

Abstract
In lupus-prone NZM2328 mice, a locus Cgnz1 on chromosome 1 was linked to chronic glomerulonephritis, severe proteinuria, and early mortality in females. A locus Adnz1 on chromosome 4 was linked to antinuclear antibody (ANA) and anti–double stranded DNA (dsDNA) antibody (Ab) production. In this investigation, two congenic strains, NZM2328.C57L/Jc1 (NZM.C57Lc1) and NZM2328.C57L/Jc4 (NZM.C57Lc4), were generated by replacing the respective genetic intervals containing either Cgnz1 or Adnz1 with those from C57L/J, a nonlupus-prone strain. The NZM.C57Lc1 females had markedly reduced incidence of chronic glomerulonephritis and severe proteinuria. NZM.C57Lc4 females had chronic glomerulonephritis and severe proteinuria without circulating ANA, anti-dsDNA, and antinucleosome Ab. These data confirm the linkage analysis. Unexpectedly, NZM.C57Lc1 females had little anti-dsDNA and related Ab, suggesting the presence of a second locus Adnz2 on chromosome 1. The diseased NZM.C57Lc4 kidneys had immune complexes by immunofluorescence and electron microscopy. The eluates from these kidneys did not contain ANA, anti-dsDNA, and antinucleosome Ab, indicative of the presence of non–anti-dsDNA nephritogenic Ab. Thus, breaking tolerance to dsDNA and chromatin is not required for the pathogenesis of lupus nephritis. These results reaffirm that anti-dsDNA and related Ab production and chronic glomerulonephritis are under independent genetic control. These findings have significant implications in the pathogenesis of systemic lupus erythematosus.
Keywords: lupus autoantibodies, glomerulonephritis, NZM2328, genetics
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease with multiple autoantibodies as a hallmark. In particular, the presence of antinuclear Ab (ANA) is a dominant feature of this disorder and is considered one of the diagnostic criteria (1, 2). Chronic glomerulonephritis with severe proteinuria and renal failure is a major complication of SLE. Anti–double stranded DNA (dsDNA) and antinucleosome Ab have been said to play major roles in its pathogenesis (3–10). From a series of experiments using NZM2410 as a mouse model for SLE, others have concluded that loss of tolerance to nuclear Ags such as dsDNA and nucleosome manifested by the production of ANA is an initial and central step in the pathogenesis of lupus glomerulonephritis (11–16). These and other studies have led to the current dogma that ANA, anti-dsDNA, and antinucleosome autoantibodies are of paramount importance in the pathogenesis of SLE (17, 18).
Recently, our laboratory has characterized a new lupus-prone strain NZM2328, which shows female bias in chronic glomerulonephritis with both males and females producing ANA, anti-dsDNA Ab, and acute glomerulonephritis. Five lupus susceptibility genetic intervals were identified in NZM2328 in the analysis of the backcross (C57L/JXNZM2328)F1XNZM2328 (19). In this study, acute glomerulonephritis was characterized by hypercellular glomeruli with little or no glomerulosclerosis, and without interstitial fibrosis or tubular atrophy/dilatation. In contrast, extensive glomerulosclerosis, interstitial fibrosis, and tubular atrophy/dilatation were diagnostic of chronic glomerulonephritis. Our previous study has documented association of severe proteinuria with chronic but not with acute glomerulonephritis (19). Based on these diagnostic criteria, a single locus Cgnz1 was significantly linked to chronic glomerulonephritis, severe proteinuria, and early mortality in female mice. Three genetic intervals, Agnz1 distal to Cgnz1 on chromosome 1, and the H-2 complex and Agnz2 distal to H-2 on chromosome 17, were suggestively linked to acute glomerulonephritis. A single locus Adnz1 on chromosome 4 was suggestively linked to elevated levels of ANA and anti-dsDNA Ab.
In this investigation, two congenic strains of NZM2328 were generated by the microsatellite marker-assisted method (20, 21). NZM2328.C57L/Jc1 (NZM.C57Lc1) was generated by replacing the segment of chromosome 1, which contains Cgnz1 and Agnz1 in NZM2328 with that of C57L/J. NZM2328.C57L/Jc4 (NZM.C57Lc4) was similarly generated. Serological, histological, and kidney elution studies provided convincing evidence that chronic lupus nephritis could be initiated without detectable ANA, antinucleosome, and anti-dsDNA autoantibody production. Thus, an alternative hypothesis to the current dogma that breaking tolerance to nuclear Ags is the initial and the most important step in lupus pathogenesis should be reconsidered.
Materials and Methods
Mice.
NZM 2328Aeg breeding pairs were obtained from The Jackson Laboratory. The NZM2328 line has been maintained in our colony for more than 10 generations by brother-sister mating with no change in serological characteristics or the incidence of renal disease. C57L/J females were obtained from The Jackson Laboratory to generate (C57L/JXNZM2328) F1. All mice were housed under pathogen-free conditions at the University of Virginia Animal Care Facility. NZM.C57Lc1 and NZM.C57Lc4 were generated using a microsatellite marker-assisted speed congenic generation protocol (20, 21). In brief, female (C57L/JXNZM2328) F1 mice were backcrossed to male NZM2328 mice. Four additional backcrosses were performed. This was followed by inbreeding through brother-sister mating. The number of backcrosses was found to be sufficient to generate heterozygotes for either the chromosome 1 or chromosome 4 regions on NZM2328 background. Genotypes were determined by PCR amplification of polymorphic microsatellite markers from genomic DNA using map pairs oligonucleotides (Research Genetics). The markers used have been described (19). After each backcross and intercross, breeders were chosen for their carrying the largest portion of the interested intervals of either chromosome 1 or chromosome 4 from C57L/J and the least amounts of donor DNA throughout the rest of the genome.
Clinical and Histological Assessment.
Screening for severe proteinuria (≥300 mg/dL on two occasions) and histological and immunofluorescence studies of kidneys were performed as previously described (19). An electron microscopic (EM) study of the kidneys was performed by the University of Virginia Central EM Facility. Kidney slices were fixed in 2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, at 4°C overnight. The tissue was washed in buffer followed by incubation at room temperature with 2% osmium tetroxide in buffer for 1 h. The tissue was then dehydrated with acetone and embedded in EPON. One-half micron sections were cut and stained with toludine blue and examined under light microscopy. Ultrathin sections of ∼70–80 nm in thickness from the area of interest were counterstained with lead citrate and uranyl acetate. They were examined using a JEOL 100-CX transmission electron microscope.
Serological Assays.
ANAs were detected by indirect immunofluorescence microscopy using either HeLa or NIH/3T3 cells as substrates and anti-dsDNA Abs were detected by ELISA as previously described (19). Serial dilutions of a monoclonal Ab (R4A) to dsDNA (provided by B. Diamond, Albert Einstein College of Medicine, Bronx, NY) were used as standard and data are presented as units/microgram IgG. Antinucleosome Ab was assayed by ELISA with histone/dsDNA as the substrate according to the procedure described by Mohan et al. (10). Sera were assayed at 1/50 dilution for ANA and 1/100 dilution for ELISA. Rheumatoid factor activity was assayed with either human or rabbit IgG as the substrate as described by Fang et al. (22). Antihistone H1 ELISA was performed with purified H1 as the substrate. Western blot analysis was performed as previously described (23). WEHI 7.1 lymphoma cells, kidney, and liver extracts from 8-wk-old NZM2328 were used as substrates in Western blot analysis.
Acid Elution of Igs from Kidney.
Igs were eluted from the kidney as previously described by Woodroffe and Wilson (24). The homogenate was centrifuged at 1,200 g for 10 min at 4°C and washed in chilled PBS, followed by vortexing and centrifugation to remove serum proteins. The washings were repeated until the OD280 of the supernatant was <0.05. The pellet was then suspended in 0.1 M glycine with 1% BSA, pH 2.8 (10 ml/g kidney), and mixed gently at 4°C for 20 min. The suspension was centrifuged and supernatant was immediately neutralized with 1 M Tris base. Anti-dsDNA Ab and IgG concentrations in the sera and the renal eluate were determined by ELISA and expressed as dsDNA Ab units/microgram IgG.
Results
Generation of NZM.C57Lc1 and NZM.C57Lc4 Congenic Lines.
A microsatellite marker-assisted protocol was used to facilitate the generation of both NZM.C57Lc1 and NZM.C57Lc4. A panel of 91 informative markers was used for the selection of the breeding pairs. As shown in Fig. 1 , the C57L/J alleles were maintained throughout the genomic interval flanked by D1Mit15 (87.9 cM) and D1Mit155 (112.0 cM) for the derivation of NZM.C57Lc1 and D4Mit91 (15.6 cM) and D4Mit72 (59.9 cM) for NZM.C57Lc4. Each of these genetic intervals contains the 95% confidence interval for the loci linked to acute and chronic glomerulonephritis (Agnz1 and Cgnz1) on chromosome 1 and to ANA and anti-dsDNA Ab production (Adnz1) on chromosome 4.
Figure 1.

NZM.C57Lc1 and NZM.C57Lc4 congenic lines were derived by replacing the genetic intervals in NZM2328 with those from C57L/J (hatched bars). The genetic intervals with SLE susceptibility genes in NZM2328 delineated by informative microsatellite markers are shown (open bars). Chromosome intervals are not drawn to scale.
Severe Proteinuria and Chronic Glomerulonephritis in NZM.C57Lc4 but Not in NZM.C57Lc1 Female Mice.
Cohorts of NZM.C57Lc1 and NZM.C57Lc4 females were observed for the development of severe proteinuria and acute and chronic glomerulonephritis. As shown in Fig. 2 A, 70% (24/34) of NZM.C57Lc4 female mice developed severe proteinuria and only 3 of the 41 NZM.C57Lc1 females (7.3%) had severe proteinuria by 12 mo of age. The incidence of severe proteinuria for NZM.C57Lc4 was similar to that of parental stain NZM2328. The incidence of severe proteinuria in NZM.C57Lc1 female mice was markedly reduced. The kinetics of proteinuria development was similar between NZM2328 and its congenic NZM.C57Lc4. There was a delay of 4 mo in the development of severe proteinuria in the three NZM.C57Lc1 mice.
Figure 2.
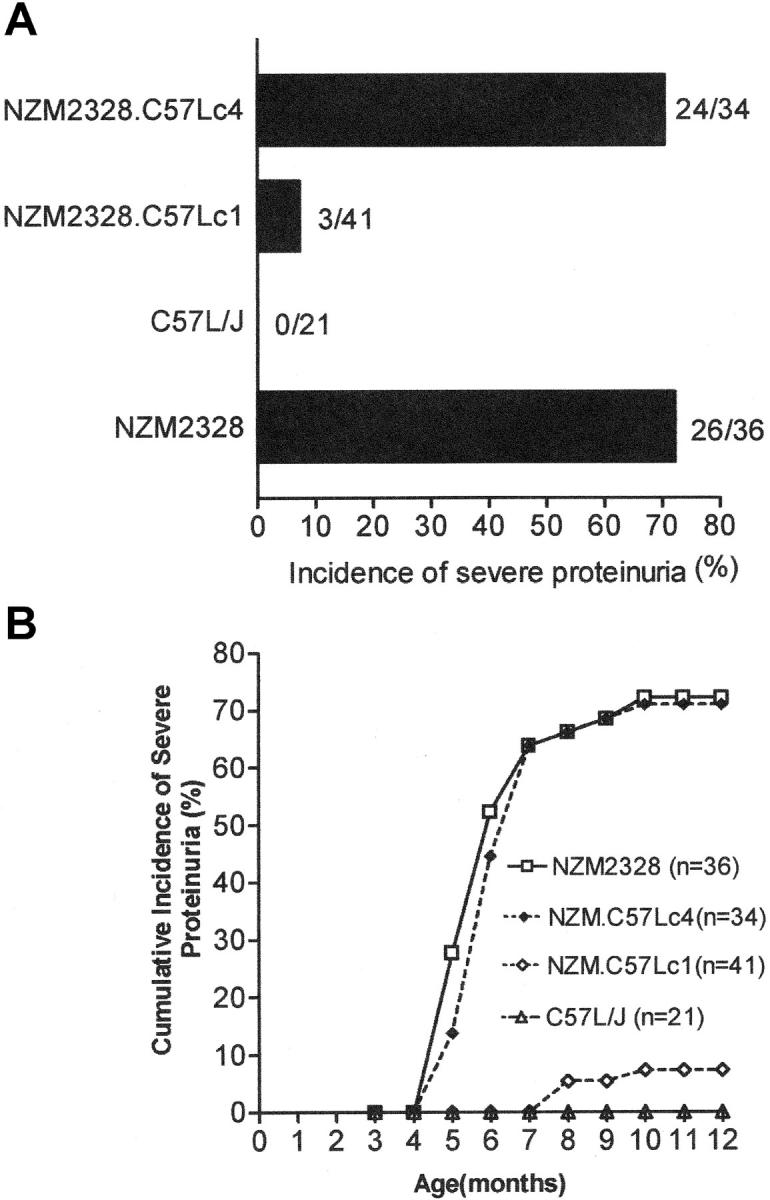
Development of severe proteinuria in females of NZM2328 and NZM.C57Lc4 but not in those of NZM.C57Lc1. (A) Incidence of severe proteinuria in each strain of mice. (B) Kinetics of proteinuria development.
The kidneys from 14 females in each congenic line and from 44 females of the parental line NZM2328 were evaluated for histological evidence of glomerulonephritis. Acute glomerulonephritis was defined by the presence of hypercellularity, mesangial proliferation, mononuclear cellular infiltration, and thickening of the capillary loops. Chronic glomerulonephritis was scored by the presence of glomerulosclerosis tubular atrophy and interstitial fibrosis (16). The incidences of acute and chronic glomerulonephritis in NZM2328 at 12 mo of age were 81.8 (36/44) and 52.2% (23/44), respectively. These incidences were not significantly different from those for NZM.C57Lc4 female cohort, which had 78.5 (11/14) and 50.0% (7/14) incidences for acute and chronic glomerulonephritis, respectively. The three NZM.C57Lc1 females, who developed severe proteinuria, had both acute and chronic glomerulonephritis. Three of the other NZM.C57Lc1 female mice had acute glomerulonephritis without chronic glomerulonephritis. Thus, there was marked reduction in renal disease in the congenic line NZM.C57Lc1. At the age of 12 mo, no significant glomerulonephritis was detected in C57L/J (19).
Histological, immunofluorescence, and EM studies of representative kidneys of the NZM2328 congenic lines are shown in Fig. 3 . The glomeruli of NZM.C57Lc1 are normal in size without cellular infiltration and similar to those seen in kidneys of C57L/J (Fig. 3 A). In contrast, the glomeruli of the NZM.C57Lc4 congenic female were enlarged with hypercellularity, mesangial proliferation, thickened capillary loops, and segmental glomerulosclerosis (Fig. 3 B). Although not shown, there were tubular atrophy and interstitial fibrosis in the diseased NZM.C57Lc4 kidneys. Immunofluorescence studies on NZM.C57Lc4 kidneys showed intense staining for IgG and C3 (Fig. 3, D and F). The thickened capillary loops were more evident with anti-IgG staining. Immunofluorescence staining of NZM.C57Lc1 kidneys showed limited staining for IgG in the mesangia and some C3 deposit in some mesangia and the Bowman capsule (Figs. 3, C and E). These limited staining patterns were often seen in aged C57L/J mice and are considered of no pathological consequences. The IgG and C3 staining patterns of NZM.C57Lc4 kidneys were suggestive of immune complex–mediated glomerulonephritis. This was confirmed by the EM study, showing electron-dense deposits in the subendothelial and subepithelial areas (Fig. 3 H). In contrast, a representative EM of NZM.C57c1 kidneys did not show such electron-dense deposits (Fig. 3 G).
Figure 3.
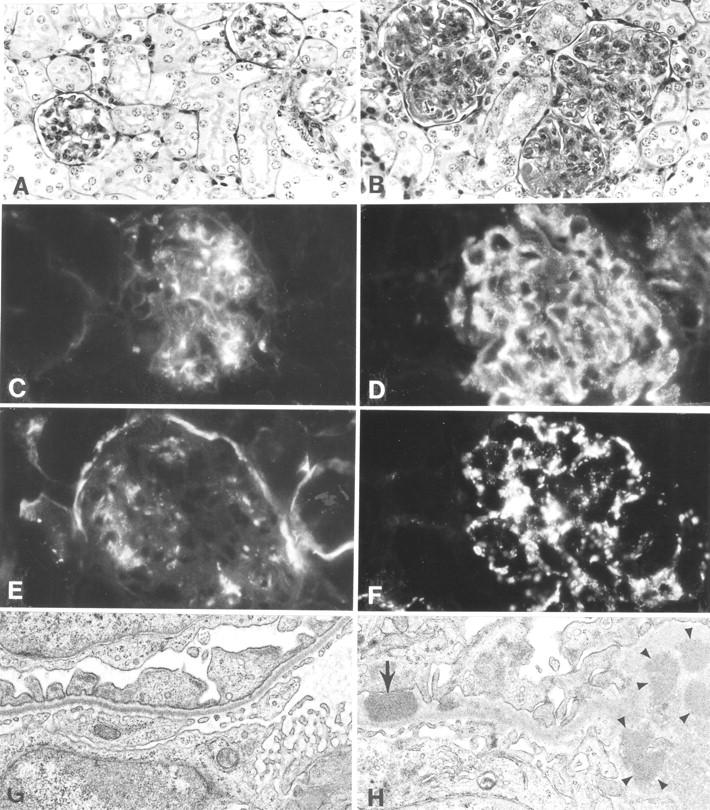
Histological, immunofluorescence, and EM studies of representative kidneys from NZM.C57Lc1 and NZM.C57Lc4 female mice. (A) Normal glomeruli (hematoxylin and eosin staining, ×200) are seen in NZM.C57Lc1. (B) In contrast, in the NZM.C57Lc4 congenic, enlarged glomeruli with mesangial proliferation, hypercellularity, obliterated capillary loops, and glomerulosclerosis are evident. (C) Immunofluorescence studies show some mesangial IgG deposits in NZM.C57Lc1, similar to the pattern seen in aged C57L/J. (D) A coarsely granular staining pattern of IgG deposits in both the mesangia and peripheral capillary walls of the glomeruli of NZM.C57Lc4. (E) Staining of the Bowman capsule and mesangia with anti-C3 Ab are seen in NZM.C57Lc1. (F) Coarsely granular staining by anti-C3 Ab throughout the glomeruli is seen in NZM.C57Lc4. (G) EM study shows normal glomeruli without electron-dense deposits in the subepithelial or subendothelial spaces (×10,000) in the kidney of NZM.C57Lc1. (H) In comparison, electron-dense deposits in both subendothelial space (arrow) and the mesangia (arrowheads) in the glomeruli of NZM.C57Lc4 are readily detected.
Lack of Circulating ANA, Anti-dsDNA, and Antinucleosome Ab in Both NZM2328 Congenic Lines.
Sera from the terminal bleeds of female NZM2328 and its two congenic lines were analyzed for ANA, anti-dsDNA, and antinucleosome Ab. As shown in the top of Fig. 4 , ANAs were readily detected in the sera of NZM2328 and not in those of NZM.C57Lc1 and NZM.C57Lc4. The quantitative data of this study are summarized in the bottom of Fig. 4. At 1/50 dilution, 19.2% (5/26) of NZM.C57Lc1 sera and 4.5% (1/22) of NZM.C57Lc4 sera were positive for ANAs. 12% of the C57L/J sera at 12 mo of age were positive for ANAs. For comparison, 53.3% (24/45) of NZM2328 sera were positive for ANAs. Thus, there were marked reductions of ANAs in both NZM congenics. Similar marked reductions in circulating anti-dsDNA and antinucleosome (histone–DNA complexes) Abs were also detected in these two congenics. It is important to note that the incidences of these three autoantibodies were not significantly different from those in the nonlupus–prone strain C57L/J. In addition, the serum IgG concentrations of C57L/J, NZM2328, and its two congenics were similar.
Figure 4.
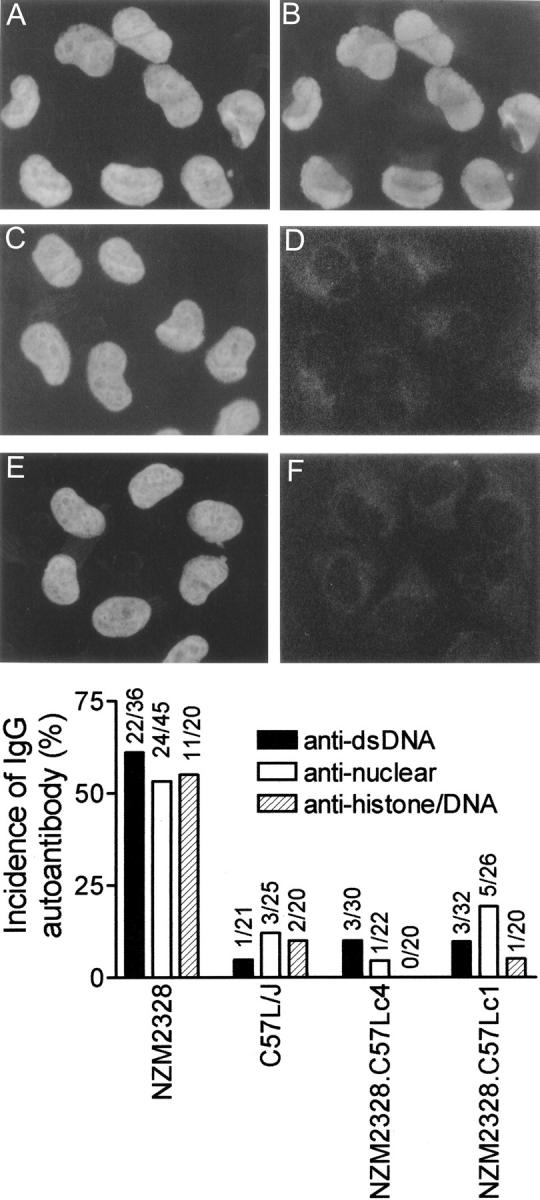
Marked reduction of circulating anti-dsDNA, antinuclear, and antinucleosome Ab in NZM.C57Lc1 and NZM.C57Lc4 congenic lines in comparison with NZM2328. Staining of HeLa cell nuclei by DAPI are seen in A, C, and E. The right side of the figure shows the presence of ANA in the serum of NZM2328 (B) but not in the sera of NZM.C57Lc1 (D) and NZM.C57Lc4 (F). Although not shown, the majority of the sera from C57L/J were not positive for ANA. On the bottom, frequencies of the presence of anti-dsDNA, antinuclear, and antihistone/DNA Ab in these strains are summarized.
RF and Other Autoantibody Activities in Sera of NZM2328 and Its Congenic Lines.
The sera from NZM2328, NZM.-C57Lc1, and NZM.C57Lc4 females were screened for RF. As shown in Fig. 5 , there were little anti-IgG Ab activities in the sera of NZM2328 and its congenic lines. The lack of RF in C57L/J was also demonstrated. In contrast, RF activities were readily demonstrated in sera from MRL/lpr+/+females. These results were obtained with either rabbit IgG or human IgG as the substrate and with sera either at 1/50 or 1/250 dilutions.
Figure 5.

Lack of RF in sera of NZM2328 and its congenics. Human IgG was used as the substrate in A and C to detect anti-IgG activities. Mouse sera were used at a dilution of 1:50 for A and 1:250 for C. Rabbit IgG was used as the substrate in B and D with mouse sera diluted at 1:50 and 1:250, respectively. A pool of MRL/lpr+/+sera was used as a positive control showing readily detectable RF in this strain of mice.
The presence of autoantibodies against cytoplasmic constituents in sera of NZM2328 and its congenics was studied by Western blot analysis with WEHI 7.1 lymphoma cell extract as the substrate. Individual serum was used at a 1/50 dilution. By the ages of 4–5 mo, autoantibodies of multiple specificities were readily detected in NZM2328 females (Fig. 6 , top, lanes 16–22), whereas autoantibodies of limited reactivities were found in NZM.C57Lc4 (Fig. 6, top, lanes 9–15). Fewer autoantibodies were detected in the sera of NZM.C57Lc1 (Fig. 6, top, lanes 1–8). By the time the mice developed fatal glomerulonephritis, fewer circulating autoantibodies were detected in NZM2328 (Fig. 6, bottom, lanes 14–20). The sera of NZM.C57Lc4 remained limited in their autoreactivties (Fig. 6, bottom, lanes 7–13). By the age of 12 mo, sera of some of the NZM.C57Lc1 and C57L/J showed autoreactivities, whereas others remained nonautoreactive (Fig. 6, bottom, lanes 1–6 and 21–24). In Fig. 6, a MRL/lpr + / + serum was used as the positive control to show that both top and bottom panels were developed to a similar extent in chemiluminescence.
Figure 6.

Western blot analysis to detect autoantibodies to cellular constituents in sera of NZM2328 and its congenics. Cell lysate of WEHI 7.1 lymphoma cell line was used as the substrate with sera at the dilution of 1:50. On the top, sera from mice at 4–5 mo of age were used with MRL/lpr+/+(MRL) pooled sera as the positive control. Lanes 1–8, NZM.C57Lc1; lanes 9–15, NZM.C57Lc4; lanes 16–22, NZM2328. On the bottom, sera at death or at death at the age of 12 mo were used. Lanes 1–6, NZM.C57Lc1; lanes 7–13, NZM.C57Lc4; lanes 14–20, NZM2328; lanes 21–24, C57L/J.
In Fig. 6, some of the sera from both congenic lines and NZM2328 reacted with proteins of molecular weights between 30 and 46 kD. Histone H1 with molecular weights in low 30 kD might be a candidate protein for the target Ag as revealed by the Western blot analysis. However, in an ELISA specific for histone H1, no specific anti-H1 Abs were detected in the sera of NZM2328 and its congenics as well as in those of C57L/J (unpublished data).
Lack of Anti-dsDNA Abs in the Eluates from NZM.C57Lc4 Kidneys Afflicted with Glomerulonephritis.
The lack of detectable circulating autoantibodies to dsDNA and other nuclear Ags in NZM.C57Lc4 despite the presence of immune complex–mediated nephritis is not a definitive proof that these Abs do not play a pathological role because these autoantibodies may have been fixed in the kidneys, rendering their absence in the blood. To rule out this possibility, NZM2328, NZM.C57Lc1, NZM.C57Lc4, and C57L/J females 10–12 mo old were killed. At death, sera were collected and their kidneys were frozen in liquid nitrogen and stored for elution. Many of the NZM2328 and NZM.C57Lc4 mice have moderate to severe proteinuria. The kidneys were homogenized and acid eluates were collected and neutralized. NZM2328 and NZM.C57Lc4 kidney eluates had comparable IgG levels, whereas those of NZM.C57Lc1 and C57L/J had significantly lower levels of IgG (unpublished data). Anti-dsDNA Ab activity was expressed as units/microgram of IgG. As shown in Fig. 7 , anti-dsDNA Abs were detected in the majority of the NZM2328 sera. There was a concentration of these anti-dsDNA Abs in the NZM2328 kidneys. The enrichment of these Abs in the kidneys was ∼20-fold. As expected, few anti-dsDNA Abs were detected in the sera of NZM.C57Lc1, NZM.C57Lc4, and C57L/J. More importantly, the kidney eluates of NZM.C57Lc4 rarely had demonstrable anti-dsDNA Abs. It is of interest to note that there was little correlation of anti-dsDNA Ab concentration in the kidney eluate with serum dsDNA Ab titer in an individual mouse. In addition, the kidney eluates of NZM.C57Lc1, NZM.C57Lc4, and C57L/J did not show antinuclear reactivity on HeLa cells. There was staining for ANA with NZM2328 kidney eluates when they were used without dilution. As with sera, the sera and their paired kidney eluates were also studied regarding RF. The kidney eluates of NZM2328, NZM.C57Lc1, NZM.C57Lc4, and C57L/J did not have detectable RF. These results support our hypothesis that ANA and RF play no significant role in the kidney disease in NZM2328 and its congenic NZM.C57Lc4.
Figure 7.

Anti-dsDNA Ab in sera and kidney eluates from NZM2328, NZM.C57Lc1, NZM.C57Lc4, and C57L/J females at 11–12 mo. Abs to dsDNA were present in sera of NZM2328 and they were enriched in their kidney eluates. These Abs were rarely detected in the sera and the kidney eluates of the other three strains. P-values indicate significant differences of the respective strain as compared with NZM2328.
Eluates from NZM.C57Lc4 Diseased Kidneys Identified Several Target Ags.
Kidney and liver from 8-wk-old NZM2328 were perfused with cold PBS. Extracts of them were used as substrates for Western blot analysis. Kidney eluates from NZM2328 and NZM.C57Lc4 were used at either 30 or 10 μg/ml for assaying Ab activity to these extracts. The results are summarized in Fig. 8 . Both eluates from NZM2328 and NZM.C57Lc4 kidneys stained several bands in the region between molecular weights of 25 and 40 kD in the kidney extract. The eluate from NZM2328 also had activities against proteins of higher molecular weights. Although the bands were not distinct, proteins of a lower molecular weight of <21 kD were also reactive with the NZM2328 eluate. It appears that the reactive Ags are also present in the liver extract. Similar results were obtained in another experiment.
Figure 8.

Abs eluted from nephritic kidneys of NZM2328 and NZM.C57Lc4 mice are reactive with proteins within the kidney and liver extracts. Proteins in the kidney (left) and liver (right) extracts were separated on a 12% SDS-PAGE, transferred to nitrocellulose paper, and used for analyzing the reactivity of Abs eluted from the nephritic kidneys of NZM2328 (lanes 2 and 3) and NZM.C57Lc4 (lanes 4 and 5) mice. Each lane is equivalent to 60 μg total protein. Abs were used at a concentration of 30 (lanes 2 and 4) and 10 μg/ml (lanes 3 and 5). Lane 1 in both panels represents reactivity of the goat anti–mouse IgG–horseradish peroxidase conjugate with the extracts. Numbers on the left represent molecular weights in kilodaltons.
Discussion
Breaking Tolerance to dsDNA and Related Nuclear Ags Is Not Required for the Pathogenesis of Lupus Glomerulonephritis.
The most significant finding in this investigation is that despite the lack of circulating ANA, anti-dsDNA and antinucleosome Ab, chronic glomerulonephritis, severe proteinuria, and early mortality are found in a high proportion of NZM.C57Lc4 females. The eluates of kidneys from both congenics at 12 mo of age did not contain significant amounts of anti-dsDNA Ab, whereas the eluate from the NZM2328 kidneys had a significant amount of such Ab. There were no significant ANA titers in the sera and kidney eluates of these congenic lines. These data would support the conclusion that tolerance to dsDNA and related nuclear Ags remains intact in aged NZM.C57Lc4 despite the development of fatal glomerulonephritis. Thus, it is reasonable to conclude that lupus glomerulonephritis can occur in the absence of these autoantibodies and that breaking tolerance to nuclear Ags and dsDNA need not be the initial or necessary step in the pathogenesis of lupus nephritis as postulated by others (11–16).
Target Ags for Nephritogenic Ab in NZM.C57Lc4.
Lupus nephritis has been considered to be a prototype of immune complex–mediated disease. The Ab–Ag systems involved have been an important area of investigation for both basic and clinical investigators. Shortly after the demonstration by Holman and Kunkel (25) that DNA is a target Ag in ANA, the importance of the dsDNA and anti-dsDNA Ab system in the pathogenesis of SLE was illustrated in two seminal papers from the laboratory of the late Henry Kunkel (3, 4). The involvement of RF in lupus nephritis was shortly demonstrated thereafter (26). Recently, Mannik et al. (27) have reported that anti-dsDNA, anti-snRNP, anti-Ro/La, anti-IgG, and antihistone autoantibodies and autoantibodies of other specificities account for <25% of IgG eluted from kidneys of SLE patients with severe glomerulonephritis. These studies suggest that more attention to Ag–Ab systems other than that of dsDNA and anti-dsDNA Ab might be warranted for our further understanding of SLE pathogenesis. In this regard, the NZM.C57Lc4 line, which contains high levels of glomerular IgG but not glomerular anti-dsDNA Ab and RF, will serve as an excellent model for further investigation of this important issue.
The role of Ab and B cells in the pathogenesis of lupus nephritis has been recently reviewed by Shlomchik and Madaio (28). Crossreactive and direct reactive Ab to renal Ag has been postulated to be an important mechanism for IgG deposition. A 108-kD renal mesangial Ag was identified to be the renal target for a monoclonal Ab to dsDNA (29). The 100-kD α-actinin has also been recently identified as the target Ag for other anti-dsDNA Abs (30, 31). In our investigation, several Ags in the range of molecular weight 25–40 kD were reactive with the NZM.C57Lc4 kidney eluate. The identity of these Ags is currently under investigation. The kidney eluate from the parental line NZM2328 had Ab activity to proteins of higher molecular weight. One of these might be α-actinin. In addition to Ab-mediated injury, evidence for a role of T cells in the pathogenesis of lupus nephritis is accumulating. Bloom et al. (32) have shown that intrathymically injected kidney cells delay the onset of lupus nephritis in MRL lpr/lpr mice. More directly, Chan et al. (33) have reported a unique form of nephritis in the MRL lpr/lpr mice, which have B cells without circulating Ig. These mice have a shortened lifespan, intermediate between the wild-type and that without B cells. These studies provide evidence that T cells play a crucial role by direct participation in end organ damage. In our preliminary studies, lymphocytes in the draining lymph nodes of the kidneys were found to be selectively activated by the age of 8 wk in NZM2328. This suggests that an immune response involving both T and B cells to kidney Ags may play an important role in the pathogenesis of lupus nephritis. This result also suggests that the kidney should be considered an active participant in this process.
SLE Susceptibility Genes.
In NZM2328, SLE susceptibility genes Agnz1 and Cgnz1 controlling acute and chronic glomerulonephritis, respectively, were identified at the distal distinct regions of chromosome 1 (19). The congenic line NZM.C57Lc1 has markedly reduced incidence of severe proteinuria and chronic glomerulonephritis although the incidence of acute glomerulonephritis is reduced to a lesser degree. These phenotypes are expected, confirming the quantitative genetic trait analytical data and the importance of a single gene Cgnz1 in controlling chronic glomerulonephritis. The marked reduction of circulating ANA, anti-dsDNA, and antinucleosome Ab in NZM.C57Lc1 is unexpected. This finding implicates a second locus, Adnz2 (Anti-dsDNA antibody in NZM2328 locus 2) within this region, to be linked to ANA, anti-dsDNA, and antinucleosome production. The location of Adnz2 remains to be determined. Because NZM.C57Lc1 expresses Adnz1, which is located on chromosome 4 (19), Adnz1 and Adnz2 must interact with each other to express the phenotype of anti-dsDNA Ab production, providing evidence for the complexity of genetic control of this seemingly simple phenotype.
The congenic line NZM.C57Lc1, in which the interval of interest was replaced by that of C57L/J, has provided us the capability to generate intrachromosomal recombinants to determine whether distinct subregions within this interval control these three phenotypes. Several such intrachromosomal recombinants have been generated and their phenotypes are being determined. These recombinants will also provide us data to determine whether Adnz2 represents several clusters of genes, each of which can independently control ANA, anti-dsDNA, and antinucleosome Ab production.
Proposed Model for the Pathogenesis of Lupus Nephritis.
The findings reported here have significant implications on our thinking of the pathogenesis of SLE. An alternative and a less anti-dsDNA– and antinucleosome Ab–centric model for the pathogenesis of SLE is put forth and depicted in Fig. 9 . In this model, autoantibody production and autoreactive T cell generation (Fig. 9, pathway I) and activation of genes leading to end organ damage (Fig. 9, pathway II) are independent events. Activation of the autoimmune response is either as a result of genetic predisposition or environmental factors such as chemical exposure and infections. Genes controlling end organ susceptibility exert their influence on the targeted end organs by causing differential expression of genes and/or increased apoptosis in target organs, which may lead to overexpression of organ-specific Ag. The initiation of these two pathways might be independent of each other. This is supported by both clinical and experimental data. Clinically, many patients with significant titers of ANA do not have symptoms and signs of SLE. Specifically, they do not have end organ damage. In experimental models such as NZM2328, male mice have significant elevations of ANA and anti-dsDNA Ab without chronic glomerulonephritis. The recent work by Jacob et al. (34), shows that Stat4-deficient NZM2328 developed accelerated nephritis and increased mortality with lower titers of anti-DNA Ab. In contrast, Stat6-deficient NZM2328 mice had a significant reduction of nephritis despite the presence of high levels of anti-DNA Ab.
Figure 9.

Interactive model for the pathogenesis of SLE. Pathway I, autoantibody production and activation of effector T cells and pathway II, activation of susceptibility genes and end organ damage, can be initiated independently while they interact at different levels as indicated by pathways III and IV. The interactions between these pathways lead to end organ damage.
The recent work by Singh et al. (35) shows that cytokines have differential effects on kidney disease in NZM2410. It is plausible that enhanced cytokines and chemokine expression as a result of activation of the innate and adaptive immune systems can increase target organ susceptibility. Despite the independence of the initiation of these two pathways, they do interact with each other (Fig. 9, pathway III). These interactions may influence differential gene expression and induce organ-specific Ab production as well as tissue-specific effector T cell generation (Fig. 9, pathway IV). In this regard, the importance of cell death in autoimmune thyroid disease has been well documented (36) and the administration of a caspase inhibitor has been shown to inhibit the development of lupus nephritis without affecting autoantibody production or skin inflammation (37).
The proposed model accommodates most of the data accumulated both by clinical observations and animal investigations. This model also implicitly states that dampening the ongoing inflammatory response present in SLE patients influences both the activation states of immune cells and the susceptibility of end organ damage and should be a major target in the therapeutic approach to SLE.
Acknowledgments
This work was supported in part by grants RO1 AR-42027, RO1 AR-42465, RO1 AR-47988, and P50 AR-45222 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. S.T. Waters was supported by minority student supplements to RO1 AR-42027 and P50AR-45222.
This work was presented in abstract form at the ACR 64th Annual Scientific Meeting at Philadelphia, PA in November, 2000 (Arthritis Rheum. 43:S361).
Abbreviations used in this paper: ANA, antinuclear Ab; dsDNA, double stranded DNA; EM, electron microscopic; NZM.C57Lc1, NZM2328.C57L/Jc1; NZM.C57Lc4, NZM2328.C57L/Jc4; SLE, systemic lupus erythematosus.
References
- 1.Tan, E.M., A.S. Cohen, J.F. Fries, A.T. Masi, D.J. McShane, N.F. Rothfield, J.G. Schaller, N. Talal, and R.J. Winchester. 1982. The 1982 revised criteria for the classification of systemic lupus erythematosus (SLE). Arthritis Rheum. 25:1271–1277. [DOI] [PubMed] [Google Scholar]
- 2.Hochberg, M.C. 1977. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus (letter). Arthritis Rheum. 40:1725. [DOI] [PubMed] [Google Scholar]
- 3.Koffler, D., P.H. Schur, and H.G. Kunkel. 1967. Immunological studies concerning the nephritis of systemic lupus erythematosus. J. Exp. Med. 126:607–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koffler, D., R. Carr, V. Agnello, R. Thoburn, and H.G. Kunkel. 1971. Antibodies to polynucleotides in human sera: antigenic specificity and relation to disease. J. Exp. Med. 134:294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emlen, W., D.S. Pisetsky, and R.P. Taylor. 1986. Antibodies to DNA. Arthritis Rheum. 29:1417–1425. [DOI] [PubMed] [Google Scholar]
- 6.Khalil, M., L. Spatz, and B. Diamond. 1999. Anti-DNA antibodies. Systemic Lupus Erythematosus. R.G. Lahita, editor. Academic Press, San Diego. 197–217.
- 7.Hahn, B.H., and B.P. Tsao. 1997. Antibodies to DNA. Dubois' Lupus Erythematosus. D.T. Wallace and B.H. Hanh, editors. Williams & Wilkins, Baltimore. 407–422.
- 8.Fritzler, M.J. 1997. Antibodies to histone and the role of the nucleosome. Dubois' Lupus Erythematosus. D.T. Wallace and B.H. Hahn, editors. Williams & Wilkins, Baltimore. 423–441.
- 9.Burlingame, R.W., R.I. Rubin, R.S. Balderas, and A.N. Theofilopoulos. 1993. Genesis and evolution of anti-chromatin autoantibodies in murine lupus implicates T-dependent immunization with self-antigens. J. Exp. Med. 91:1687–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohan, C., S. Adams, V. Stanik, and S.K. Datta. 1993. Nucleosome: a major immunogen for pathogenic autoantibodyinducing cells of lupus. J. Exp. Med. 177:1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morel, L., U.H. Rudofsky, J.A. Longmate, J. Schiffenbauer, and E.K. Wakeland. 1994. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1:219–229. [DOI] [PubMed] [Google Scholar]
- 12.Mohan, C., E. Alas, L. Morel, P. Yang, and E.K. Wakeland. 1998. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance H2A/H2B/DNA subnucleosomes. J. Clin. Invest. 101:1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohan, C., L. Morel, P. Yang, and E.K. Wakeland. 1997. Genetic dissection of systemic lupus erythematosus pathogenesis: Sle2 on murine chromosome 4 leads to B cell hyperactivity. J. Immunol. 159:454–465. [PubMed] [Google Scholar]
- 14.Mohan, C., Y. Yu, L. Morel, P. Yang, and E.K. Wakeland. 1999. Genetic dissection of Sle pathogenesis: Sle3 on murine chromosome 7 impacts T cell activation, differentiation, and cell death. J. Immunol. 162:6492–6502. [PubMed] [Google Scholar]
- 15.Morel, L., B.P. Croker, K.R. Blenman, C. Mohan, G. Huang, G. Gilkeson, and E.K. Wakeland. 2000. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc. Natl. Acad. Sci. USA. 97:6670–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morel, L., K.R. Blenman, B.P. Croker, and E.K. Wakeland. 2001. The major murine systemic lupus erythematosus susceptibility locus, Sle, is a cluster of functionally related genes. Proc. Natl. Acad. Sci. USA. 98:1781–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakeland, E.K., K. Liu, R.R. Graham, and T.W. Behrens. 2001. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 15:397–408. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen, C., N. Limaye, and E.K. Wakeland. 2002. Suscetibility genes in the pathogenesis of murine lupus. Arthritis Res. 4:S255–S263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waters, S.T., S.M. Fu, F. Gaskin, U.S. Deshmukh, S.S.J. Sung, C.C. Kannapell, K.S.K. Tung, S.B. McEwen, and M. McDuffie. 2001. NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clin. Immunol. 100:372–383. [DOI] [PubMed] [Google Scholar]
- 20.Wakeland, E.K., L. Morel, K. Achey, M. Yui, and J.A. Longmate. 1997. Speed congenics: a classic technique moves into the fast lane (relatively speaking). Immunol. Today. 18:473–477. [DOI] [PubMed] [Google Scholar]
- 21.Markel, P., P. Shu, C. Ebeling, G.A. Carlson, D.L. Nagle, J.S. Smutko, and K.J. Moore. 1997. Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nat. Genet. 17:280–284. [DOI] [PubMed] [Google Scholar]
- 22.Fang, Q., C.C. Kannapell, F. Gaskin, A. Solomon, W.J. Koopman, and S.M. Fu. 1994. Human rheumatoid factors with restrictive specificity for rabbit IgG: auto- and multi-reactivity, diverse VH gene segment usage and preferential usage of VλIIIb. J. Exp. Med. 179:1445–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deshmukh, U.S., J.L. Lewis, F. Gaskin, C.C. Kannapell, S.T. Waters, Y.-h. Lou, K.S.K. Tung, and S.M. Fu. 1999. Immune responses to Ro60 and its peptides in mice. I. The nature of the immunogen and endogenous autoantigen determine the specificities of the induced autoantibodies. J. Exp. Med. 189:531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodroffe, A.J., and C.B. Wilson. 1977. An evaluation of elution techniques in the study of immune complex glomerulonephritis. J. Immunol. 118:1788–1794. [PubMed] [Google Scholar]
- 25.Holman, R.H., and H.G. Kunkel. 1957. Affinity between the lupus erythematosus serum factor and cell nuclei and nucleoprotein. Science. 126:162–163. [DOI] [PubMed] [Google Scholar]
- 26.Agnello, V., D. Joffler, and H.G. Kunkel. 1973. Immune complex systems in the nephritis of systemic lupus erythematosus. Kidney Int. 3:90–99. [DOI] [PubMed] [Google Scholar]
- 27.Mannik, M., C.E. Merrill, L.D. Stamps, and M.H. Wener. 2003. Multiple autoantibodies form the glomerular immune deposits in patients with systemic lupus erythematosus. J. Rheumatol. 30:1495–1504. [PubMed] [Google Scholar]
- 28.Shlomchik, M.J., and M.P. Madaio. 2003. The role of autoantibodies and B cells in the pathogenesis of lupus nephritis. Springer Semin. Immunopathol. 24:363–375. [DOI] [PubMed] [Google Scholar]
- 29.D'Andrea, D.M., B. Coupaye-Gerard, T.R. Kleyman, M.H. Foster, and M.P. Madaio. 1996. Lupus autoantibodies interact directly with distinct glomerular and vascular cell surface antigens. Kidney Int. 49:1213–1221. [DOI] [PubMed] [Google Scholar]
- 30.Mostoslavsky, G., R. Fischel, N. Yachimovich, Y. Yarkoni, E. Rosenmann, M. Monestier, M. Baniyash, and D. Eliat. 2001. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: a case for tissue injury by molecular mimicry. Eur. J. Immunol. 31:1221–1227. [DOI] [PubMed] [Google Scholar]
- 31.Deocharan, B., X. Qing, J. Lichauco, and C. Putterman. 2002. α-Actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J. Immunol. 168:3072–3078. [DOI] [PubMed] [Google Scholar]
- 32.Bloom, R.D., T. O'Connor, B. Cizman, R. Kalluri, A. Naji, and M.P. Madaio. 2002. Intrathymic kidney cells delays the onset of lupus nephritis in MRL-lpr/lup mice. Intern. Immunol. 14:867–871. [DOI] [PubMed] [Google Scholar]
- 33.Chan, O.T., L.G. Hannum, A.M. Haberman, M.P. Madaio, and M.J. Shlomchik. 1999. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in lupus nephritis. J. Exp. Med. 189:1639–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacob, C.O., S. Zang, L. Li, V. Ciobanu, F. Quismorio, A. Mizutani, M. Satoh, and M. Koss. 2003. Pivotal role of Stat4 and Stat6 in the pathogenesis f the lupus-like disease in the New Zealand mixed 2328 mice. J. Immunol. 171:1564–1571. [DOI] [PubMed] [Google Scholar]
- 35.Singh, R.R., V. Saxena, S. Zang, L. Li, F.D. Finkelman, D.P. Witte, and C.O. Jacob. 2003. Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J. Immunol. 170:4818–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stassi, G., and R. De Maria. 2002. Autoimmune thyroid disease: new models of cell death in autoimmunity. Nat. Rev. Immunol. 2:195–204. [DOI] [PubMed] [Google Scholar]
- 37.Seery, J.P., V. Cattell, and F.M. Watt. 2001. Cutting edge: amelioration of kidney disease in a transgenic mouse model of lupus nephritis by administration of the caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-(β-o-methyl)-fluoromethylketone. J. Immunol. 167:2452–2455. [DOI] [PubMed] [Google Scholar]