodstream forms. Nucleic Acids Res. 1993;21:191–5. doi: 10.1093/nar/21.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brooks DR, McCulloch R, Coombs GH, Mottram JC. Stable transformation of trypanosomatids through targeted chromosomal integration of the selectable marker gene encoding blasticidin S deaminase. FEMS Microbiol Lett. 2000;186:287–91. doi: 10.1111/j.1574-6968.2000.tb09119.x. [DOI] [PubMed] [Google Scholar]
- 48.Baba Y, Nakano M, Yamada Y, Saito I, Kanegae Y. Practical range of effective dose for Cre recombinase-expressing recombinant adenovirus without cell toxicity in mammalian cells. Microbiol Immunol. 2005;49:559–70. doi: 10.1111/j.1348-0421.2005.tb03753.x. [DOI] [PubMed] [Google Scholar]
- 49.Patnaik PK, Kulkarni SK, Cross GAM. Autonomously replicating single-copy episomes in Trypanosoma brucei show unusual stability. EMBO J. 1993;12:2529–38. doi: 10.1002/j.1460-2075.1993.tb05908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng B, Sage M, Sheppeard EA, Jurecic V, Bradley A. Engineering mouse chromosomes with Cre-loxP: range, efficiency, and somatic applications. Mol Cell Biol. 2000;20:648–55. doi: 10.1128/mcb.20.2.648-655.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Z, Lutz B. Cre recombinase-mediated inversion using lox66 and lox71: method to introduce conditional point mutations into the CREB-binding protein. Nucleic Acids Res. 2002;30:e90. doi: 10.1093/nar/gnf089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oberdoerffer P, Otipoby KL, Maruyama M, Rajewsky K. Unidirectional Cre-mediated genetic inversion in mice using the mutant loxP pair lox66/lox71. Nucleic Acids Res. 2003;31:e140. doi: 10.1093/nar/gng140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horn D, Barry JD. The central roles of telomeres and subtelomeres in antigenic variation in African trypanosomes. Chromosome Res. 2005;13:525–33. doi: 10.1007/s10577-005-0991-8. [DOI] [PubMed] [Google Scholar]
- 54.Araki K, Araki M, Yamamura K. Targeted integration of DNA using mutant lox sites in embryonic stem cells. Nucleic Acids Res. 1997;25:868–72. doi: 10.1093/nar/25.4.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Call LM, Moore CS, Stetten G, Gearhart JD. A cre-lox recombination system for the targeted integration of circular yeast artificial chromosomes into embryonic stem cells. Hum Mol Genet. 2000;9:1745–51. doi: 10.1093/hmg/9.12.1745. [DOI] [PubMed] [Google Scholar]
- 56.Araki K, Araki M, Yamamura K. Site-directed integration of the cre gene mediated by Cre recombinase using a combination of mutant lox sites. Nucleic Acids Res. 2002;30:e103. doi: 10.1093/nar/gnf102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carvalho TG, Thiberge S, Sakamoto H, Menard R. Conditional mutagenesis using site-specific recombination in Plasmodium berghei. Proc Natl Acad Sci U S A. 2004;101:14931–6. doi: 10.1073/pnas.0404416101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peacock L, Ferris V, Bailey M, Gibson W. Dynamics of infection and competition between two strains of Trypanosoma brucei brucei in the tsetse fly observed using fluorescent markers. Kinetoplastid Biol Dis. 2007;6:4. doi: 10.1186/1475-9292-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oberholzer M, Morand S, Kunz S, Seebeck T. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol. 2006;145:117–20. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 60.Kelly S, Reed J, Kramer S, Ellis L, Webb H, Sunter J, Salje J, Marinsek N, Gull K, Wickstead B, Carrington M. Functional genomics in Trypanosoma brucei: a collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol Biochem Parasitol. 2007;154:103–9. doi: 10.1016/j.molbiopara.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boeke JD, Trueheart J, Natsoulis G, Fink GR. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:164–75. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- 62.Grimm C, Kohli J, Murray J, Maundrell K. Genetic engineering of Schizosaccharomyces pombe: a system for gene disruption and replacement using the ura4 gene as a selectable marker. Mol Gen Genet. 1988;215:81–6. doi: 10.1007/BF00331307. [DOI] [PubMed] [Google Scholar]
- 63.Conway C, Proudfoot C, Burton P, Barry JD, McCulloch R. Two pathways of homologous recombination in Trypanosoma brucei. Mol Microbiol. 2002;45:1687–700. doi: 10.1046/j.1365-2958.2002.03122.x. [DOI] [PubMed] [Google Scholar]
- 64.Troyanskaya OG, Arbell O, Koren Y, Landau GM, Bolshoy A. Sequence complexity profiles of prokaryotic genomic sequences: a fast algorithm for calculating linguistic complexity. Bioinformatics. 2002;18:679–88. doi: 10.1093/bioinformatics/18.5.679. [DOI] [PubMed] [Google Scholar]
- 65.Black ME, Newcomb TG, Wilson HM, Loeb LA. Creation of drug-specific Herpes simplex virus type 1 thymidine kinase mutants for gene therapy. Proc Natl Acad Sci USA. 1996;93:3525–9. doi: 10.1073/pnas.93.8.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alsford S, Kawahara T, Glover L, Horn D. Tagging a T .brucei RRNA locus improves stable transfection efficiency and circumvents inducible expression position effects. Mol Biochem Parasitol. 2005;144:142–8. doi: 10.1016/j.molbiopara.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]