

Abstract
The clinical picture of experimental autoimmune encephalomyelitis (EAE) is critically dependent on the nature of the target autoantigen and the genetic background of the experimental animals. Potentially lethal EAE is mediated by myelin basic protein (MBP)–specific T cells in Lewis rats, whereas transfer of S100β- or myelin oligodendrocyte glycoprotein (MOG)–specific T cells causes intense inflammatory response in the central nervous system (CNS) with minimal disease. However, in Dark Agouti rats, the pathogenicity of MOG-specific T cells resembles the one of MBP-specific T cells in the Lewis rat. Using retrovirally transduced green fluorescent T cells, we now report that differential disease activity reflects different levels of autoreactive effector T cell activation in their target tissue. Irrespective of their pathogenicity, the migratory activity, gene expression patterns, and immigration of green fluorescent protein+ T cells into the CNS were similar. However, exclusively highly pathogenic T cells were significantly reactivated within the CNS. Without local effector T cell activation, production of monocyte chemoattractants was insufficient to initiate and propagate a full inflammatory response. Low-level reactivation of weakly pathogenic T cells was not due to anergy because these cells could be activated by specific antigen in situ as well as after isolation ex vivo.
Keywords: autoimmunity of the CNS, disease model, retroviral gene transfer, reactivation in the CNS, multiple sclerosis
Introduction
Autoimmune responses in the central nervous system (CNS) are notoriously unpredictable in their course and development of neurological deficits. In multiple sclerosis (MS), for example, myelin-directed autoimmunity may produce a disease that rapidly progresses to cause severe disability; in other patients, the course is relapsing–remitting, whereas in other cases the disease remains mild and self-contained.
A similar diversity of clinical outcomes can be observed in variants of experimental autoimmune encephalomyelitis (EAE) animal models, which represent inflammatory aspects of MS. EAE can be induced by a broad spectrum of T cells that react against a diversity of CNS antigens. But the resulting neurological disease can be radically different in character, depending on the genetics of the experimental animals and on the specific antigen used for induction (1). The mechanisms determining these different disease manifestations are still largely unknown. They could include reduced capacity of encephalitogenic T cells to infiltrate into the CNS, different migration patterns within the target organ, partial tolerization events due to autoantigen expression outside of the CNS, and intrinsic properties of the T cells themselves (e.g., different cytokine pattern).
In this paper, we made use of T cell lines (TCLs) known to transfer either strong or mild monophasic clinical EAE. In Lewis rats, the adoptive transfer of activated myelin basic protein (MBP)–specific T cells (TMBP) triggers a severe, potentially lethal neurological disease associated with the invasion of the CNS by T cells and large numbers of activated macrophages (2). In this model, the neurological deficit is mediated by soluble factors produced by activated macrophages (3). In contrast, in the same strain of rats, T cells specific for either myelin oligodendrocyte glycoprotein (MOG) (TMOG) or the astrocyte protein S100β (TS100β) fail to induce severe clinical disease despite inducing an intense inflammatory response in the CNS (4–6). However, in the context of a different genotype, in the Dark Agouti (DA) rat, MOG-specific T cells can induce a severe MBP-like EAE (6). Disease activity in these EAE models is strictly controlled by the pathogenic T cells and is reflected by inflammation rather than demyelination, thus representing the earliest inflammatory events in MS (4–6). Antimyelin autoantibodies are not generated after T cell transfer alone. Immunopathological studies revealed that the inability of TMOG and TS100β cells to induce disease in Lewis rats was associated with a failure to recruit activated macrophages into the CNS, but the molecular/cellular mechanisms involved are unknown (4–6).
Using retrovirally engineered T cells that express the gene of green fluorescent protein (GFP), we investigated the behavior of these differently pathogenic TCLs in the course of adoptive transfer EAE (tEAE). We report that reactivation of autoreactive T cells in the target organ crucially determines the onset and severity of clinical autoimmune disease. Highly pathogenic T cells are intensely reactivated after entry into the CNS, as demonstrated by changes in their cell surface phenotype and up-regulation of proinflammatory cytokines. In contrast, weakly pathogenic cells undergo only partial activation in the target organ and fail to up-regulate the expression of proinflammatory cytokines, although they infiltrate the CNS at equivalent numbers.
Materials and Methods
Animals and Antigens.
Lewis rats were obtained from the animal facility of the Max-Planck Institute for Biochemistry, and DA rats were obtained from Charles River Breeding Laboratories. The animals were kept under standardized conditions at constant temperature (22°C), controlled lighting, and free access to food and water. The animals were used at 6–8 wk of age. MBP was purified from guinea pig brains as described previously (7). Bovine S100β protein was purchased from Sigma-Aldrich. Recombinant MOG (amino acids 1–120) was produced as described previously (8). Mycobacterial purified protein derivative (PPD) was purchased from the State Serum Institute. All breeding and experiments were performed in accordance with the guidelines of the committee on animals of the Max-Planck Institute for Neurobiology.
Generation and Characterization of T Cells.
Generation of GFP-transduced T cells was performed as reported previously (9). As retroviral vector, we used the Moloney leukemia virus derivative pLXSN (11) in which the enhanced green fluorescent protein gene cDNA (CLONTECH Laboratories, Inc.) had been integrated (pLGFPSN). Packaging cell lines producing GFP-carrying retrovirus were established by transferring the pLGFPSN construct into the GP+E 86 cells (10). T lymphocyte blasts were expanded in IL-2–containing growth medium after 3 d of coculture. Selection with G418 and amplification of the T cells was performed as described previously (2, 10).
Induction of tEAE and Animal Preparation.
Adoptive transfer of the encephalitogenic TCLs was performed by intraperitoneal injection. The dose of T cells injected was adjusted to 5 × 106–107 T cell blasts/animal. Animals were monitored daily by measuring weight and examining disease scores (Table I).
Table I.
Characterization of Antigen-specific TGFP Cell Lines
|
|
TMBP-GFP cells
|
TS100β-GFP cells
|
TDA-MOG-GFP cells
|
TLE-MOG-GFP cells
|
|---|---|---|---|---|
| Phenotype | CD4+ (>95%) | CD4+ (>95%) | CD4+ (>95%) | CD4+ (>95%) |
| Cytokine release a | ||||
| IFNγ (ng/ml) | 63 | 97 | 228 | 74 |
| TNFα (ng/ml) | 20 | 8 | 23 | 14 |
| IL-4 (ng/ml) | 0 | 0 | 0 | 0 |
| IL-10 (ng/ml) | 21 | 14 | 18 | 14 |
| MCP-1 (ng/ml) | 6 | 6 | 5 | 8 |
| MIP-1α (ng/ml) | 0.9 | 0.8 | 1.7 | 1.6 |
| IFNγ-ELISPOTb | ||||
| Blast T cells (SD) | 282 (52) | 1,767 (83) | n.d. | n.d. |
| Resting T cells (SD) | 1.75 (1.5) | 5 (2.5) | n.d. | n.d. |
| Specificityc | ||||
| Specific Ag: cpm (SD) | 2,223 (229) | 8,942 (393) | 12,529 (234) | 10,852 (465) |
| Control Ag: cpm (SD) | 112 (51) | 53 (75) | 636 (120) | 339 (54) |
TGFP cell lines were CD4+, Th-1–type cells, highly specific for their respective antigen.
Cytokine release was measured in 24-h supernatant of T cell blasts. Cell concentration: 1 × 106 cells/ml; the SD of the duplicate measurements never exceeded 20%.
Specific dots per 100,000 cells are shown.
Cpm incorporated with specific antigen versus cpm incorporated with control antigen (PPD) are shown. n.d., not done.
Immunohistochemistry.
Tissue preparation for histology and immunohistochemical staining using mouse monoclonal antibodies directed against ED1 (Camon) and W3/13 (Becton Dickinson) were performed as described previously (12). For immunohistochemical quantification, spinal cords were analyzed 4 (MBP-, DA-MOG-EAE) or 5 d (S100β-, LE-MOG-EAE) after transfer. After paraformaldehyde fixation, the tissues were treated as described previously (13). Immunostained T cells (W3/13) and monocytes/macrophages (ED1) were quantified on three randomly selected complete spinal cord cross sections from the lower thoracic level. The section area was determined using a morphometrical grid. The values represent the mean ± SD of at least two individual animals/group.
Cell Isolation, Cytofluorometry, and FACS®.
Single cell suspensions from organs were obtained as described previously (9, 12, 14). Cytofluorometric analysis and cell sorting were performed as described previously using FACSort™ operated by CELL- Quest™ software (Becton Dickinson; references 9, 12). The following monoclonal antibodies were used for surface membrane analysis: W3/25 (CD4; Serotec), R73 (αβTCR), OX-6 (rat MHC class II), OX-40 antigen (CD134), and OX-39 (CD25, IL-2Rα chain) (all obtained from Becton Dickinson).
ELISAs.
The cytokine profile of TGFP cell lines was controlled by IFNγ, IL-10, IL-2, TNFα (Biosource International), and MCP-1 (Becton Dickinson) ELISAs using the recommended protocols. Mip-1α ELISA was performed using a polyclonal rabbit anti–Mip-1α (Biosource International) and biotin-conjugated anti–Mip-1α (Serotec) antibody combination and evaluated by streptavidin–peroxidase conjugate and orthophenylen diamine as substrate (Sigma-Aldrich). As standard, we used recombinant rat Mip-1α (Biosource International).
Intracellular Staining for IFNγ and IFNγ ELISPOT Assay.
Intracellular IFNγ staining was performed with anti–mouse/rat IFNγ antibody (clone DB-1; Becton Dickinson). Control IgG (mouse IgG MOPC31) was purchased from Sigma-Aldrich. Staining was performed in 96-round-well plates. After ex vivo isolation, the cells were incubated for 5 h with 1 μM monensin in supplemented DMEM 1% rat serum (no additional growth factors). After centrifugation (300 g for 10 min at 4°C), the cells were washed with PBS and fixed with 2% paraformaldehyde for 20 min. After washing in PBS, they were permeabilized using a commercially available permeabilization buffer (Becton Dickinson) and incubated with primary antibody for 30 min at 4°C (dilution, 1:100). After washing twice in permeabilization buffer, secondary RPE-Cy5–labeled goat anti–mouse antiserum (dilution, 1:25) was added to the cells for 30 min at 4°C. Stimulation of cells with PMA/ionomycin (5 μg/ml and 1 μM, respectively; Sigma-Aldrich) was performed for 3 h in supplemented DMEM containing 10% horse serum followed by a 3-h incubation with 1 μM monensin (Biosource International).
ELISPOT analysis was performed according to commercially available protocols using polyclonal goat anti–rat-IFNγ and biotinylated goat anti–rat-IFNγ antiserum (R&D Systems). The ELISPOT assays were analyzed with an automated imaging system and appropriate computer software (KS ELISPOT automated image analysis system; Carl Zeiss MicroImaging, Inc.). The frequency of cytokine-producing cells was expressed as the difference between the mean number of spots and the mean background for each experiment. A value equal to zero was assigned to spot frequencies smaller than the mean background of the individual assay plus a standard deviation of two. All standard deviations were <20% of the mean.
Quantitative PCR.
Ex vivo–isolated TMBP-GFP and TS100β-GFP cells were centrifuged (300 g for 10 min at 4°C) and immediately shock frozen. Spinal cord homogenates were frozen 4 and 8 h after intrathecal S100β/MOG injections in TRI reagent (Sigma-Aldrich). mRNA extraction and cDNA preparation was performed using standard protocols (Sigma-Aldrich). Taqman analysis was performed as reported using sequence detector “Taqman” (ABI Prim 7700; Applied Biosystems; reference 12). For quantification of cytokine mRNAs, the expression of a housekeeping gene (β-actin) was set in relation to the cytokine mRNA. The analyzed mRNA was extracted from >0.5 × 106 ex vivo–sorted TMBP-GFP or TS100β-GFP cells that had been isolated and pooled from organs of three animals. Representative data of at least two independent experiments are shown. All PCR data were obtained by two independent measurements. The cycle threshold value of the measurements did not differ >0.5 amplification cycles.
Restimulation of Ex Vivo–isolated TGFP Cells.
TGFP cells sorted from spleens 4 d after transfer. (5 × 104 cells/well) were cultured without growth factors (DMEM 1% rat serum) in 96-well plates together with irradiated thymocytes (5,000 rad, 106 cells/well) in the presence of no antigen, 2.5 μg/ml Con A, 10 μg/ml of specific antigen (MBP, S100β, and MOG), or 10 μg/ml of irrelevant antigen PPD, respectively. At the same time, parallel held TGFP cell cultures were restimulated using identical conditions. Proliferation was evaluated measuring [3H]dT (2 Ci/mmol; Amersham Biosciences) incorporation (9).
Intrathecal Soluble Antigen Injection.
Intrathecal injection was performed 4 (S100β EAE) or 5 d (LE-MOG EAE) after T cell transfer. Under narcosis using 300 mg/kg chloralhydrate (Merck), the antigens (20 μg S100β, MOG, or OVA, respectively) were stereotactically injected into the cisterna magna. Spinal cords and spleens were harvested 4 h after intrathecal injection, and TGFP cells were analyzed cytofluorometrically for the expression of activation markers as aforementioned. For histology, spinal cords were prepared 24 h after injection. Subsequent tissue preparation and antibody staining was performed as aforementioned.
RT/PCR Dot-blot Hydridization Analysis of Chemokine mRNA Expression in CNS Tissues.
RT-PCR dot-blot hydridization (15–17) was performed to quantitate MCP-1 and MIP-1α mRNA levels before and at the beginning of clinical EAE induced by TS100β and TMBP cells. The assays were performed as reported previously (18). The results of initial experiments were confirmed by analysis of a second set of samples obtained from rats that received T cell transfers on a separate occasion. Data shown were derived from the initial experiment.
Online Supplemental Material.
Fig. S1 shows the “migratory phenotype” of the used GFP+ TCLs, which is characterized by down-regulation of activation markers IL-2R and OX-40 antigen and up-regulation of MHC class II. Intrathecal injection of soluble antigen locally induced mRNA of proinflammatory cytokines (IFNγ and IL-2) and monocyte chemoattractants (MCP-1 and Mip-1α; Fig. S2 A), followed by increased recruitment of monocytes/macrophages into the CNS (Fig. S2 B). Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20031064/DC1.
Results
Retroviral Transduction with GFP Does Not Modify the Functional Phenotype of Neuroantigen-specific TCLs.
MBP-, S100β-, and MOG-specific T cells from Lewis rats (TMBP-GFP, TS100β-GFP, and TLE-MOG-GFP cells) and MOG-specific T cells from DA rats (TDA-MOG-GFP cells) were retrovirally engineered to express GFP as genetic marker (9). After one to two rounds of restimulation in vitro and expansion in the presence of G418, these GFP-expressing TCLs were exclusively specific for their selecting antigen and were defined as members of the CD4+ Th-1 T cell subset by their cell surface phenotype and cytokine profile (Table I) . Like their nonmanipulated counterparts, TMBP-GFP cells and TDA-MOG-GFP cells transferred severe EAE to syngeneic naive recipients, whereas TS100β-GFP cells and TLE-MOG-GFP cells were only weakly pathogenic (Table II).
Table II.
TGFP Cell–induced EAE
|
|
TMBP-GFP
cells |
TS100β-GFP
cells |
TDA-MOG–GFP
cells |
TLE-MOG-GFP
cells |
|---|---|---|---|---|
| Injected T cells | 5 × 106 | 107 | 5 × 106 | 107 |
| Disease course | ||||
| Onset (d) | 3 | 3 | 3 | 4 |
| Maximum (d) | 6 | 6 | 5 | 6 |
| Duration (d) | 8 | 8 | 8 | 6 |
| Disease severity | ||||
| Maximal score (SD) | 2.7 (0.6) | 0.6 (0.5) | 3.7 (0.5) | 0.3 (0.5) |
| % Weight loss (SD) | 16 (3) | 8 (4) | 19 (3) | 9 (4) |
| Incidence of clinical disease | 52/52 | 21/45 | 32/32 | 6/41 |
| Histological analysis | ||||
| W3/13+ cells (SD) | 427 (271) | 147 (57) | 250 (134) | 664 (439) |
| ED1+ cells (SD) | 275 (137) | 19 (26) | 285 (91) | 83 (50) |
| ED1/W3/13 ratio | 1/1.6 | 1/7.9 | 1/0.9 | 1/8 |
Disease score: 0, no disease; 1, flaccid tail; 2, gait disturbance; 3, complete hind limb paralysis; 4, tetraparesis; and 5, death. (Disease course) Time period of weight loss and/or clinical paresis. (Disease severity, Maximal score) A representative EAE experiment involving at least five animals/group is indicated. (Weight loss) In percent of the body weight before tEAE induction. (Incidence of clinical disease) Animals with pareses/animals with pure weight loss. (Histological analysis) Numbers of W3/13+, and ED1+ cells in spinal cord lesions (perivascular and parenchymal) per 1 mm2.
Neuroantigen-specific T Cells Accumulate in the CNS Irrespective of Their Ability to Induce Clinical Disease.
TMBP-GFP cells acquire a migratory phenotype in the periphery before invading the CNS of Lewis rats (12). This phenotypic change is characterized by the down-regulation of the activation markers CD25 and OX-40, and the passive adsorption of MHC class II protein (12). This phenotypic change was seen in all neuroantigen-specific T cells transferred, irrespective of their antigen-specificity and pathogenic potential. TMBP-GFP, TS100β-GFP, TLE-GFP, and TDA-MOG-GFP cells showed similar patterns of CD25, OX-40, and MHC class II expression after a sojourn of 84 h in the spleens of syngeneic recipients (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20031064/DC1) (12).
FACS® analysis further revealed that large numbers of these GFP-labeled T cells invaded the CNS irrespective of their autoantigen specificity (Fig. 1) . 4 d after the transfer of TMBP-GFP, TS100β-GFP, TLE-MOG-GFP, or TDA-MOG-GFP effector T cells, the majority (75–94%) of CD4+ T cells infiltrating the CNS expressed GFP (Fig. 1). In all cases, this very high proportion of GFP-expressing effector T cells initially present in the infiltrates decreased to <10% within the following 72 h (unpublished data).
Figure 1.
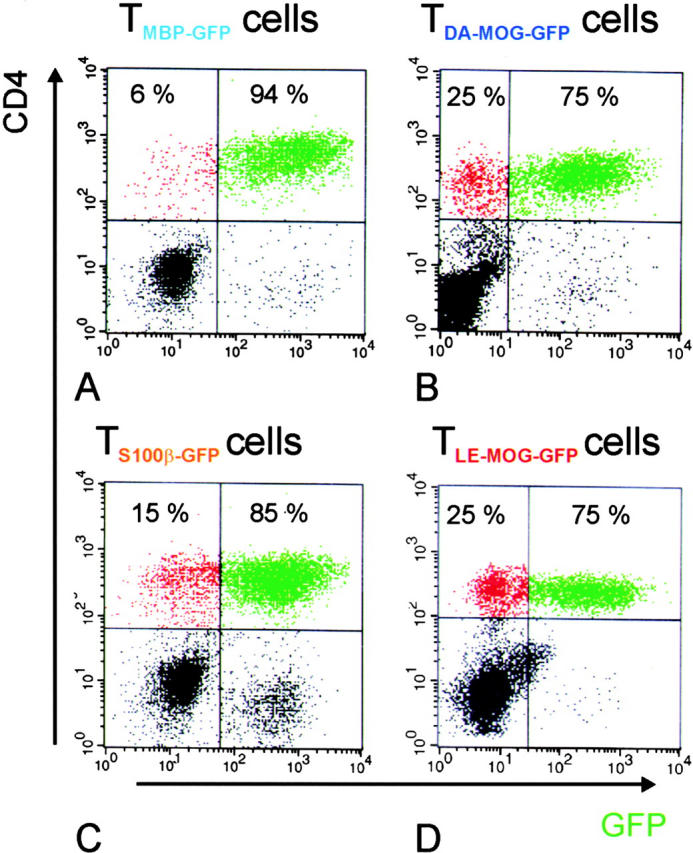
Relative numbers of TGFP cells infiltrating the CNS in the course of tEAE. The number of CNS-infiltrating GFP-positive T cells in the CD4+ cell population was analyzed 4 d after transfer. TMBP-GFP (A), TDA-MOG-GFP (B), TS100β-GFP (C), and TLE-MOG-GFP (D) cells are shown. The majority of CD4+ T cells were GFP positive in all cell lines tested. Representative data of at least three independent experiments/TCLs are shown.
Invasion of GFP-expressing effector T cells into the CNS was verified by confocal immunofluorescence microscopy. Beyond confirming the FACS® data, morphology showed similar migration patterns for all types of neuroantigen-specific GFP+ cells. The majority of cells did not remain in the perivascular space, but penetrated deeply into the parenchyma (Fig. 2 and Table II). Therefore, the inability of TS100β-GFP and TMOG-GFP cells to induce a significant neurological deficit in the Lewis rat cannot be attributed to a failure to cross the blood brain barrier (BBB) and infiltrate into the target tissue.
Figure 2.

Infiltration of W3/13+ and ED1+ cells in the course of tEAE induced by TMBP-GFP, TS100β-GFP, TDA-MOG-GFP, and TLE-MOG-GFP cells. Lesions in the lumbosacral spinal cord 4 d after transfer of TMBP-GFP (A and E), TS100β-GFP (B and F), TDA-MOG-GFP (C and G), and TLE-MOG-GFP (D and H) cells mediated by tEAE are shown. TGFP cells (green), immunohistochemical staining with T cell marker W3/13 (A–D, red), and monocyte/macrophage marker ED1 (E–H, red). (Arrowheads) Location of TGFP cells (A–D) and monocytes/macrophages (E–H). Magnification bars, 10 μm. TGFP cells of all specificities infiltrate the CNS in high numbers at the onset of clinical symptoms. They can be found as well in the meningeal and perivascular areas as deep as the CNS parenchyma (A–H). ED-1–positive macrophages in TMBP-GFP and TDA-MOG-GFP cell–tEAE enter the CNS in high numbers where they are distributed throughout the tissue (E and G). In contrast, TS100β-GFP and TLE-MOG-GFP cells recruit less ED1+ monocytes/macrophages, which are mainly restricted to the meningeal and perivascular areas (F and H). Representative histological sections of lumbo-sacral spinal cords of at least two independent experiments/TCLs are shown.
Activation and Proinflammatory Cytokine Expression by Effector T Cells Is a Prerequisite for Disease Induction.
Upon invasion of the CNS, encephalitogenic Lewis TMBP-GFP effector cells underwent reactivation as indicated by increased levels of OX-40 antigen and IL-2R (Fig. 3) and partial down-modulation of the CD3–TCR complex on their surface (not depicted; reference 12). Similar changes were also seen in highly pathogenic TDA-MOG-GFP cells when analyzed isolated from the CNS shortly after onset of EAE 4 d after transfer (Fig. 3). In striking contrast, the weakly pathogenic TS100β-GFP and TLE-MOG-GFP cells that infiltrated the CNS in equally high numbers (Figs. 1 and 2) did not show significant up-regulation of IL-2R and OX-40 antigen (Fig. 3), down-modulation of the CD3–TCR complex (not depicted), nor did they induce clinical disease (Table II).
Figure 3.

Weakly pathogenic TGFP cells show minimal reactivation after infiltrating the target organ. 84 h after transfer of TMBP-GFP, TDA-MOG-GFP, TS100β-GFP, and TLE-MOG-GFP cells, the spinal cords and spleens of recipient rats were prepared. TGFP cells were analyzed cytofluorometrically for the expression of the surface membrane molecules OX-40 antigen (OX-40), IL-2R, and CD4. (IgG) Isotype control. Shaded histograms represent spleen-derived TGFP cells, and unshaded overlay histograms show TGFP cells isolated simultaneously from the CNS. Spinal cord–derived TMBP-GFP cells (blue histograms) and TDA-MOG-GFP cells (black histograms), but not TS100β-GFP cells (red histograms) and TLE-MOG-GFP cells (yellow histograms), up-regulate OX-40 antigen and IL-2R. Representative data of at least four independent experiments/TCLs are shown.
The activation status of TMBP-GFP and TS100β-GFP cells recovered from the CNS was investigated further by real-time RT-PCR. Activation of rat CD4+ Th-1 TCLs in vitro leads to the rapid up-regulation of cytokines (IL-2, IFNγ, and IL-10) and membrane proteins (OX-40 antigen and IL-2R). We compared the mRNA transcription of IL-2, IFNγ, IL-10, and IL-2R between TMBP-GFP and TS100β-GFP cells cytofluorometrically sorted from either spleen or CNS 4 d after transfer, and the same TCLs maintained in parallel in IL-2–containing cultures (Fig. 4) .
Figure 4.

IFNγ, IL-10, and IL-2R mRNA of TGFP cells within the CNS. mRNAs of TMBP-GFP and TS100β-GFP cells from spleens (SPL) and spinal cords (CNS) 84 h after transfer and from parallel cultures (CUL) were quantitatively analyzed for the expression of IL-2, IFNγ, IL-10, and IL-2R. TMBP-GFP cells (blue columns), but not TS100β-GFP cells (orange columns), up-regulated the expression of IFNγ, IL-10, and IL-2R upon infiltration into the spinal cord. Specific copies of mRNA in relation to the housekeeping β-actin mRNA are shown. Ex vivo TGFP cells were obtained from three animals and measured in two independent quantitative PCR reactions.
Upon entry into the CNS, the pathogenic TMBP-GFP cells markedly increased IL-2, IL-2R, IFNγ, and IL-10 mRNA transcription relative to TMBP-GFP cells isolated from the periphery. In contrast, in nonpathogenic TS100β-GFP effector T cells isolated from CNS infiltrates, we detected a significant increase in the expression of mRNA transcripts only in the case of IL-2, but not IFNγ, IL-10, or IL-2R (Fig. 4).
We compared the expression of IFNγ protein in TMBP-GFP and TS100β-GFP cells on a single cell level. FACS® analysis of TMBP-GFP cells recovered from the CNS revealed that 15% of the cells stained positive for intracellular IFNγ. In contrast, brain-infiltrating TS100β-GFP effector T cells did not express IFNγ in measurable amounts, although they clearly did so after stimulation with PMA/ionomycin (Fig. 5) . ELISPOT analysis of TMBP-GFP and TS100β-GFP cells during the 24-h period directly after isolation from spleen and CNS confirmed the distinct expression patterns of IFNγ by TMBP-GFP cells on protein secretion levels, and again showed that brain-infiltrating TS100β-GFP effector T cells failed to release IFNγ in measurable amounts (Fig. 5 I).
Figure 5.

IFNγ production of TGFP cells within the CNS. Intracellular staining for IFNγ of (A) TMBP-GFP cells isolated from spleens (blue shaded histogram) and spinal cords (unshaded red overlaid histogram) and (E) TS100β-GFP cells isolated from spleens (orange shaded histogram) and spinal cords (unshaded blue overlaid histogram) 4 d after transfer. For control, resting (shaded histograms) and blast T cells (unshaded overlaid histograms, 24 h after restimulation with specific antigen) of (B) TMBP-GFP cells and (F) TS100β-GFP cells were stained. Both TMBP-GFP cells (42%) and TS100β-GFP cells (54%) up-regulated IFNγ expression in vitro upon stimulation with specific antigen (B and F). However, exclusively TMBP-GFP cells produced IFNγ upon infiltration into the spinal cord (A, 15%). (C, D, G, and H) TMBP-GFP and TS100β-GFP cells isolated from spleens (C, TMBP-GFP cells, 53%; G, TS100β-GFP cells, 72%) and spinal cords (D, TMBP-GFP cells, 43%; H, TS100β-GFP cells, 63%) could be stimulated to produce IFNγ in the presence of PMA/ionomycin. (Shaded histograms) Nonstimulated ex vivo TGFP cells. (Overlaid open histograms) PMA/ionomycin-stimulated ex vivo TGFP cells. (I) (IFNγ–ELISPOT) Spontaneous IFNγ release of TMBP-GFP cells (MBP) and TS100β-GFP cells (S100β) isolated from spleens (blue bars) and spinal cords (pink bars) was determined after 24 h of culture. The values represent means of quadruplicate measurements involving four (MBP-tEAE) and nine (S100β-tEAE) animals, respectively. IFNγ-positive dots per 100,000 sorted cells were determined. Both TMBP-GFP cells and TS100β-GFP cells isolated from spleens and spinal cords could be stimulated to produce IFNγ in the presence of PMA/ionomycin (not depicted).
Neuroantigen-specific T Cells Are Not Anergized.
The failure of the CNS to activate infiltrating S100β and MOG-specific T cells in the Lewis rat could be explained if T cells with these specificities were rendered functionally unresponsive. Therefore, we isolated TS100β−GFP, TMBP-GFP, TLE-MOG-GFP, and TDA-MOG-GFP cells from the spleen 4 d after transfer and immediately confronted them with their cognate antigen in vitro (Table III). Irrespective of their antigen specificity or ability to induce clinical EAE, all four TCLs retained their ability to proliferate in response to their cognate antigen in vitro (Table III). Furthermore, intracellular IFNγ staining revealed that TS100β−GFP and TLE-MOG-GFP cells isolated from spleen and CNS can be induced to express IFNγ after stimulation with PMA/ionomycin in vitro (Fig. 5, G and H, and Fig. 6 D). Therefore, the inability of certain T cell specificities to induce clinical EAE in the Lewis rat cannot be attributed to anergy.
Table III.
Ex Vivo–isolated TGFP Cells Are Not Anergic
| Proliferation | TMBP-GFP cells | TS100β-GFP cells | TDA-MOG-GFP cells | TLE-MOG-GFP cells |
|---|---|---|---|---|
| Specific Ag: cpm (SD) | 628 (43) | 8,358 (320) | 11,574 (672) | 9,166 (465) |
| Control Ag: cpm (SD) | 26 (8) | 10 (7) | 181 (238) | 10 (2) |
Proliferation occurred when TGFP cells were isolated from spleens (n = 3) 4 d after transfer and exposed to specific/control antigen (PPD) in the presence of irradiated thymic APCs. [3H]thymidine incorporation was determined 72 h after culturing began. Triplicate values ± SD are shown.
Figure 6.

Intrathecal injection of antigen activates weakly pathogenic TGFP cells and aggravates clinical disease. Intrathecal injection of 20 μg of specific antigen (A, S100β; B, MOG, closed squares and black bars) or 20 μg of control antigen (OVA, open triangles) was performed at day 4 (A, S100β-EAE) or day 5 (B, LE-MOG-EAE) after T cell transfer. Control animals, which did not receive intrathecal injection, are shown (black circles). Weight loss was monitored daily, at day 4 (A) or 5 (B) in closer time intervals (4/5a: 1 h before; 4/5b: 4 h after; and 4/5c: 8 h after intrathecal S100β/MOG injection). Animals that had received 20 μg of specific antigen intrathecally showed enhanced weight loss starting 8 h after antigen injection. Animals of TLE-MOG-GFP cell–induced EAE developed clinical symptoms reaching maximal scores of three (B, black bars). Mean value and standard deviation of five independent experiments (S100β) and three independent experiments (MOG) are shown, including eight animals/treatment group. (C) TS100β-GFP (orange histograms) or TLE-MOG-GFP cells (yellow histograms) were isolated from spinal cords of control antigen (shaded histograms, 20 μg OVA) or specific antigen (overlay histograms, 20 μg S100β or MOG, respectively)–treated animals 4 h after intrathecal injection, and were analyzed cytofluorometrically for the expression of IL-2R and OX-40 antigen (OX-40). TGFP cells from specific antigen-treated but not OVA-treated animals up-regulated OX-40 antigen and IL-2R. (D) Intracellular IFNγ staining of TS100β-GFP (orange histograms) or TLE-MOG-GFP cells (yellow histograms) after intrathecal treatment with control antigen (shaded histograms, 20 μg OVA) or specific antigen (overlay histograms, 20 μg S100β or MOG, respectively). 4 h after intrathecal antigen injection TGFP cells in the CNS (top left histogram, TS100β-GFP cells, 24% of cells were IFNγ+; middle left histogram, TLE-MOG-GFP cells, 10% of cells were IFNγ+) but not in the spleen (top right histogram, TS100β-GFP cells, 2% of cells were IFNγ+; middle right histogram, TLE-MOG-GFP cells, 1% of cells were IFNγ+) up-regulated IFNγ production. Upon stimulation with PMA/ionomycin (unshaded overlay histograms), a high percentage of TLE-MOG-GFP cells isolated from CNS (bottom left histogram), and spleen (bottom right histogram) produced IFNγ. Representative results of two independent sets of experiments/antigen are shown.
Activation of CNS-infiltrated TGFP Cells by Intrathecal Antigen Injection Aggravates EAE.
To test whether enhanced activation of weakly pathogenic TS100β-GFP or TLE-MOG-GFP cells within the CNS increases their pathogenicity, we injected soluble S100β or MOG antigen, respectively. Intrathecal injection of 20 μg of specific antigen, but not of control antigen ovalbumin, clearly exacerbated clinical disease; S100β animals showed a marked increase in weight loss (Fig. 6 A) and LE-MOG animals additionally developed pareses (Fig. 6 B). A significant number of TS100β-GFP and TLE-MOG-GFP cells isolated from spinal cords 4 h after S100β/MOG injection up-regulated OX-40 antigen, IL-2R (Fig. 6 C), and produced IFNγ (Fig. 6 D). These activation-related changes were restricted to TGFP cells within the CNS (Fig. 6 D). Quantitative mRNA analyses from spinal cords treated with specific antigen confirmed a strong up-regulation of IFNγ and IL-2R within 4 h after intrathecal antigen injections (Fig. S2 A, available http://www.jem.org/cgi/content/full/jem.20031064/DC1).
Partial T Cell Activation Is Insufficient to Induce the Expression of MCP-1 and MIP-1α and Fails to Recruit Macrophages into the CNS.
The neurological deficits in EAE seem to be caused by activated macrophages recruited to the CNS (19, 20). Indeed, CNS infiltrates induced by highly encephalitogenic TMBP-GFP and TDA-MOG-GFP cells contained large numbers of activated ED1+ macrophages, whereas these cells were rare in the CNS infiltrates induced by weakly pathogenic TS100β-GFP and TLE-MOG-GFP cells (Fig. 2 and Table II). Therefore, we compared the levels of macrophage-attracting CC chemokines MCP-1 and MIP-1α (21, 22) with both types of lesion. Quantification of MCP-1 and MIP-1α mRNA transcripts by RT-PCR in Lewis rats injected with either TMBP or TS100β cells revealed that, in animals injected with TMBP cells, MCP-1 and MIP-1α transcripts increased dramatically by day 4 coincident with the onset of clinical EAE (Fig. 7) . In contrast, in rats injected with S100β-specific T cells, there was no corresponding increase in the signals for MCP-1 and MIP-1α, which remained ∼10-fold lower than in animals with MBP-induced EAE (Fig. 7). Therefore, partial activation of T cells within the CNS can trigger the sustained recruitment of T cells into the lesions, but it is insufficient to activate MCP-1 and MIP-1α expression, which mediate the recruitment of macrophages into the CNS. Intrathecal injection of specific antigen induced a strong increase of MCP-1 and MIP-1α mRNA within the CNS (Fig. S2 A) followed by concomitant recruitment of ED1+ monocytes/macrophages into the CNS (Fig. S2 B) and aggravation of clinical disease (Fig. 6, A and B).
Figure 7.
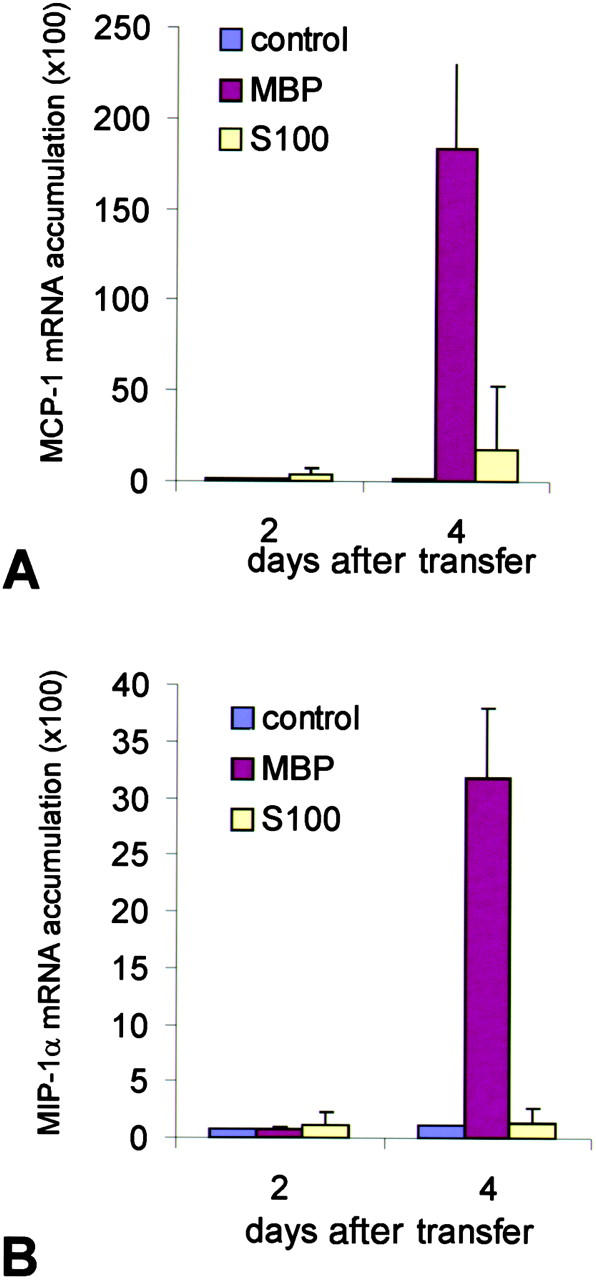
Quantitative chemokine mRNA expression in the CNS. The relative amount of mRNA transcripts for (A) MCP-1 and (B) MIP-1α was analyzed from spinal cord tissue of MBP-EAE (MBP), S100β-EAE (S100), and control animals (control) by dot blot hybridization at days 2 and 4 after transfer. Note the massive up-regulation of MCP-1 and MIP-1α mRNA in the CNS of TMBP cell–treated, but not in the TS100β cell–treated, animals. Amplification of β-tubulin and analysis on ethidium bromide–stained agarose gels demonstrated intact RNA in all samples (not depicted). The data were confirmed in a second set of independently prepared samples.
Discussion
We recently used retrovirally engineered encephalitogenic T cells to trace the fate of autoimmune effector T cells before, during, and beyond clinical EAE (9, 12). These studies were performed in the classical Lewis rat EAE model of adoptively transferred MBP-specific T cells, which produce an acute, potentially lethal clinical course. Here, we used the same methodological approach to learn why CD4+ T cells specific for alternative brain autoantigens, or from different rat strains, cause autoimmune responses of strikingly different character. T cells specific for the astrocyte-derived S100β antigen, for example, transfer mild clinical EAE, yet they produce massive inflammatory infiltrates in the recipient CNS (22). A similar pattern is seen with MOG-specific T cells in the Lewis rat (5), whereas, in contrast, in DA rats, MOG-specific T cell–mediated EAE is as violent as MBP-induced EAE in the Lewis strain (23).
We compared the behavior of both groups of encephalitogenic T cells on several levels. We analyzed their in vitro responsiveness, their migratory behavior, and functional state upon transfer in vivo; we also measured their capacity to infiltrate the target CNS tissues and, finally, local reactivation within the CNS. The TCLs were all members of the same subset of CD4+, CD8−, α/βTCR Th-1 T cells. Irrespective of their neuroantigen specificity, all four TCLs produced similar profiles and quantities of Th-1–associated cytokines in response to antigen presented by professional APCs in vitro. Differences in antigen specificity neither influenced phenotypic T cell maturation in vivo nor the kinetics of antigen-specific effector T cell recruitment into the CNS. All the GFP-expressing T cells assumed the same migratory phenotype in vivo, in particular down-regulation of activation markers and proinflammatory mediators (12).
The undistinguishable behavior of highly versus weakly autoaggressive T cells before migration into the CNS may not be unexpected because a similar behavior has been observed also in T cells specific for a foreign antigen, such as ovalbumin (12). In fact, it appears that the prodromal phase likely excludes contacts with the specific target autoantigen. It was more surprising that each type of brain-specific T cells invaded the brain with a very similar time course and intensity. In all models, invasion of the CNS by migratory autoimmune T cells always started abruptly (∼3–4 d p.i.), and also in all models, during the initial phases of invasion, brain-specific T cells formed the majority of all CD4+-infiltrating T cells, though with rapid decrease of this high proportion. In contrast, migratory T cells specific for a foreign antigen (e.g., ovalbumin) failed to accumulate in the CNS (12). Irrespective of their pathogenic potential, the majority of neuroantigen-specific TGFP cells migrated deeply into the CNS parenchyma (Table II). These data are in contrast with earlier studies of a chronic relapsing mouse model, which located brain-specific T cells in the perivascular area (24).
Migratory encephalitogenic T cells readily crossed the BBB, provided it had been activated during the three to four prodromal days preceding EAE, but they are excluded by a resting BBB (12). The processes involved in BBB activation are still unknown. It has been speculated (25, 26) that a few highly activated T cells, which enter the CNS within the first few hours after transfer, act on the CNS milieu by secreting proinflammatory cytokines. Such activation would enable glial cells to process and present protein antigen, and render BBB endothelium permissive for invasion by migratory autoimmune T cells. Unexpectedly, our results suggest that, irrespective of their pathogenic potential, all CNS autoimmune T cells affect the BBB in a similar fashion.
However, the response patterns of highly and weakly encephalitogenic T cells within the CNS tissues were profoundly full of discrepancy. TMBP-GFP and TDA-MOG-GFP cells, which transfer severe clinical EAE to syngeneic hosts, up-regulated two classical activation markers, OX-40 antigen and IL-2R (Fig. 3), and they turned on production of cytokines (Fig. 4, IFNγ, IL-10, and IL-2). These functional changes closely resemble antigen-dependent T cell activation in vitro. This and the finding that CNS-infiltrating activated effector T cells show partly down-modulated TCR–CD3 complexes (12) are compatible with antigen-dependent activation (27). In striking contrast, weakly encephalitogenic effector T cells (TLE-MOG-GFP or TS100β-GFP cells) isolated from inflammatory infiltrates in the Lewis rat were neither demonstrably reactivated (the only notable change was some increased production of IL-2) (Fig. 4), nor did they show signs of TCR modulation (not depicted).
Antigen presentation by CNS cells could have led to disparate effects, activation in the case of highly encephalitogenic T cells, and anergy in the case of the weakly encephalitogenic T cells. Antigen-dependent T cell activation is critically determined by the circumstances of antigen presentation within the immune synapse (28). Availability and affinity of peptide/MHC surface to TCR, the presence of accessory molecules, and the local cytokine milieu all influence the level of T cell activation, and, thus, determine the character of the subsequent immune response. Depending on the relative strength of the signal, the responding T cell unfolds its repertoire of gene expression in a graded fashion, ranging from complete activation with proliferation and secretion of a full mediator spectrum to the other extreme, a state of anergy (29).
These general principles govern immune reactions within and outside of the CNS, but note that immune reactivity in the CNS adheres to additional rules. The resting CNS tissues constitute a milieu hostile to immune responses. Production of MHC determinants, costimulatory factors, cell adhesion molecules, proinflammatory mediators, and other structures required for productive immune responses are normally suppressed in the healthy CNS (30). Neurons play a pivotal role in this down-regulation (31). However, suppression of immune genes can be overcome, either by application of overwhelming proinflammatory stimuli or by compromised neuronal function (30).
Thus, within the CNS, several diverse factors may modulate antigen presentation. One of these relates to the activation of locally available APCs. For example, astrocytes are inducible to express MHC class II determinants, but they do not normally produce costimulatory molecules in levels required for full T cell activation. Presentation of autoantigen by partly activated astrocytes may suppress T cell activation (32), induce anergy (33), or may divert T cells to the Th2 activation pathway (34). Indeed, studies involving transgenic mice carrying brain-reactive T cells reported on tolerance induction of the transgenic T cell population within the CNS (35). However, in our model, there was no evidence of anergy induction or immune deviation. When isolated from CNS infiltrates, the weakly pathogenic T cells responded with cytokine production (Fig. 5) and full proliferation (not depicted) upon exposure with specific antigen.
Because availability of autoantigen for antigen presentation within the target organ may influence the intensity of T cell responses in the CNS, we locally supplied soluble S100β or MOG protein to spinal cords infiltrated by TS100β-GFP or TLE-MOG-GFP cells, respectively. Within hours, the resting infiltrate T cells became activated, followed by up-regulation of proinflammatory cytokines (Fig. 6 and Fig. S2 A) and monocyte chemoattractants (Fig. S2 A). At present, it is not clear why intrathecal antigen injection aggravated clinical disease in LE-MOG-EAE, but much less in S100β-EAE. It remains to be shown whether activation-induced cell death (AICD) of T cells (36, 37) is involved after activation through soluble antigen. This mechanism is known to accelerate naturally occurring cell death of effector T cells within the CNS (38–40). Higher susceptibility of TS100β-GFP cells toward AICD might explain the transient nature of the clinical effects after S100β injection (Fig. 6 A). A less plausible explanation would involve different types of antigen-presenting cells in S100β- versus MOG-EAE.
Peripheral antigen expression might modulate the reactivation capacity of encephalitogenic T cells when they enter the CNS. It should be noted that S100β differs significantly from MBP in terms of tissue distribution and physiochemical properties. MBP and S100β are both major components of the CNS, where they are produced by oligodendrocytes and astrocytes, respectively. Additionally, both proteins can be detected in immune organs where they may modulate the composition and functional activity of their respective T cell repertoires. In this respect, S100β is ubiquitously expressed throughout the body and can be detected in many different tissues and cell types (41–48), suggesting that the S100β repertoire may be exposed to a higher level of tolerogenic signaling. Therefore, the inability of active immunization or the adoptive transfer of TS100β cells to initiate classical EAE could be mediated by tolerogenic signals in the periphery. However, the only available evidence for stimulation of S100β cells in the periphery are slight increases in the expression of IL-2 and IFNγ mRNA transcripts within the spleen (Fig. 4). This does not result in classical anergy because TS100β-GFP cells recovered from the spleen still produce IFNγ (Fig. 5) and proliferate in response to S100β in vitro (Table III).
Unequal availability or distribution of autoantigen does not explain why MOG-specific T cells are highly encephalitogenic in DA rats, but only weakly so in the Lewis strain. There is evidence that genetic factors codetermine the result of antigen-dependent T cell activation in the CNS. Indeed, studies of MOG-specific T cells from Lewis rats congenic for diverse MHC haplotypes indicate that genes located within the MHC determine the level of pathogenicity of these myelin autoimmune T cells (reference 49 and unpublished data). Highly pathogenic MOG-specific T cell responses were obtained in LEW.1A rats that express the same class II (RT1.B/Da) MHC haplotype as DA rats. In contrast, MOG-specific TCLs derived from LEW.1N (RT1.B/Dn) and LEW.1W (RT1.B/Du) were only weakly pathogenic, reproducing the results obtained using Lewis MOG-specific TCLs. We are currently investigating the role of the MHC–peptide–TCR complex interactions in controlling the level of T cell activation in the CNS in more detail in these models.
The intensity of early T cell activation within the CNS milieu profoundly affects the cellular composition of the inflammatory infiltrate. As described previously, EAE models with severe and mild clinical disease strikingly differ by the frequency of activated macrophages within the CNS infiltrates (50). Activated macrophages are the crucial effector cell population in EAE; selective depletion and inactivation of macrophages profoundly suppresses clinical disease, but leaves the T cell component of the inflammatory infiltrate relatively intact (51, 52). Several observations suggest that the CC chemokines MCP-1 and MIP-1α play a crucial role in mediating macrophage recruitment into the CNS in EAE (53). There is a positive correlation between the temporal expression of MCP-1 and MIP-1α in the CNS and the inflammatory response in animals with EAE (21). Moreover, antibodies directed against these chemokines suppress disease in adoptive transfer and active EAE (54, 55). MCP-1 and MIP-1α are both expressed in similar levels by our TGFP cell lines (Table I). Experiments using transgenic mice deficient for MCP-1 revealed that, whereas mutant MCP-1−/−–derived T cells induced disease in wild-type recipients, the converse was not true, indicating that the MCP-1 necessary for disease induction is not produced by the effector T cells, but rather by other cells within the CNS parenchyma (56). Other works support this view (16, 57).
Our finding that weakly pathogenic TLE-MOG-GFP and TS100β-GFP cells fail to induce strong MCP-1 and MIP-1α responses in the CNS correlates well with the low numbers of activated macrophages recruited into the CNS and may be due to the failure of the T cells to express IFNγ and TNFα in the CNS. These proinflammatory cytokines exert pleiotropic effects in EAE and can act on endothelial cells, microglial cells, and astrocytes. Critically important, they are both strong inducers of MCP-1 and MIP-1α and induce chemokine expression in a wide variety of cell types (58–59).
Our present work identifies the level of T cell activation within the CNS as a critical checkpoint that determines whether or not T cell infiltration initiates macrophage recruitment and subsequently clinical disease in EAE. This regulatory cascade may be extended to other tissue-specific autoimmune diseases. Indeed, a similar checkpoint has been postulated in the NOD mouse diabetes model that determine progression from the infiltration of the pancreas by T cells to the actual destruction of the islet cells and onset of clinical diabetes (60). Our results imply that the level of activation reached by an infiltrating effector T cell within the target organ determines the detrimental character of the inflammatory infiltrate. This critical level of activation can be influenced by the nature of the target autoantigen (e.g., MBP vs. S100β in the Lewis rat) as well as genetic factors as demonstrated by the differential pathogenicity of the MOG-specific T cell response in Lewis and DA rats. Understanding these mechanisms may lead to novel therapeutic strategies that selectively disable the signaling pathways that lead to clinical autoaggression, while leaving intact a benign T cell infiltrate that may actually promote regeneration and tissue repair (61).
Acknowledgments
The authors want to thank S. Bauer, P. Grämmel and I. Haarmann for excellent technical assistance and Dr. J. Chalcroft for his help with the artwork.
We are grateful to Drs. K. Dornmair and E. Meinl for critical reading. This work was supported by the Deutsche Forschungsgemeinschaft (SFB455) and the European Community (Mechanisms of Brain Inflammation: QLG3-CT-2002-00712).
The online version of this article includes supplemental material.
The present address of S. Lassmann is the Institute of Pathology, University of Freiburg, 79104 Freiburg, Germany.
The present address of C. Linington is the Dept. of Medicine and Therapeutics, Institute of Medical Sciences, University of Aberdeen, Aberdeen AB25 2ZD, UK.
Abbreviations used in this paper: BBB, blood brain barrier; CNS, central nervous system; DA, Dark Agouti; EAE, experimental autoimmune encephalomyelitis; GFP, green fluorescent protein; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; PPD, purified protein derivative; TCL, T cell line; tEAE, adoptive transfer EAE.
References
- 1.Wekerle, H., K. Kojima, J. Lannes-Vieira, H. Lassmann, and C. Linington. 1994. Animal models. Ann. Neurol. 36:S47–S53. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Nun, A., H. Wekerle, and I.R. Cohen. 1981. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur. J. Immunol. 11:195–199. [DOI] [PubMed] [Google Scholar]
- 3.Benveniste, E.N. 1997. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J. Mol. Med. 75:165–173. [DOI] [PubMed] [Google Scholar]
- 4.Kojima, K., H. Wekerle, H. Lassmann, T. Berger, and C. Linington. 1997. Induction of experimental autoimmune encephalomyelitis by CD4+ T cells specific for an astrocyte protein, S100β. J. Neural Transm. 49(Suppl.):43–51. [DOI] [PubMed] [Google Scholar]
- 5.Linington, C., T. Berger, L. Perry, S. Weerth, D. Hinze-Selch, Y. Zhang, H.-C. Lu, H. Lassmann, and H. Wekerle. 1993. T cells specific for the myelin oligodendrocyte glycoprotein (MOG) mediate an unusual autoimmune inflammatory response in the central nervous system. Eur. J. Immunol. 23:1364–1372. [DOI] [PubMed] [Google Scholar]
- 6.Storch, M.K., A. Stefferl, U. Brehm, R. Weissert, E. Wallström, M. Kerschensteiner, T. Olsson, C. Linington, and H. Lassmann. 1998. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 8:681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eylar, E.H., P.J. Kniskern, and J.J. Jackson. 1974. Myelin basic protein. Methods Enzymol. 32:323–341. [PubMed] [Google Scholar]
- 8.Adelmann, M., J. Wood, I. Benzel, P. Fiori, H. Lassmann, J.-M. Matthieu, M.V. Gardinier, K. Dornmair, and C. Linington. 1995. The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J. Neuroimmunol. 63:17–27. [DOI] [PubMed] [Google Scholar]
- 9.Flügel, A., M. Willem, T. Berkowicz, and H. Wekerle. 1999. Gene transfer into CD4+ T lymphocytes: Green fluorescent protein engineered, encephalitogenic T cells used to illuminate immune responses in the brain. Nature Med. 5:843–847. [DOI] [PubMed] [Google Scholar]
- 10.Markowitz, D., S. Goff, and A. Bank. 1988. A safe packaging line for gene transfer: Separating viral genes on two different plasmids. J. Virol. 62:1120–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller, A.D., and G. Rosman. 1989. Improved retroviral vectors for gene transfer and expression. Biotechniques. 7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 12.Flügel, A., T. Berkowicz, T. Ritter, M. Labeur, D. Jenne, Z. Li, J. Ellwart, M. Willem, H. Lassmann, and H. Wekerle. 2001. Migratory activity and functional changes of green fluorescent effector T cells before and during experimental autoimmune encephalomyelitis. Immunity. 14:547–560. [DOI] [PubMed] [Google Scholar]
- 13.Vass, K., H. Lassmann, H. Wekerle, and H.M. Wisniewski. 1986. The distribution of Ia antigen in the lesions of rat acute experimental allergic encephalomyelitis. Acta. Neuropathol. 70:149–160. [DOI] [PubMed] [Google Scholar]
- 14.Cohen, J.A., D.M. Essayan, B. Zweiman, and R.P. Lisak. 1987. Limiting dilution analysis of the frequency of antigen-reactive lymphocytes isolated from the central nervous system of Lewis rats with experimental allergic encephalomyelitis. Cell. Immunol. 108:203–213. [DOI] [PubMed] [Google Scholar]
- 15.Glabinski, A.R., M. Tani, R.M. Strieter, V.K. Tuohy, and R.M. Ransohoff. 1997. Synchronous synthesis of α- and β-chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic autoimmune encephalomyelitis. Am. J. Pathol. 150:617–30. [PMC free article] [PubMed] [Google Scholar]
- 16.Glabinski, A.R., V. Balasingam, M. Tani, S.L. Kunkel, R.M. Strieter, V.W. Yong, and R.M. Ransohoff. 1996. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J. Immunol. 156:4363–4368. [PubMed] [Google Scholar]
- 17.Tani, M., A.R. Glabinski, V.K. Tuohy, M.H. Stoler, M.L. Estes, and R.M. Ransohoff. 1996. In situ hybridization analysis of glial fibrillary acidic protein mRNA reveals evidence of biphasic astrocyte activation during acute experimental autoimmune encephalomyelitis. Am. J. Pathol. 148:889–896. [PMC free article] [PubMed] [Google Scholar]
- 18.McTigue, D.M., M. Tani, K. Krivacic, A. Chernovsky, G.S. Kelner, D. Maciejewski, R. Maki, R.M. Ransohoff, and B.T. Stokes. 1998. Selective chemokine mRNA accumulation in the rat spinal cord after contusion injury. J. Neurosci. Res. 53:368–376. [DOI] [PubMed] [Google Scholar]
- 19.Brosnan, C.F., M.B. Bornstein, and B.R. Bloom. 1981. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. J. Immunol. 126:614–620. [PubMed] [Google Scholar]
- 20.Huitinga, I., N. Van Rooijen, C.J.A. De Groot, B.M.J. Uitdehaag, and C.D. Dijkstra. 1990. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J. Exp. Med. 172:1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ransohoff, R.M. 1999. Mechanisms of inflammation in MS tissue: adhesion molecules and chemokines. J. Neuroimmunol. 98:57–68. [DOI] [PubMed] [Google Scholar]
- 22.Kojima, K., T. Berger, H. Lassmann, D. Hinze-Selch, Y. Zhang, J. Gehrmann, H. Wekerle, and C. Linington. 1994. Experimental autoimmune panencephalitis and uveoretinitis in the Lewis rat transferred by T lymphocytes specific for the S100β molecule, a calcium-binding protein of astroglia. J. Exp. Med. 180:817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stefferl, A., A. Schubart, M. Storch, A. Amini, I.H. Mather, H. Lassmann, and C. Linington. 2000. Butyrophilin, a milk protein, modulates the encephalitogenic T cell response to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis. J. Immunol. 165:2859–2865. [DOI] [PubMed] [Google Scholar]
- 24.Cross, A.H., B. Cannella, C.F. Brosnan, and C.S. Raine. 1990. Homing to central nervous system vasculature by antigen-specific lymphocytes. I. Localization of 14C-labelled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Lab. Invest. 63:162–170. [PubMed] [Google Scholar]
- 25.Wekerle, H., C. Linington, H. Lassmann, and R. Meyermann. 1986. Cellular immune reactivity within the CNS. Trends Neurosci. 9:271–277. [Google Scholar]
- 26.Hickey, W.F., B.L. Hsu, and H. Kimura. 1991. T lymphocyte entry into the central nervous system. J. Neurosci. Res. 28:254–260. [DOI] [PubMed] [Google Scholar]
- 27.Liu, H., M. Rhodes, D.L. Wiest, and D.A.A. Vignali. 2000. On the dynamics of the TCR:CD3 complex cell surface expression and downmodulation. Immunity 13:665–675. [DOI] [PubMed] [Google Scholar]
- 28.Grakoui, A., S.K. Bromley, C. Sumen, M.M. Davis, A.S. Shaw, P.M. Allen, and M.L. Dustin. 1999. The immunological synapse: a molecular machine controlling T cell activation. Science. 285:221–227. [DOI] [PubMed] [Google Scholar]
- 29.Itoh, Y., and R.N. Germain. 1997. Single cell analysis reveals regulated hierarchical T cell antigen receptor signaling thresholds and intraclonal heterogeneity for individual cytokine responses of CD4+ T cells. J. Exp. Med. 186:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neumann, H., and H. Wekerle. 1998. Neuronal control of the immune response in the central nervous system: linking brain immunity to neurodegeneration. J. Neuropathol. Exp. Neurol. 58:1–9. [DOI] [PubMed] [Google Scholar]
- 31.Neumann, H., T. Misgeld, K. Matsumuro, and H. Wekerle. 1998. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc. Natl. Acad. Sci. USA. 95:5779–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meinl, E., F. Aloisi, B. Ertl, F. Weber, R. De Waal Malefijt, H. Wekerle, and R. Hohlfeld. 1994. Multiple sclerosis: immunomodulatory effects of human astrocytes on T cells. Brain. 117:1323–1332. [DOI] [PubMed] [Google Scholar]
- 33.Xiao, B.-G., A. Diab, J. Zhu, P. Van der Meide, and H. Link. 1998. Astrocytes induce hyporesponsiveness of myelin basic protein-reactive T and B cell function. J. Neuroimmunol. 89:113–121. [DOI] [PubMed] [Google Scholar]
- 34.Aloisi, F., F. Ria, and L. Adorini. 2000. Regulation of T cell responses by CNS antigen presenting cells: different roles for microglia and astrocytes. Immunol. Today. 21:141–147. [DOI] [PubMed] [Google Scholar]
- 35.Brabb, T., P. Von Dassow, N. Ordonez, B. Schnabel, B. Duke, and J. Goverman. 2000. In situ tolerance within the central nervous system as a mechanism for preventing autoimmunity. J. Exp. Med. 192:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Critchfield, J.M., M.K. Racke, J.C. Zuñiga-Pflücker, B. Cannella, C.S. Raine, J. Goverman, and M.J. Lenardo. 1994. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 263:1139–1143. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi, N., D. Liu, B. Min, S. Ben-Sasson, and W.E. Paul. 2002. Antigen challenge leads to in vivo activation and elimination of highly polarized TH1 memory T cells. Proc. Natl. Acad. Sci. USA. 99:6187–6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pender, M.P., K.B. Nguyen, P.A. McCombe, and J.F.R. Kerr. 1991. Apoptosis in the nervous system in experimental allergic encephalomyelitis. J. Neurol. Sci. 104:81–87. [DOI] [PubMed] [Google Scholar]
- 39.Schmied, M., H. Breitschopf, R. Gold, H. Zischler, G. Rothe, H. Wekerle, and H. Lassmann. 1993. Apoptosis of T lymphocytes–a mechanism to control inflammation in the brain. Am. J. Pathol. 143:446–452. [PMC free article] [PubMed] [Google Scholar]
- 40.Flügel, A., F.-W. Schwaiger, H. Neumann, I. Medana, M. Willem, H. Wekerle, G.W. Kreutzberg, and M.B. Graeber. 2000. Neuronal FasL induces cell death of encephalitogenic T lymphocytes. Brain Pathol. 10:353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritz, R.B., and I. Kalvakolanu. 1995. Thymic expression of the golli-myelin basic protein gene in the SJL/J mouse. J. Neuroimmunol. 57:93–99. [DOI] [PubMed] [Google Scholar]
- 42.Fritz, R.B., and M.-L. Zhao. 1996. Thymic expression of myelin basic protein (MBP). Activation of MBP-specific T cells by thymic cells in the absence of exogenous MBP. J. Immunol. 157:5249–5253. [PubMed] [Google Scholar]
- 43.Kalwy, S.A., M.-C. Marty, P. Bausero, and B. Pessac. 1998. Myelin basic protein-related proteins in mouse brain and immune tissues. J. Neurochem. 70:435–438. [DOI] [PubMed] [Google Scholar]
- 44.Mor, F., G.L. Boccaccio, and T. Unger. 1998. Expression of autoimmune disease-related antigens by cells of the immune system. J. Neurosci. Res. 54:254–262. [DOI] [PubMed] [Google Scholar]
- 45.Kojima, K., H. Lassmann, H. Wekerle, and C. Linington. 1997. The thymus and self tolerance: co-existence of encephalitogenic S-100β specific T cells and their nominal autoantigen, S-100β, in the normal rat thymus. Int. Immunol. 9:897–904. [DOI] [PubMed] [Google Scholar]
- 46.Lauriola, L., F. Michetti, V.M. Stolfi, G. Tallini, and D. Cocchia. 1984. Detection by S-100 immunolabelling of interdigitating reticulum cells in human thymuses. Virchows Arch. 45:187–195. [DOI] [PubMed] [Google Scholar]
- 47.Kondo, H., T. Iwanaga, and T. Nakajima. 1983. An immunocytochemical study on the localization of S-100 protein in the retina of rats. Cell Tiss. Res. 231:527–532. [DOI] [PubMed] [Google Scholar]
- 48.Kondo, K., K. Mukai, Y. Sato, Y. Matsuno, Y. Shimosato, and Y. Monden. 1990. An immunohistochemical study of thymic epithelial tumors. III. The distribution of interdigitating reticulum cells and S-100β-positive small lymphocytes. Am. J. Surg. Pathol. 14:1139–1147. [PubMed] [Google Scholar]
- 49.Iglesias, A., J. Bauer, T. Litzenburger, A. Schubart, and C. Linington. 2001. T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia. 36:220–234. [DOI] [PubMed] [Google Scholar]
- 50.Berger, T., S. Weerth, K. Kojima, C. Linington, H. Wekerle, and H. Lassmann. 1997. Experimental autoimmune encephalomyelitis: the antigen specificity of T-lymphocytes determines the topography of lesions in the central and peripheral nervous system. Lab. Invest. 76:355–364. [PubMed] [Google Scholar]
- 51.Tran, E.H., K. Hoekstra, N. Van Rooijen, C.D. Dijkstra, and T. Owens. 1998. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage depleted mice. J. Immunol. 161:3767–3775. [PubMed] [Google Scholar]
- 52.Martiney, J.A., A.J. Rajan, P.C. Charles, A. Cerami, P.C. Ulrich, S. Macphail, K.J. Tracey, and C.F. Brosnan. 1998. Prevention and treatment of experimental autoimmune encephalomyelitis by CNI-1493, a macrophage deactivating agent. J. Immunol. 160:5588–5595. [PubMed] [Google Scholar]
- 53.Izikson, L., R.S. Klein, A.D. Luster, and H.L. Weiner. 2002. Targeting monocyte recruitment in CNS autoimmune disease. Clinical Immunology. 103:125–131. [DOI] [PubMed] [Google Scholar]
- 54.Karpus, W.J., N.W. Lukacs, B.L. McRae, R.M. Strieter, S.L. Kunkel, and S.D. Miller. 1995. An important role for the chemokine macrophage inflammatory protein-1α in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J. Immunol. 155:5003–5010. [PubMed] [Google Scholar]
- 55.Youssef, S., G. Wildbaum, and N. Karin. 1999. Prevention of experimental autoimmune encephalomyelitis by MIP-1α and MCP-1 naked DNA. J. Autoimmun. 13:21–29. [DOI] [PubMed] [Google Scholar]
- 56.Huang, D.R., J. Wang, P. Kivisäkk, B.J. Rollins, and R.M. Ransohoff. 2001. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 193:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glabinski, A.R., B. Bielicki, and R.M. Ransohoff. 2003. Chemokine upregulation follows cytokine expression in chronic relapsing experimental autoimmune encephalomyelitis. Scand. J. Immunol. 58:81–88. [DOI] [PubMed] [Google Scholar]
- 58.McManus, C.M., C.F. Brosnan, and J.W. Berman. 2000. Cytokine induction of MIP-1 alpha and MIP-1 beta in human fetal microglia. J. Immunol. 160:1449–1455. [PubMed] [Google Scholar]
- 59.Oh, J.-W., L.M. Schwiebert, and E.N. Benveniste. 1999. Cytokine regulation of CC and CXC chemokine expression by human astrocytes. J. Neurovirol. 5:82–94. [DOI] [PubMed] [Google Scholar]
- 60.André, I., A. Gonzalez, B. Wang, J. Katz, C. Benoist, and D. Mathis. 1996. Checkpoints in the progression of autoimmune disease: lessons from diabetes mellitus. Proc. Natl. Acad. Sci. USA. 93:2260–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwartz, M., G. Moalem, R. Leibowitz-Amit, and I.R. Cohen. 1999. Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci. 22:295–299. [DOI] [PubMed] [Google Scholar]