

Abstract
In budding yeast, phosphate starvation triggers inhibition of the Pho80-Pho85 cyclin–cyclin-dependent kinase (CDK) complex by the CDK inhibitor Pho81, leading to expression of genes involved in nutrient homeostasis. We isolated myo-d-inositol heptakisphosphate (IP7) as a cellular component that stimulates Pho81-dependent inhibition of Pho80-Pho85. IP7 is necessary for Pho81-dependent inhibition of Pho80-Pho85 in vitro. Moreover, intracellular concentrations of IP7 increased upon phosphate starvation, and yeast mutants defective in IP7 production failed to inhibit Pho80-Pho85 in response to phosphate starvation. These observations reveal regulation of a cyclin-CDK complex by a metabolite and suggest that a complex metabolic network mediates signaling of phosphate availability.
In response to nutrient limitation, cells increase transcription of starvation response genes. Many proteins that function in nutrient response systems have been identified, but the molecular details of their regulation are not well understood (1). The Saccharomyces cerevisiae Pho80-Pho85 cyclin-CDK complex is a key regulator in the phosphate (Pi)–responsive (PHO) signaling pathway (2, 3). When cells are grown in medium that is rich in Pi, Pho80-Pho85 is active and phosphorylates the transcription factor Pho4 (4), resulting in its export from the nucleus (5). Upon Pi starvation, Pho80-Pho85 kinase activity is decreased, leading to nuclear accumulation of Pho4 and transcription of PHO genes. In vivo, the Pho81 CDK inhibitor (CKI) is bound to Pho80-Pho85 regardless of Pi conditions and is required for the inhibition of Pho80-Pho85 activity in response to Pi-limitation (6).
We used a biochemical strategy to identify cellular components that influence Pho80-Pho85-Pho81 activity. The Pho80-Pho85-Pho81 complex immunopurified from cells grown in Pi-rich conditions actively phosphorylated Pho4 in vitro, whereas the complex isolated from Pi-starved cells was less active (6) (Fig. 1A). To identify regulators of Pho80-Pho85-Pho81, we added extracts from cells grown in Pi-rich and Pi-starved conditions to active (from Pi-rich) and inactive (from Pi-starved) immunopurified Pho80-Pho85-Pho81 complexes and performed kinase assays with recombinant Pho4 serving as the substrate. Extracts from Pi-starved cells efficiently inactivated Pho80-Pho85-Pho81, whereas the Pi-rich cell extract had little effect (Fig. 1B). When the extract of Pi-starved cells was fractionated into fractions of large (>3 kD) and small (<3 kD) molecular size, most inhibitory activity was found in the fractions of small molecular size. This activity had little effect on Pho80-Pho85 purified from pho81Δ cells (Fig. 1C), indicating that, as is true in vivo (6), the inhibitory activity requires Pho81 for its function.
Fig. 1.
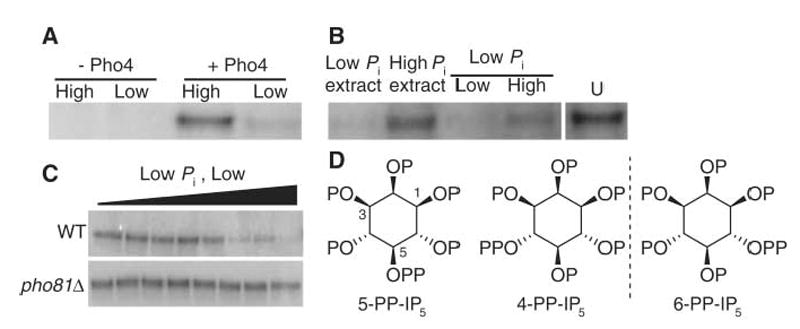
Purification of a cellular component that inactivates Pho80-Pho85 in a Pho81-dependent manner. (A) Phosphorylation of recombinant Pho4 by Pho80-Pho85-Pho81 immunopurified from Pi-rich (High) and Pi-starved (Low) cells expressing Pho80–hemagglutinin tag (3HA). (B) Effects of cell fractions on Pho80-Pho85 activity. Extracts from cells (3 × 109 cells per ml extract) grown in high-Pi medium (High Pi extract) or starved for phosphate (Low Pi extract) were added either directly or after filtration (molecular weight cut-off 3 kD; Low and High indicate low– and high–molecular weight fractions, respectively) to Pho80-Pho85-Pho81 immunopurified from Pi-rich cells, and kinase assays were done as in (A). U, untreated Pho80-Pho85-Pho81 from the same gel. (C) The low-Pi low–molecular weight fraction in (B) was serially diluted and added to Pho80-Pho85 immunopurified from wild-type (WT) and pho81Δ cells that had been grown in Pi-rich medium. Kinase assays were then performed using recombinant Pho4 as a substrate. (D) Structures of relevant IP7 isomers. 4-PP-IP5 and 6-PP-IP5 are mirror images of each other. P, phosphate; PP, pyrophosphate.
To determine the identity of the inhibitory activity, we enriched it through several fractionation steps and characterized its properties (fig. S1) (7). The low–molecular weight inhibitor contained acid-labile Pi (8), and characterization by nuclear magnetic resonance and mass spectrometry suggested that it was inositol heptakisphosphate (9), also known as diphospho-myo-d-inositol pentakisphosphate, PP-InsP5, PP-IP5, InsP7, or IP7 (Fig. 1D).
Inositol polyphosphates are signaling molecules found in all eukaryotic cells. They are produced through the action of a conserved set of inositol polyphosphate kinases from inositol 1,4,5-trisphosphate (IP3) (9). IP3, which regulates Ca2+ release from the endoplasmic reticulum (10), can be sequentially phosphorylated to generate IP4, IP5, inositol hexakisphosphate (IP6), and inositol pyrophosphates such as IP7. Loss-of-function studies have implicated IP7 in recombination (11), vacuole function (12), gene expression, endo- and exocytosis (13, 14), osmotic stress (12), and telomere length control (15, 16), but it is unclear whether IP7 directly regulates these processes. One well-characterized role for IP7 is the regulation of chemotaxis in Dictyostelium, in which intracellular concentrations of IP7 increase upon chemoattractant addition and IP7 competes with phosphatidylinositol 3,4,5-trisphosphate (PIP3) to bind to proteins containing a pleckstrin homology domain (17).
To test whether the inhibitory activity was IP7, we synthesized IP7 by a nonenzymatic method (8) and tested its effect on the Pho80-Pho85 complex. When the Pho80-Pho85-Pho81 complex that had been immunopurified from Pi-rich cells was treated with synthetic IP7, kinase activity was inhibited with an apparent median inhibitory concentration (IC50) of ~50 μM (Fig.2A). IP7 had little or no effect on the kinase purified from pho81Δ cells, suggesting that IP7-mediated inactivation of Pho80-Pho85 requires Pho81. Inactivation of Pho80-Pho85-Pho81 by IP7 appears to be specific, given that inositol and several inositol derivatives did not affect kinase activity (Fig. 2B).
Fig. 2.

Inhibition of Pho80-Pho85-Pho81 by synthetic IP7. (A) Synthetic IP7 was serially diluted and added to immunopurified Pho80-Pho85 isolated from wild-type (WT) or pho81Δ cells that had been grown in Pi-rich medium (Fig. 1C). Kinase activity was then measured. (B) Serial dilutions of inositol, myo-d-inositol 1,4,5-triphosphate (IP3), IP6, or IP7 were added to Pho80-Pho85-Pho81 immunopurified from wild-type cells grown in Pi-rich medium, and kinase activity was measured. (C) Synthetic IP7 was added to recombinant Pho80-Pho85 or Pho80-Pho85–Pho81-MD complex (50 nM Pho80-Pho85 and 250 nM Pho81-MD) and incubated 20 min at room temperature. Kinase activity was then measured.
We next tested whether IP7-mediated inactivation of Pho80-Pho85-Pho81 is direct or whether it requires other factors from yeast. Neither synthetic IP7 nor the activity from Pi-starved cell extract could inactivate recombinant Pho80-Pho85 purified from Escherichia coli (Fig. 2C). Similarly, addition of recombinant Pho81 minimum domain (Pho81-MD) (18), a fragment that functionally substitutes for full-length Pho81 in vivo, did not cause inhibition of recombinant Pho80-Pho85 in vitro, even in two- to threefold molar excess (Fig. 2C). Because Pho81-MD binds to Pho80-Pho85 (apparent dissociation constant Kd ~ 170 nM, fig. S2), we concluded that neither Pho81-MD nor IP7 is sufficient for the inactivation of Pho80-Pho85. However, when both IP7 and Pho81-MD were added, Pho80-Pho85–Pho81-MD was inactivated (Fig. 2C). Therefore, IP7 directly inactivates Pho80-Pho85-Pho81 and this inactivation requires Pho81.
To test whether the cellular concentration of IP7 increases when cells are starved of Pi, we monitored inositol polyphosphate levels in cells labeled with [3H]-inositol and grown in Pi-rich or Pi-limiting conditions (12). In Pi-rich conditions, the IP7 concentration was approximately 3% or less of the IP6 concentration (Fig. 3) (12). However, upon Pi limitation, IP7 levels increased to more than 20% of the levels of IP6 (Fig. 3). This increase in abundance of IP7 is not due to Pho4-dependent transcriptional activation (fig. S3) (7), and the time course and dependence of the IP7 increase on extracellular Pi are comparable to those observed for PHO pathway regulation (fig. S3). Assuming that the IP6 concentration is ~100 μM in cells (9) and that the IP6 concentration does not change in response to Pi limitation, the concentration of IP7 may reach ~30 μM during Pi limitation (fig. S3), a value similar to the apparent IC50 measured in vitro (Fig. 2).
Fig. 3.
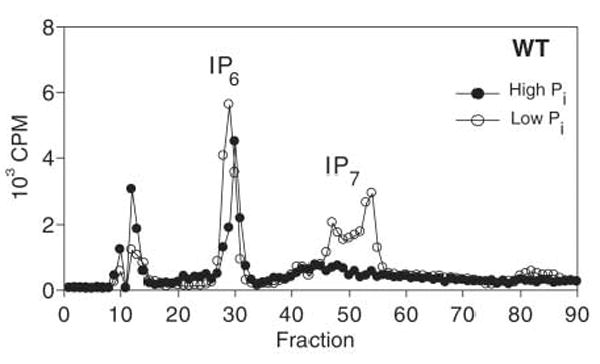
Increase in IP7 upon Pi starvation. Strong anion exchange chromatograms of extracts derived from wild-type (WT) cells labeled with [3H]-inositol grown in medium containing high Pi (10 mM) or low Pi (5 μM). Cells were starved for 2 hours at 30°C before lysis and analysis. CPM, counts per minute.
Many gene products involved in yeast inositol polyphosphate metabolism are known (Fig. 4A) (9). Kcs1 is a homolog of IP6 kinases in other organisms (19) and is a major IP6 kinase in yeast (12). However, IP7 is still observable in kcs1Δ cells (16, 20), suggesting that another IP6 kinase exists. A second yeast IP6 kinase, Vip1, generates an isomer of IP7 distinct from that produced by the mammalian IP6 kinase and likely distinct from that produced by Kcs1 (21). To determine which IP6 kinase is involved in the PHO pathway, we assayed the ability of cellular extracts from Pi-starved wild-type, kcs1Δ, and vip1Δ cells to inactivate Pho80-Pho85-Pho81 in vitro (Fig. 4B). The extract from kcs1Δ cells inactivated Pho80-Pho85 in a Pho81-dependent manner, whereas extract from vip1Δ cells did not, suggesting that Vip1 mediates synthesis of IP7 relevant to the PHO system.
Fig. 4.
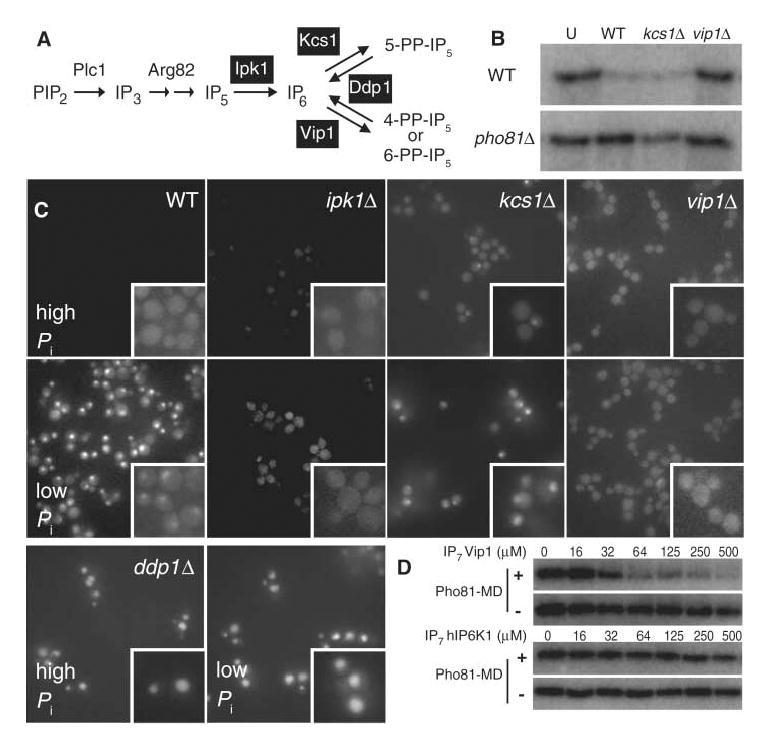
Effect of disrupted inositol polyphosphate metabolism on Pho80-Pho85 activity, monitored by Pho4 localization. (A) Schematic diagram showing inositol polyphosphate metabolism in S. cerevisiae. (B) Wild-type (WT), kcs1Δ, or vip1Δ cell extracts from Pi-starved cells were added to immunopurified Pho80-Pho85 from Pi-rich wild-type (top) or pho81Δ (bottom) cells, and kinase activity was measured. U, untreated. (C) Fluorescence microscopic (100× magnification) analysis of wild-type, ipk1Δ, kcs1Δ, vip1Δ, or ddp1Δ cells expressing Pho4–green fluorescent protein, grown in medium containing high (top) or low (bottom) concentrations of Pi. (D) Isomer specificity of the IP7-mediated Pho80-Pho85 inactivation. Pho80-Pho85 (8 nM) in the presence (top) and the absence (bottom) of His6–Pho81-MD (250 nM) was incubated with IP7 isomers synthesized with recombinant Vip1 (top) and human IP6 kinase 1 (bottom), and kinase activity was measured.
To further assess the physiological role of Vip1-generated IP7 in Pi signaling in vivo, we determined whether yeast strains with mutations in enzymes involved in inositol polyphosphate metabolism (Fig. 4A) (9) had defects in the regulation of Pho80-Pho85-Pho81. We monitored the activity of Pho80-Pho85 by measuring the subcellular localization of its substrate, Pho4 (Fig. 4C). In the ipk1Δ and vip1Δ strains, Pho4 was localized to the cytoplasm even in conditions of Pi starvation, suggesting that the kinase cannot be inactivated. In the kcs1Δ strain, Pho4 became localized to the nucleus upon Pi starvation, consistent with the model that Kcs1 does not generate the IP7 that inhibits Pho80-Pho85-Pho81. A fraction of the kcs1Δ cells grown in high-Pi medium exhibited nuclear localization of Pho4; the cause of this phenotype is not understood (22, 23). Pho4 was constitutively nuclear in ddp1Δ cells (16), which have high levels of IP7, but was constitutively cytoplasmic in kcs1Δvip1Δddp1Δ and arg82Δ cells, which cannot produce IP7 and IP5, respectively (fig. S4A). Pho4 was constitutively nuclear in a vip1Δpho80Δ strain and was constitutively cytoplasmic in ddp1Δpho81Δ cells, suggesting that IP7 acts upstream of Pho80-Pho85-Pho81 (fig. S4B). Additionally, IP7 enzymatically synthesized by recombinant Vip1 inhibited Pho80-Pho85–Pho81-MD in a manner dependent on Pho81-MD, but IP7 enzymatically synthesized by the human homolog of Kcs1, hIP6K1, did not (Fig. 4D; apparent IC50 ~ 50 μM; fig. S4C) (21).
We conclude that IP7 produced by Vip1 acts as a signaling molecule that controls the yeast PHO signaling pathway: IP7 concentrations increase in response to Pi limitation, and IP7 is required for inhibition of Pho80-Pho85-Pho81 in vivo and in vitro. This work illuminates the molecular mechanisms underlying previously reported genetic interactions between inositol polyphosphate metabolism and the phosphate-responsive signaling pathway (22, 24). Our work reveals regulation of a cyclin-CDK-CDK inhibitor complex by a small molecule, IP7. It is unclear whether IP7 acts by allosterically regulating Pho80-Pho85-Pho81 or by covalently modifying one or more of these components, as it has been demonstrated to do for other yeast and mammalian proteins (25). IP7 may be a more general signal for nutrient limitation given that Pi starvation of budding yeast and nutrient limitation of Dictyostelium (17) leads to elevation of IP7 levels. In addition, considering that a mammalian IP6 kinase is known to stimulate Pi uptake (26), a phenotype similar to Pi-starved yeast cells, it is possible that connections between phosphate metabolism and inositol polyphosphate are conserved in other organisms.
Supplementary Material
www.sciencemag.org/cgi/content/full/316/582/109/DC1
Materials and Methods
SOM Text
Figs. S1 to S5
Tables S1 and S2
References
References and Notes
- 1.Wilson WA, Roach PJ. Cell. 2002;111:155. doi: 10.1016/s0092-8674(02)01043-7. [DOI] [PubMed] [Google Scholar]
- 2.Lenburg ME, O’Shea EK. Trends Biochem Sci. 1996;21:383. [PubMed] [Google Scholar]
- 3.Carroll AS, O’Shea EK. Trends Biochem Sci. 2002;27:87. doi: 10.1016/s0968-0004(01)02040-0. [DOI] [PubMed] [Google Scholar]
- 4.Kaffman A, Herskowitz I, Tjian R, O’Shea EK. Science. 1994;263:1153. doi: 10.1126/science.8108735. [DOI] [PubMed] [Google Scholar]
- 5.O’Neill EM, Kaffman A, Jolly ER, O’Shea EK. Science. 1996;271:209. doi: 10.1126/science.271.5246.209. [DOI] [PubMed] [Google Scholar]
- 6.Schneider KR, Smith RL, O’Shea EK. Science. 1994;266:122. doi: 10.1126/science.7939631. [DOI] [PubMed] [Google Scholar]
- 7.Materials and methods are available as supporting material on Science Online.
- 8.Mayr GW, Radenberg T, Thiel U, Vogel G, Stephens LR. Carbohydr Res. 1992;234:247. [Google Scholar]
- 9.Irvine RF, Schell MJ. Nat Rev Mol Cell Biol. 2001;2:327. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- 10.Streb H, Irvine RF, Berridge MJ, Schulz I. Nature. 1983;306:67. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- 11.Luo HR, et al. Biochemistry. 2002;41:2509. doi: 10.1021/bi0118153. [DOI] [PubMed] [Google Scholar]
- 12.Dubois E, et al. J Biol Chem. 2002;277:23755. doi: 10.1074/jbc.M202206200. [DOI] [PubMed] [Google Scholar]
- 13.Ye W, Ali N, Bembenek ME, Shears SB, Lafer EM. J Biol Chem. 1995;270:1564. [PubMed] [Google Scholar]
- 14.Fleischer B, et al. J Biol Chem. 1994;269:17826. [PubMed] [Google Scholar]
- 15.Saiardi A, Resnick AC, Snowman AM, Wendland B, Snyder SH. Proc Natl Acad Sci USA. 2005;102:1911. doi: 10.1073/pnas.0409322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.York SJ, Armbruster BN, Greenwell P, Petes TD, York JD. J Biol Chem. 2005;280:4264. doi: 10.1074/jbc.M412070200. [DOI] [PubMed] [Google Scholar]
- 17.Luo HR, et al. Cell. 2003;114:559. doi: 10.1016/s0092-8674(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 18.Huang S, Jeffery DA, Anthony MD, O’Shea EK. Mol Cell Biol. 2001;21:6695. doi: 10.1128/MCB.21.19.6695-6705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Curr Biol. 1999;9:1323. doi: 10.1016/s0960-9822(00)80055-x. [DOI] [PubMed] [Google Scholar]
- 20.Seeds AM, Bastidas RJ, York JD. J Biol Chem. 2005;280:27654. doi: 10.1074/jbc.M505089200. [DOI] [PubMed] [Google Scholar]
- 21.Mulugu S, et al. Science. 2007;316:106. doi: 10.1126/science.1139099. [DOI] [PubMed] [Google Scholar]
- 22.Auesukaree C, Tochio H, Shirakawa M, Kaneko Y, Harashima S. J Biol Chem. 2005;280:25127. doi: 10.1074/jbc.M414579200. [DOI] [PubMed] [Google Scholar]
- 23.Huang S, O’Shea EK. Genetics. 2005;169:1859. doi: 10.1534/genetics.104.038695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flick JS, Thorner J. Genetics. 1998;148:33. doi: 10.1093/genetics/148.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH. Science. 2004;306:2101. doi: 10.1126/science.1103344. [DOI] [PubMed] [Google Scholar]
- 26.Schell MJ, et al. FEBS Lett. 1999;461:169. doi: 10.1016/s0014-5793(99)01462-3. [DOI] [PubMed] [Google Scholar]
- 27.We thank Y. Liu for preparation of strains; P. Fridy for reagents; B. Stern, H. Kim, C. Leimkuhler, and D. Schwarz for comments on the manuscript; members of the J.D.Y. laboratory for unpublished data, helpful discussions, and comments on the manuscript; and D. Kahne and his laboratory members for access to equipment. This work was supported by NIH R01 GM051377 (E.K.O.), DK070272 (J.D.Y.), and HL055672 (J.D.Y.); the David and Lucile Packard Foundation (E.K.O.); and the Howard Hughes Medical Institute (E.K.O. and J.D.Y.). The authors have no conflicting financial interest.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencemag.org/cgi/content/full/316/582/109/DC1
Materials and Methods
SOM Text
Figs. S1 to S5
Tables S1 and S2
References