

Abstract
The low number of CD4+ CD25+ regulatory T cells (Tregs), their anergic phenotype, and diverse antigen specificity present major challenges to harnessing this potent tolerogenic population to treat autoimmunity and transplant rejection. In this study, we describe a robust method to expand antigen-specific Tregs from autoimmune-prone nonobese diabetic mice. Purified CD4+ CD25+ Tregs were expanded up to 200-fold in less than 2 wk in vitro using a combination of anti-CD3, anti-CD28, and interleukin 2. The expanded Tregs express a classical cell surface phenotype and function both in vitro and in vivo to suppress effector T cell functions. Most significantly, small numbers of antigen-specific Tregs can reverse diabetes after disease onset, suggesting a novel approach to cellular immunotherapy for autoimmunity.
Keywords: autoimmunity, tolerance, CD4+CD25+ T cells, NOD mice, immunoregulation
Introduction
It has become increasingly clear that the balance of pathogenic and immune regulatory pathways underlies disease progression in many autoimmune settings. The loss of regulatory pathways such as CTLA-4, TGF-β, and FoxP3 leads to lethal autoimmunity (1–7). This is best exemplified in type 1 diabetes (T1D) in nonobese diabetic (NOD) mice and humans, where genetic or biologic loss of function of these immunoregulatory pathways exacerbates disease development (8–10). Increasingly, these pathways have pointed to a novel CD4+ regulatory T cell (Treg) lymphocyte subset as the central controller of autoimmunity in a variety of experimental animal models as well as an intrinsic regulator of spontaneous autoimmunity (for review see references 11–15). Although the most widely used markers for Tregs are the expression of CD4 and CD25, other molecules such as CD62L, CTLA-4, glucocorticoid-induced TNF receptor (GITR), and FoxP3 have emerged as additional markers of this unique T cell lineage (5–7, 16–20). Importantly, Treg therapy can effectively delay and cure mice of a variety of immunological diseases including diabetes, colitis, gastritis, and graft-versus-host disease (8, 21–24). Several studies have suggested that the Tregs are antigen specific, relying on TCR engagement to fully acquire suppressive activity in vivo. The regulatory cells appear to function preferentially at the site of inflammation to effect proliferation and/or cytokine production by the pathogenic T cells (17, 25, 26).
Recent studies have suggested that Tregs function via the production of immunosuppressive cytokines, particularly TGF-β and IL-10 (21, 27–29), whereas other studies indicate that suppressive function requires cell–cell contact and cannot be attributed to soluble inhibitors (30–36). Barthlott et al. (37) and Stockinger et al. (38) suggested that Tregs function to “take up space,” thus blocking the pathogenic cells from filling up their appropriate niche. The Treg population is reduced in autoimmune-prone animals and patients (8, 39). It appears that Tregs may be defective in NOD mice (8, 39). For instance, Tregs constitute only ∼5% of the circulating CD4+ T cells in NOD mice, significantly lower than that observed in other strains (8). Moreover, a large number of Tregs (1:1 Treg/Teff ratio) are required to suppress ongoing disease in this model and other autoimmune models (8, 16, 17). Finally, recent studies have suggested that it might be impossible to reverse ongoing autoimmune diabetes due to the autoreactive T cells becoming resistant to suppression during the active phase of the disease. However, the studies were limited to in vitro analyses presumably due to limited cell numbers (40). In spite of this complexity, the potential for Tregs to actively regulate autoimmunity and induce long-term tolerance has great potential applications both for understanding immune homeostasis and as a strategy for inducing long-lived tolerance.
It has been reported that Tregs preferentially respond to dendritic cells to proliferate in vitro and in vivo, but the in vitro Treg expansion induced by dendritic cells was still very limited (41). In fact, taking advantage of Tregs has been complicated by the difficulty in expanding and characterizing this minor T cell subset. In this study, we developed a robust technique for expanding antigen-specific Tregs from autoimmune NOD mice. The expanded Tregs retained all the quintessential characteristics of this subset including expression of CD25, CD62L, FoxP3, and GITR. The ability of expanded NOD Tregs to suppress diabetes in prediabetic and diabetic mice in vivo was significantly enhanced using the autoantigen-specific T cells when compared with polyclonal Tregs. Antigen-specific Tregs effectively suppressed the development of diabetes in Treg-deficient CD28−/− mice, blocked syngeneic islet graft rejection in chronically diabetic animals, and in contrast to previous reports (40), Tregs are shown to reverse diabetes in mice with new onset disease.
Materials and Methods
Mice.
NOD mice (Taconic), BALB/c mice (Charles River Laboratories), BDC2.5 TCR transgenic (Tg) mice, glutamic acid decarboxylase (GAD)286 TCR Tg mice (42), NOD.CD28−/− mice, NOD.RAG−/− mice, and NOD.TCR-α−/− mice were housed and bred under specific pathogen-free conditions at the University of California San Francisco Animal Barrier Facility.
Antibodies and Other Reagents.
FITC-labeled mAbs against CD4 (GK1.5) and GITR (DTA-1; reference 19) were purified from hybridoma culture supernatant and conjugated in our lab. R-PE–conjugated anti-CD25 (7D4) mAbs were purchased from Southern Biotechnology Associates, Inc. Allophycocyanin (APC)-labeled mAbs against CD4 (RM4-5) and CD62L were purchased from BD Biosciences or eBioscience. The p31/I-Ag7mIgG2a was generated in our lab (43). Carboxyfluorescein diacetate succinimidyl ester (CFSE) was purchased from Molecular Probes.
Cell Sorting and Flow Cytometry.
CD4+ T cells were enriched from pooled LNs and spleens by negative selection using an AutoMACS (Miltenyi Biotec). The cells were then stained with anti–CD4-FITC, anti–CD25-PE, and anti–CD62L-APC, and the Tregs and CD4+ CD62L+ CD25− T effector cells (Teffs) were sorted on a Mo-Flo cytometer™ (DakoCytomation) based on the expression of CD4, CD25, and CD62L to >98% purity. Flow cytometric analyses were performed on a FACScalibur™ flow cytometer with CELLQuest™ software (Becton Dickinson).
In Vitro Expansion of T Cells.
FACS®-purified T cells were stimulated with anti-CD3 and anti-CD28 coupled to 4.5-μm paramagnetic beads (provided by Xcyte Therapeutics Inc.) supplemented with 2,000 IU/ml rhIL-2 (Chiron Corp.) in complete medium, which consisted of 10% heat-inactivated fetal bovine serum (Biosource International), nonessential amino acids, 0.5 mM sodium pyruvate, 5 mM Hepes, 1 mM glutaMax I (all from Invitrogen), and 55 μM β-mercaptoethanol (Sigma-Aldrich) in DMEM base. The cultures were monitored daily and maintained at 0.7–1 × 106/ml by diluting with IL-2–supplemented complete medium for 8–12 d. At the end of the culture, the anti-CD3 and anti-CD28 beads were removed using AutoMACS, and the cells were routinely assayed for CD4, CD62L, and CD25 expression by flow cytometry and for suppressive activity in vitro. It should be pointed out that conventional anti-CD3 plus anti-CD28–coated 6-μm polystyrene beads can be adapted for use in this procedure.
In Vitro Suppression Assays.
Graded numbers of expanded or fresh sorted Tregs were added to 50,000 CD4+ T cells stimulated with 50,000 irradiated splenic APC (2,000 rads) and 1 μg/ml anti-CD3 in a U-bottomed 96-well plate. CD4+ T cell cultures without Tregs were stimulated in the same manner as positive controls. For some experiments, CD4+ T cells from DO11.10 TCR Tg mice were used as responders and the cocultures were stimulated with anti-CD3 as described above or with 0.1 μg/ml OVA peptide. The cultures were maintained at 37°C for a total of 64 or 72 h and pulsed with 1 μCi/well [3H]thymidine for the last 14 or 8 h, respectively. For some experiments, the responder CD4+ cells were labeled with 2.5 μM CFSE before the suppression assay, and the level of proliferation was assessed by determining the dilution of CFSE using flow cytometry 72 h after the initiation of the culture.
Real Time PCR Analysis.
Total RNA was extracted using Trizol reagent (Invitrogen) or RNAeasy (QIAGEN) from expanded T cells. cDNA was synthesized from 50 ng–2.5 μg of each RNA sample using SuperScript II RNase H reverse transcriptase and oligo dT as primer (Invitrogen), and 0.6–31.25 ng of the cDNA was used in each quantitative real time PCR reaction. The same amount of RNA and cDNA was used for each Treg and CD25− sample pair. Primers and probes for FoxP3, CTLA-4, neuropilin, PD-1, TRAIL, and HPRT were purchased as reagent kits from Applied Biosystems. Primer sequences for SOCS-2 were 5′-GCGTCTGGCGAAAGCCCT (forward) and 5′-CTTCATTAACAGTCATACTTCC (reverse), and they were ordered from Intergrated DNA Technologies Inc. The probe sequence for SOCS-2 was 5′-FAM-CGCGAGCTCAGTCAAACAGGATGGT-TAMRA-3′, which was ordered from Applied Biosystems. The real time PCR was performed on an ABI prizm 7700 using Taqman Universal PCR master mix (Applied Biosystems) in duplicates and the average threshold cycles (Ct) of the duplicates were used to calculate the fold change between expanded Tregs and CD4+ CD62L+ CD25− cells. Ct for HPRT was used to normalize the samples. Expression ratios between Tregs and CD25− cells were calculated using the following formula: Expression Ratio (Treg/CD25−)Gene × = 2n, n = (CD25− CtGene × −CD25− CtHPRT) − (Treg CtGene × −Treg CtHPRT).
Western Blot.
5 × 105 of each cell type were lysed in sample buffer (62.5 mM Tris, pH 6.8, 12.5% glycerol, 2% SDS, 30 ng/ml bromophenoblue), sonicated, and passed through 28-gauge needles. The lysates were clarified by centrifugation and boiled for 5 min before separating on a 10% SDS PAGE gel. The samples were transferred to PVDF membrane after electrophoresis and incubated with rabbit anti–FoxP3 antisera (provided by S. Ziegler, Benaroya Research Institute at Virginia Mason, Seattle, WA) followed by horseradish peroxidase–conjugated anti–rabbit Ig. The blot was developed with SuperSignal® Chemiluminescent Substrate (Pierce, Rockford, IL) and visualized on a Kodak Image Station 440CF (Eastman Kodak Co.) and quantified using Kodak Digital Science 1D Image Analysis software 3.0.
Cytokine ELISA.
The level of IL-2, IFN-γ, IL-10, and TGF-β in the culture supernatant was determined by ELISA using antibody pairs purchased from BD Biosciences. For TGF-β ELISA, the culture supernatant was first treated with acid to lower the pH to 2.0 to denature latency-associated peptide to allow the detection of active TGF-β. The supernatant was neutralized to pH 7.0 before ELISA.
Adoptive Transfer.
Expanded T cells were labeled with 2.5 μM CFSE, and 1–3 × 106 cells were transferred via retro-orbital injection. The recipient mice were killed on day 7 after cell transfer and the dilution of CFSE in splenic, peripheral LN, and pancreatic LN cell preparations was determined by flow cytometry. For adoptive transfer of diabetes to NOD.RAG−/− recipients, CD4+ CD62L+ CD25− cells from BDC2.5 TCR Tg mice were purified by cell sorting using a DakoCytomation Mo-Flo and 0.5–1 × 106 cells were transferred to each recipient mouse. For some experiments, the purified Teffs were activated with anti-CD3 and anti-CD28 for 8–10 d before transfer. To transfer diabetes with polyclonal Teffs, 25 × 106 pooled spleen and LN cells from diabetic NOD mice were injected into each recipient. When indicated, expanded Tregs were depleted of anti-CD3 and anti-CD28 beads and washed extensively before mixing with effector cells for injection. The expanded Tregs were similarly processed before transferring into NOD.CD28−/−, chronically diabetic NOD syngeneic islet transplant recipients, and NOD mice with new onset diabetes. Nonfasting blood glucose levels in recipient mice were monitored using an Accu-Check glucometer (Roche Diagnostics Corp.)
Murine Pancreatic Islet Isolation.
Murine islets were isolated using a modified previously published protocol (44). In brief, a 3-ml collagenase P (Roche Molecular Biochemicals) solution (0.75 mg/ml) was injected into the pancreatic duct of 4-wk-old NOD mice. The distended pancreases were removed and incubated at 37°C for 17 min. The liberated free islets were purified by centrifugation on Eurocollin-Ficoll gradients that comprised four different densities (1.108, 1.096, 1.069, and 1.037). After centrifugation, the islet-containing layers between densities 1.069 and 1.096 were collected and washed. Islets were then handpicked for transplantation.
Murine Islet Transplantation.
Naturally diabetic NOD mice were used as recipients. The mice were diabetic for at least 2 wk before transplantation. The recipient mice were maintained with subcutaneous insulin pellets (Lin-Shin Canada, Inc.). 1 d before transplantation, the insulin pellets were removed and hyperglycemia was confirmed on the day of transplantation. 500 isolated and handpicked islets were transplanted beneath the left renal capsule of each recipient. Nonfasting blood glucose levels were determined in all animals daily after transplantation. Return to normoglycemia within 24 h after transplant was indicative of successful surgery. Rejection of the islets grafts was considered to have occurred when nonfasting blood glucose concentration exceeded 250 mg/dl for 3 consecutive days.
Results
Expansion of Tregs from Autoantigen-specific TCR Tg NOD Mice.
Previous studies have shown that the number and function of Tregs in NOD mice decrease over time correlating with clinical disease onset between 16 and 24 wk of age (9, 40). These observations support the use of Tregs to prevent or treat diabetes even after disease onset. However, the ability to use these cells therapeutically is severely limited by the small numbers of cells resident in the circulation or lymphoid organs (<5% of CD4+ T cells in NOD mice and <2% of CD4+ T cells in humans with T1D; references 8 and 39). Moreover, a large number of cells are required for therapeutic efficacy due to an inability at present to select the cells based on antigen specificity. Therefore, we developed a technique for rapid and efficient expansion of autoantigen-specific Tregs based on observations that these cells, present in TCR Tg mice, can be driven into cell cycle with coimmobilized anti-CD3 and anti-CD28 antibodies plus exogenous IL-2. As shown in Fig. 1, A and B, FACS®-purified NOD Tregs cultured with anti-CD3/anti-CD28–coated beads in the presence of IL-2 expanded 150–225-fold in 11 d (Fig. 1 B). In general, the CD4+ CD25− T cells expanded more vigorously (ranging from 300–800-fold in multiple experiments). A purity of >98% CD4+ CD25+ CD62+ T cells was essential to enable successful Treg expansion as a small contamination of either CD25− CD4+ or CD8+ T cells significantly impacted the ability to specifically expand the Tregs (unpublished data). It should be noted that the Treg expansion is dependent on the high level of IL-2 (2,000 IU/ml). No Treg expansion was observed when 200 IU/ml IL-2 was used (unpublished data).
Figure 1.

In vitro expansion of Tregs. (A) Representative flow cytometry plots of CD25 and CD62L expression on CD4 cells from NOD (left), BDC2.5 (middle), and GAD286 (right) mice. FACS®-purified Tregs (•) and CD4+ CD62L+ CD25− cells (○) from NOD (B), BDC2.5 TCR Tg (C), or GAD286 TCR Tg (D) mice were stimulated in vitro with anti-CD3– and anti-CD28–coated beads along with IL-2. (E) T cells from BDC2.5 TCR Tg mice were expanded as described above with p31-linked IAg7-mIgG2a immobilized on latex beads. All cultures were quantitated by viable cell counting.
Previous studies have shown that CD4+ CD25+ Tregs isolated from young NOD mice suppressed the ability of Teffs from diabetic NOD mice to transfer disease in immunodeficient NOD mice (8, 16, 17). However, the process was highly inefficient and the suppressive effects of Tregs in this setting required a 0.5:1 or 1:1 ratio of Treg/Teff. This is likely due to the low precursor frequency of antigen-specific Tregs. Thus, we examined whether Tregs from two different antigen-specific TCR Tg mice (Fig. 1 A) could be expanded in vitro using the same methodology as with the polyclonal NOD Tregs. BDC2.5 TCR Tg mice express a TCR specific for an islet antigen expressed in the granules of β cells, whereas the GAD286 TCR Tg recognizes a peptide derived from the islet antigen GAD. Tregs were purified from BDC2.5 and GAD286 mice and expanded using the anti-CD3/anti-CD28 plus IL-2 cocktail (Fig. 1, C and D). The BDC2.5 cells expressed the Tg TCR αβ based on efficient staining with an MHC peptide tetramer previously shown to react with this TCR (43), and the expanded GAD286 Tregs expressed the Tg TCR-β chain (unpublished data). The CD4+ CD62L+ CD25− and Tregs from BDC2.5 TCR Tg mice can also be expanded using immobilized MHC peptide dimers (Fig. 1 E). These results suggest that a population of CD4+ CD25+ CD62L+ exists in both wild-type and TCR Tg mice that can be expanded using this protocol.
Next, we examined the phenotype of the expanded Tregs by flow cytometry, Western blot, and real time PCR. As can be seen in Fig. 2 A, the expanded Tregs maintained high levels of expression of CD25 as compared with expanded CD25− T cells, whereas the expression of CD62L remained high in both cell types. In addition, quantitative PCR showed that all of the Tregs expressed high levels of SOCS2, PD-1, and CTLA-4 as compared with similarly expanded CD25− T cells. Moreover, the recently identified markers neuropilin and TRAIL (20, 45) were also highly expressed on the expanded Tregs (Fig. 2 B). A high level of cell surface GITR expression was observed on the expanded Tregs. However, this previously identified Treg marker was also induced on the expanded CD25− T cells (19, 20, and unpublished data). It should be noted that the quantitative PCR studies were performed on five separate expanded Treg populations (including both polyclonal and BDC2.5 TCR Tg Tregs) and the relative expression of the Treg-specific genes was highly reproducible. Finally, we examined the recently identified lineage/differentiation marker for Tregs, FoxP3 (Fig. 2, B and C). As noted by both real time PCR and Western blot analyses, the expanded Tregs expressed levels of FoxP3 similar to those observed in fresh Tregs and significantly higher than those in fresh or expanded CD25− T cells. The RNA expression (10-fold) and protein amounts (20-fold) were consistent with previous studies of fresh Tregs, although there was clearly some increase in FoxP3 in CD25− Teffs, suggesting that the culture conditions may induce some Tregs within the CD25− subset, or FoxP3 is expressed at a low level in activated Teffs.
Figure 2.

Phenotype of in vitro–expanded Tregs. (A) Expression of CD25 and CD62L on expanded Tregs and CD4+ CD62L+ CD25− cells was determined by flow cytometry on day 8 after the culture initiation. Results are representative of more than 20 independent experiments. (B) Levels of mRNA for the indicated genes in expanded NOD (filled symbols) or BDC2.5 TCR Tg T cells (open symbols) were determined by real time PCR analysis on day 10 after the initiation of the cultures. The relative expression ratio (Treg/TCD25 −) for each pair of cultures was calculated from Ct values as described in Materials and Methods. The dashed line represents the ratio of 1 (i.e., identical level of gene expression in Treg and CD4+ CD62L+ CD25− cultures). (C) Western blot analysis of FoxP3 protein expression in fresh and expanded T cells. The level of tubulin expression was included as a loading control. Results are representative of three independent experiments. (D) Cytokine secretion by expanded BDC2.5 T cells 48 h after restimulation with antigenic peptide and splenic APC. Results are representative of two independent experiments.
We also examined the ability of the expanded Tregs to secrete cytokines. Unlike activated CD25− T cells, the Tregs did not produce IL-2 or IFN-γ, but rather expressed the immunosuppressive cytokines IL-10 and TGF-β (Fig. 2 D). Thus, the extensive activation and proliferation of the Tregs does not alter the phenotype of the Tregs, which remained distinct from the CD25− T cell subset.
Functional Activity of In Vitro–expanded Tregs.
Previous studies have shown that Tregs can effectively suppress proliferative responses of CD25− T cells stimulated with anti-CD3 and splenic APC. The expanded NOD Tregs efficiently suppressed proliferative responses (Fig. 3 A) and cytokine production including IL-2 and IFN-γ (unpublished data). In fact, in multiple experiments, the expanded Tregs suppressed significantly better than fresh NOD Tregs. The suppression was routinely observed at Treg/Teff ratios of <1:10. Similar results were observed using the expanded Tregs from the TCR Tg mice, as the expanded BDC2.5 Tregs were effective in suppressing the proliferative response of BDC2.5 (unpublished data) as well as polyclonal NOD T cells (Fig. 3 B). Although the expanded Tregs expressed significant levels of IL-10 and TGF-β, suppressor activity was unaffected by the addition of anti–IL-10, anti–TGF-β, or a combination of both antibodies to the in vitro cultures (unpublished data). These results are consistent with numerous models of Treg suppression where cell–cell contact is the primary means of immunosuppression in the in vitro setting (30–36).
Figure 3.

In vitro suppression by expanded Tregs. (A) Fresh and expanded Tregs were compared for their ability to suppress the proliferation of CD4+ responder T cells stimulated with anti-CD3 and T cell–depleted splenocytes. (B) Suppression by Tregs expanded from BDC2.5 TCR Tg or GAD286 TCR Tg mice was assayed as described in A. (C) Suppressive activity of BALB/c-expanded Tregs on CD4+ responder T cells from DO11.10 TCR Tg mice stimulated with anti-CD3 or an OVA peptide. Results are representative of three independent experiments.
To further assess the antigen specificity of the expanded Tregs and determine whether the expanded Tregs were constitutively suppressive, expanded Tregs from normal BALB/c mice were examined for their ability to suppress T cells from the OVA-specific DO11.10 TCR Tg mouse. Tregs and DO11.10 Tg Teffs were cocultured in the presence of OVA antigen (to activate only the Teffs) or anti-CD3 (to activate both the Teff and Tregs). Expanded BALB/c Tregs did not inhibit the proliferative response of the DO11.10 T cells stimulated by the OVA peptide. However, the anti-CD3 response was fully inhibited at low Treg/Teff ratios. These results suggest that the expanded Tregs lack constitutive suppressive activity but require TCR-specific activation for effective suppression. This result also ruled out the trivial possibility that the cells were inhibiting the cultures by consuming available IL-2 through the high level of CD25 expression.
In Vivo Survival and Activation of Expanded Tregs.
Effective suppression of immune responses in vivo by Tregs requires that the cells migrate to appropriate sites, respond to antigen, and survive long-term. We have observed recently that blockade of the CD28/B7 pathway resulted in rapid loss of Tregs in vivo and subsequent loss of critical immune regulation (8, 46). Thus, we examined the ability of expanded Tregs to survive and proliferate in vivo. Expanded Tregs were labeled with CFSE and transferred into normal nonlymphopenic syngeneic mice. At 30 d after transfer, the mice were killed and examined for the number of CFSE+ cells as an indication of cell survival. As seen in Fig. 4 A, a significant number of CFSE+ cells were recovered from mice transferred with expanded Tregs. The number of CFSE+ Tregs was equal to that observed with fresh Tregs transferred in the same manner (Fig. 4 A). In fact, Thy1.1-marked expanded Tregs were observed at least 50 d after transfer (unpublished data).
Figure 4.
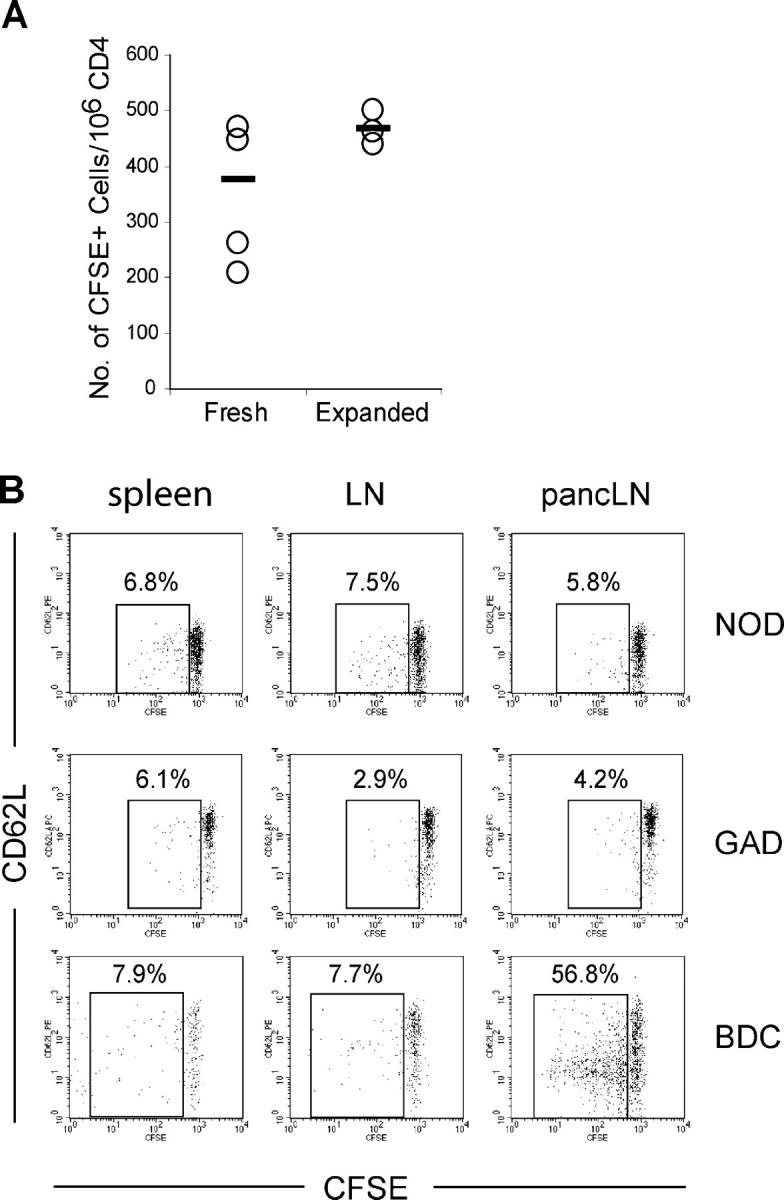
In vivo survival and activation of expanded Tregs. (A) Freshly isolated and expanded BALB/c Tregs were labeled with CFSE and then injected into normal BALB/c mice (106/mouse). All recipient mice were killed on day 30 after injection and the numbers of CFSE+ cells in the peripheral LN and spleen (not shown) were determined by flow cytometry. The data are presented as the number of CD4+ CFSE+ cells/106 endogenous CD4+ T cells. Each circle represents the value from one mouse and the black bar represents the mean of the group. (B) Expanded Tregs from NOD.Thy1.2 (top) and GAD286 TCR Tg Thy1.2 (middle) were labeled with CFSE and 3 × 106 were transferred to normal 8–12-wk-old NOD.Thy1.1 recipients. Expanded BDC2.5 TCR Tg Thy1.1 were labeled with CFSE and 3 × 106 were transferred to normal 8–12-wk-old NOD.Thy1.2 recipients. The presence of transferred cells and their activation status in spleens, LNs, and pancreatic LNs were determined by flow cytometry on day 7 after cell transfer. The dot plots shown are gated on the Thy1.2 for NOD and GAD286 cells and Thy1.1 for BDC2.5 cells. The percentages of cells with CFSE dilution are shown on the plots. Results are representative of at least three recipient mice in two separate experiments.
Next, we analyzed the ability of the adoptively transferred Tregs to respond to antigen and proliferate in vivo. Expanded Tregs from NOD, BDC2.5, and GAD286 mice were labeled with CFSE and transferred into normal nonlymphopenic NOD recipients. At 7 d after transfer, the mice were killed and examined for the dilution of CFSE to assess in vivo proliferation. As seen in Fig. 4 B, top, a small but significant number of Tregs proliferated, as indicated by CFSE dilution. However, there was no selective proliferation of the NOD Tregs in the pancreatic LNs (Fig. 4 B, pancLN), suggesting that a detectable number of islet autoantigen-specific cells did not exist within the NOD Treg repertoire. In contrast to the NOD Tregs, Tregs from BDC2.5 Tg mice proliferated extensively and selectively in the pancreatic LNs, dividing at least three to four times during the 7-d period (Fig. 4 B, bottom). Interestingly, the expression of the CD62L molecule was down-regulated on the surface of expanded Tregs. This is surprising because the cells had undergone multiple proliferative cycles in vitro before transfer and had maintained high levels of CD62L expression. In contrast to the BDC2.5 Tregs, the GAD286 Tregs did not proliferate in vivo (Fig. 4 B, middle). The results of previous studies suggest that T cells in these two TCR Tg mice differ significantly in their thymic development. The BDC2.5 Tg mice do not negatively select the islet-specific T cells in the thymus, but rather develop a small, reproducible number of Tregs. These cells have been shown to block disease manifested by potential pathogenic CD4+ CD25− T cells resident in these animals (47). By comparison, the majority of GAD286 TCR Tg T cells are deleted in the NOD thymus by negative selection. In fact, the minor population of cells that escape use alternative TCR-α chains. Thus, although the peripheral GAD286 TCR Tg cells respond to GAD peptide in vitro, the reactivity is weak and, in contrast to the BDC2.5, they are unable to induce diabetes upon adoptive transfer, suggesting the “absence” of an autoreactive repertoire (42). These results support the conclusion that the two Tg mice are resistant to diabetes for distinct reasons. The GAD286 TCR Tg mice are protected from the development of diabetes due to the potent central tolerance mechanism of clonal deletion and receptor editing. By comparison, the BCD2.5 TCR Tg have circulating autoreactive Tregs that home to peripheral target tissues where they are activated and expand after encountering autoantigen, resulting in immune suppression and homeostasis.
In Vitro–expanded Tregs Suppress Adoptive Transfer of Diabetes In Vivo.
Next, we examined the ability of the expanded BDC2.5 Tregs to suppress diabetes after in vivo cotransfer of activated BDC2.5 T cells into NOD.RAG mice. The Tregs were effective in blocking the transfer of diabetes, functioning at as low as a 1:9 ratio of Treg/Teff (Fig. 5 A), whereas the GAD286 Tregs did not protect even at a Treg/Teff ratio of 1:1 (Fig. 5 B). In fact, the expanded BDC2.5 Tregs suppressed polyclonal T cell–mediated disease. As few as 2 × 106 expanded BDC2.5 Tregs blocked the ability of 25 × 106 diabetogenic NOD spleen and LN cells to transfer disease (Fig. 5 C). The expanded antigen-specific Tregs from the BDC2.5 mice were far more efficient than expanded polyclonal NOD Tregs in preventing the onset of diabetes. The same number (2 × 106) as well as 5 × 106 expanded NOD Tregs did not confer any protection under the same conditions (Fig. 5 C). In fact, even the transfer of four times as many expanded NOD Tregs (8 × 106) only slightly delayed diabetes onset and prevented diabetes in only one diabetogenic cell recipient (Fig. 5 C). This result is consistent with previous findings suggesting that a high ratio of polyclonal Tregs to Teffs is necessary to efficiently suppress disease transfer in this setting (8, 16, 17). Importantly, these data suggest that in vitro activity of the Tregs does not predict in vivo function in this disease setting.
Figure 5.
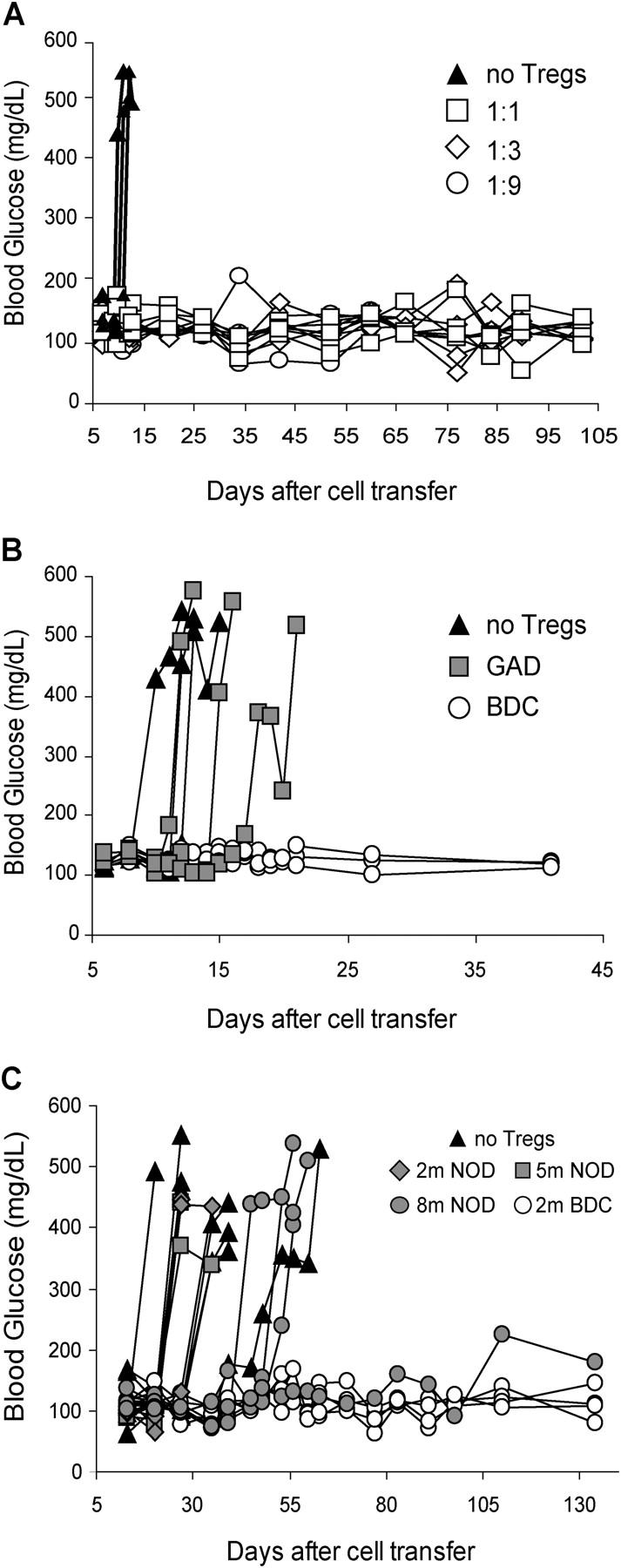
Prevention of diabetes transfer by expanded Tregs. (A) Activated diabetogenic BDC2.5 CD4+ CD62L+ CD25− cells (3.5 × 105) were cotransferred with BDC2.5-expanded Tregs to 8-wk-old NOD. RAG−/− recipients at the indicated ratio. The blood glucose for individual recipient mouse was monitored and plotted to access diabetes. n = 3 for no Tregs and 1:9 groups; n = 4 for 1:1 and 1:3 groups. Results are representative of three independent experiments. (B) Diabetes was induced in 6-wk-old NOD.RAG−/− mice in the same manner as described in A, except that the number of transferred expanded Tregs from GAD286 TCR Tg mice and BDC2.5 TCR Tg mice equaled the number of transferred Teffs (n = 3 mice/group). (C) Diabetes was induced in 5–8-wk-old NOD.RAG−/− or NOD.TCR-α−/− recipients by injection of 25 × 106 pooled spleen and LN cells from diabetic donors (n = 8). Some recipient mice were coinjected with expanded Tregs from NOD (2 × 106, n = 3; 5 × 106, n = 3; 8 × 106, n = 4) or BDC2.5 TCR Tg mice (2 × 106, n = 4). Results represent two independent experiments.
Expanded Tregs Prevent Diabetes In Vivo in a Nonlymphopenic Setting.
Although there are multiple models demonstrating the immunoregulatory activity of Tregs, many of the systems are based on adoptive transfer models that take advantage of lymphopenic mice to enhance Treg proliferation (8, 21–24). Questions have been raised whether disease suppression observed in lymphopenic settings after Treg transfer is due to active regulation or a side efffect of competition for “space” (37, 38). Therefore, we examined the ability of the expanded Tregs to prevent diabetes in a nonlymphopenic animal model. Previous studies have shown that CD28−/− NOD mice have normal numbers of T cells and Th1 responses. In fact, these mice develop exacerbated autoimmunity due to a deficiency in Th2 and Tregs, which were shown to be exquisitely CD28 dependent (8, 21–24). Thus, we examined whether wild-type expanded BDC2.5 Tregs transferred into CD28−/− NOD mice could delay or prevent onset of disease. 5 × 105 Tregs were transferred into 5-wk-old CD28−/− NOD mice and monitored for diabetes (Fig. 6). The transfer of expanded BDC2.5 Tregs prevented the development of diabetes in 100% of mice followed for as long as 20 wk after transfer. In contrast, the transfer of similar numbers of expanded NOD Tregs had no effect on disease incidence (unpublished data). These results suggest that the antigen-specific expanded Tregs functioned in vivo in the face of a fully functional pathogenic T cell response.
Figure 6.

Prevention of autoimmune diabetes in NOD.CD28−/− mice with BDC2.5-expanded Tregs. 5-wk-old prediabetic NOD.CD28−/− mice were injected with 5 × 105 BDC2.5-expanded Tregs (n = 3) or left untreated (n = 4). The development of diabetes was monitored and blood glucose levels of individual mice were plotted. Results are representative of at least five independent experiments.
Expanded Tregs Reverse Diabetes In Vivo.
The ultimate utility of Treg therapy depends on an ability to treat individuals with ongoing disease. Thus, we examined the regulatory effects of expanded BDC2.5 Tregs in NOD mice that had been diabetic for at least 2 wk to ensure total endogenous islet cell destruction. Expanded BDC2.5 Tregs were transferred into diabetic NOD mice in conjunction with 500 syngeneic NOD islet transplant. Mice reverted to normoglycemia within 24–48 h after transplantation. However, unlike the control mice that rejected the transplanted islets within 2 wk, the transfer of 2 × 106 BDC2.5-expanded Tregs blocked rejection of the syngeneic islets, consistent with an ability of the suppressor cells to block ongoing autoimmunity in this setting. The transfer of 5 × 106 polyclonal NOD Tregs had no effect in this model. More significantly, the adoptive transfer of expanded BDC2.5 Tregs reversed diabetes in new-onset diabetic NOD mice (Fig. 7 B). Previous studies have shown that NOD mice diagnosed within the first week of hyperglycemia retain sufficient insulin-producing β cell activity, such that effective immunosuppression introduced at that time can reverse diabetes. To test the efficacy of expanded BDC2.5 Tregs in this setting, 107 Tregs were transferred into NOD mice diagnosed with disease based on recently elevated blood glucose levels (>300 mg/dL). The transferred Tregs reversed diabetes in 60% of the mice. Thus, the expanded Tregs were extremely effective in blocking and reversing diabetes in an ongoing autoimmune setting.
Figure 7.

Reversal of diabetes with expanded Tregs. (A) NOD mice with chronic diabetes were transplanted with syngeneic islets under the kidney capsule. On the day of transplantation, some recipient mice received 5 × 106 NOD-expanded Tregs (n = 4) or 2 × 106 BDC2.5-expanded Tregs (n = 5), and the remaining mice (n = 3) were left untreated. Blood glucose level was monitored. All islet recipients normalized blood glucose within the first day after transplantation. Results are representative of two independent experiments. (B) NOD mice with new onset diabetes (blood glucose > 300 mg/dL, n = 7) were injected with 107 BDC2.5-expanded Tregs and blood glucose was monitored. Two consecutive readings of blood glucose of <250 mg/dL was considered remission of diabetes.
Discussion
The past few years have seen an increased interest in and understanding of the role of Tregs in immune homeostasis. As an example, we have recently shown that anti-CD3 therapy in new-onset diabetes leads to the production and expansion of TGF-β–dependent Tregs that reverse diabetes and promote long-term tolerance (9). Moreover, Edinger et al. (24) have shown that the adoptive transfer of Tregs in mice can block graft-versus-host disease without affecting graft-versus-leukemia responses. These studies and others have led investigators to conclude that these cells might be involved in human autoimmune diseases. The results have also prompted investigators to consider this cell type for immunotherapy. However, successful application of adoptive cellular immune therapy with these cells will depend on a large, reliable source of well-characterized Tregs.
In this study, we describe a robust and effective method for expanding Tregs while retaining their phenotype and suppressive activities. We demonstrate that expanded antigen-specific Tregs prevent the development of diabetes and even restore an immune regulatory state that reverses diabetes and allows the mice to maintain long-term immune homeostasis. The expanded antigen-specific Tregs survived long-term in vivo, were less dependent on CD28 costimulation (unpublished data), but required antigen exposure for functional activity. To our knowledge, this is the first example of Treg activity in a lymphocyte-sufficient diabetic animal. These results are especially important in light of a recent publication by Gregori et al. (40), suggesting that Tregs do not effectively suppress Teffs in the setting of diabetes. The difference between the two studies may reflect either of the different assay systems: in vivo versus in vitro or that the expanded Tregs might be more efficient. In this regard, we have noted that the expanded Tregs do indeed survive better in vivo than the fresh Tregs.
Among the more interesting and perhaps unexpected observations in this study was the differential dependency of antigen specificity for in vivo versus in vitro Treg functions. Studies in multiple models have shown that polyclonal Tregs are effective in blocking autoimmunity (8, 21–24). In fact, in most settings (with a few notable exceptions such as allogeneic organ transplantation; reference 28), the ability to functionally suppress in vitro has been highly predictive of in vivo efficacy and presumed to be antigen nonspecific. Yet, in this study, the islet autoantigen-specific BDC2.5 Tregs were significantly more efficient than polyclonal NOD Tregs in regulating autoimmune responses in vivo. This discriminating activity was not predicted by the in vitro studies that demonstrated equal efficacy in blocking anti-CD3 responses among the various expanded Tregs. There are several potential explanations for this observation. First, it is possible that the expansion method causes selective depletion of the autoantigen-specific Tregs in the polyclonal NOD populations. This seems unlikely as the BDC2.5 Tregs grew in vitro equally well when compared with the polyclonal NOD Tregs. Moreover, cursory analysis of the TCR usage in the expanded Tregs showed no preferential changes in the repertoire of the cells. Second, it is possible that the different expanded polyclonal Treg populations homed or functioned differently in vivo. However, analysis of a panel of cell surface markers, intracellular proteins, and soluble cytokines suggested that the various cell populations all retained the essential properties of Tregs including the high expression of CD25 and FoxP3. Most likely, the differences observed reflected the model in which we have tested the Tregs. In many models, Tregs are cotransferred with effector cells into lymphoid cell–deficient animals, in which the Tregs home to the site of inflammation and inhibit Teff response indirectly by competing for the “niche.” In fact, in one model of inflammatory bowel disease (37), CD25− T cells are as effective as CD25+ Tregs with little evidence for antigen specificity. Similarly, there has been little evidence supporting an essential role for antigen-specific cells for the treatment of graft-versus-host disease. In sharp contrast, Treg-mediated immune regulation is routinely found to be antigen specific in the nonlymphopenic setting (28, 48–50). Thus, the greater functional activity of the antigen-specific Tregs in the NOD models described herein is most likely due to the fully functional immune system in these animals and the requirement for effective antigen-mediated reactivation of Tregs at the inflammatory site.
It is important to note that in the absence of antigen-specific activation, the Tregs have no effect on regulating disease. This is most evident in the adoptive transfer studies using the Tregs isolated from Tg mice on the NOD background expressing a TCR specific for peptide epitope 286–300 (p286) of GAD65. Although lymphocytes from these TCR Tg mice proliferated and produced cytokines when stimulated in vitro with GAD65 peptide 286–300 (42), the response was weak and the T cells escaping deletion expressed alternative TCR-α chains that were unable to transfer diabetes. In fact, the only model where the GAD Tg T cells were functional was in an adoptive transfer system where the p286-tetramer+ CD4+ T cells from TCR Tg mice delayed diabetes induced in NOD.SCID mice by diabetic NOD spleen cells independent of CD25 expression, not unlike other systems where the transferred T cells suppress independent of CD25 expression or antigen-specific mechanisms (42).
Finally, the observation that the Tregs were able to reverse diabetes has important implications for clinical therapy. We imagine a scenario in a number of autoimmune settings where Tregs are isolated from patients either during remission (as would be the case for systemic lupus erythematosis or multiple sclerosis) or soon after disease onset (as would be the case for T1D). The cells would be expanded and reintroduced at the time of maximal disease activity to moderate the inflammatory response. In some cases this could be combined with rapamycin, anti-CD3, or other drugs that cause deletion of the pathogenic cells without affecting the Tregs (unpublished data). Together, these therapies could both reduce the short-term pathogenic responses while reinstating a homeostatic balance for long-term tolerance induction. Findings in this study suggest that the efficacy of Treg-based immune therapy is critically dependent on the antigen specificity of the Tregs, at least in the autoimmune diabetes setting. Therefore, it is important to develop a procedure to selectively and reproducibly expand antigen-specific Tregs from polyclonal populations for therapeutic use. Many islet-specific T cell antigens have been identified to contribute to diabetes development in both mice and humans and MHC multimer coupled with these antigenic epitopes has been developed (43, 51–53). It is possible that these MHC multimer reagents can be adapted for expanding islet-specific Tregs. Current efforts are underway to identify antigen-specific Tregs in the polyclonal population and develop a protocol to selectively expand islet antigen–specific Tregs from normal NOD mice using immobilized antigenic peptide–linked MHC multimers. In addition, we are developing similar techniques for purifying and expanding human Tregs for clinical use in these settings.
Acknowledgments
We thank Paul Wegfarht for expert assistance with the mice, Shuwei Jiang and Cliff McArthur for cell sorting, and Dr. Abul Abbas and all Bluestone Lab members for critical discussion. The authors would also like to thank Dr. Steve Ziegler for providing anti-FoxP3 antibodies for this study.
This work was supported by Juvenile Diabetes Research Foundation Center grant number 4-1999-841 and National Institutes of Health grant number R37 AI46643.
Abbreviations used in this paper: APC, allophycocyanin; CFSE, carboxyfluorescein diacetate succinimidyl ester; Ct, threshold cycle(s); GITR, glucocorticoid-induced TNF receptor; GAD, glutamic acid decarboxylase; NOD, nonobese diabetic; T1D, type 1 diabetes; Teff, T effector cell; Tg, transgenic; Treg, regulatory T cell.
References
- 1.Waterhouse, P., J.M. Penninger, E. Timms, A. Wakeham, A. Shahinian, K.P. Lee, C.B. Thompson, H. Griesser, and T.W. Mak. 1995. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 270:985–988. [DOI] [PubMed] [Google Scholar]
- 2.Tivol, E.A., F. Borriello, A.N. Schweitzer, W.P. Lynch, J.A. Bluestone, and A.H. Sharpe. 1995. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 3:541–547. [DOI] [PubMed] [Google Scholar]
- 3.Kulkarni, A.B., C.G. Huh, D. Becker, A. Geiser, M. Lyght, K.C. Flanders, A.B. Roberts, M.B. Sporn, J.M. Ward, and S. Karlsson. 1993. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA. 90:770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shull, M.M., I. Ormsby, A.B. Kier, S. Pawlowski, R.J. Diebold, M. Yin, R. Allen, C. Sidman, G. Proetzel, D. Calvin, et al. 1992. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 359:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 6.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Scurfin in CD4+ CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 7.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 8.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+ CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]
- 9.Belghith, M., J.A. Bluestone, S. Barriot, J. Megret, J.F. Bach, and L. Chatenoud. 2003. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat. Med. 9:1202–1208. [DOI] [PubMed] [Google Scholar]
- 10.Ueda, H., J.M. Howson, L. Esposito, J. Heward, H. Snook, G. Chamberlain, D.B. Rainbow, K.M. Hunter, A.N. Smith, G. Di Genova, et al. 2003. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 423:506–511. [DOI] [PubMed] [Google Scholar]
- 11.Sakaguchi, S., N. Sakaguchi, J. Shimizu, S. Yamazaki, T. Sakihama, M. Itoh, Y. Kuniyasu, T. Nomura, M. Toda, and T. Takahashi. 2001. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol. Rev. 182:18–32. [DOI] [PubMed] [Google Scholar]
- 12.Chatenoud, L., B. Salomon, and J.A. Bluestone. 2001. Suppressor T cells–they're back and critical for regulation of autoimmunity! Immunol. Rev. 182:149–163. [DOI] [PubMed] [Google Scholar]
- 13.Wood, K.J., and S. Sakaguchi. 2003. Regulatory T cells in transplantation tolerance. Nat. Rev. Immunol. 3:199–210. [DOI] [PubMed] [Google Scholar]
- 14.Singh, B., S. Read, C. Asseman, V. Malmstrom, C. Mottet, L.A. Stephens, R. Stepankova, H. Tlaskalova, and F. Powrie. 2001. Control of intestinal inflammation by regulatory T cells. Immunol. Rev. 182:190–200. [DOI] [PubMed] [Google Scholar]
- 15.Curotto de Lafaille, M.A., and J.J. Lafaille. 2002. CD4(+) regulatory T cells in autoimmunity and allergy. Curr. Opin. Immunol. 14:771–778. [DOI] [PubMed] [Google Scholar]
- 16.Herbelin, A., J.M. Gombert, F. Lepault, J.F. Bach, and L. Chatenoud. 1998. Mature mainstream TCR alpha beta+ CD4+ thymocytes expressing l-selectin mediate “active tolerance” in the nonobese diabetic mouse. J. Immunol. 161:2620–2628. [PubMed] [Google Scholar]
- 17.Lepault, F., and M.C. Gagnerault. 2000. Characterization of peripheral regulatory CD4+ T cells that prevent diabetes onset in nonobese diabetic mice. J. Immunol. 164:240–247. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi, T., T. Tagami, S. Yamazaki, T. Uede, J. Shimizu, N. Sakaguchi, T.W. Mak, and S. Sakaguchi. 2000. Immunologic self-tolerance maintained by CD25+ CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J. Exp. Med. 192:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu, J., S. Yamazaki, T. Takahashi, Y. Ishida, and S. Sakaguchi. 2002. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3:135–142. [DOI] [PubMed] [Google Scholar]
- 20.McHugh, R.S., M.J. Whitters, C.A. Piccirillo, D.A. Young, E.M. Shevach, M. Collins, and M.C. Byrne. 2002. CD4(+) CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 16:311–323. [DOI] [PubMed] [Google Scholar]
- 21.Read, S., V. Malmstrom, and F. Powrie. 2000. Cytotoxic T lymphocyte–associated antigen 4 plays an essential role in the function of CD25+ CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor, P.A., C.J. Lees, and B.R. Blazar. 2002. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 99:3493–3499. [DOI] [PubMed] [Google Scholar]
- 23.Hoffmann, P., J. Ermann, M. Edinger, C.G. Fathman, and S. Strober. 2002. Donor-type CD4+ CD25+ regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J. Exp. Med. 196:389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edinger, M., P. Hoffmann, J. Ermann, K. Drago, C.G. Fathman, S. Strober, and R.S. Negrin. 2003. CD4+ CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat. Med. 9:1144–1150. [DOI] [PubMed] [Google Scholar]
- 25.Lohr, J., B. Knoechel, S. Jiang, A.H. Sharpe, and A.K. Abbas. 2003. The inhibitory function of B7 costimulators in T cell responses to foreign and self-antigens. Nat. Immunol. 4:664–669. [DOI] [PubMed] [Google Scholar]
- 26.Graca, L., S.P. Cobbold, and H. Waldmann. 2002. Identification of regulatory T cells in tolerated allografts. J. Exp. Med. 195:1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asseman, C., S. Mauze, M.W. Leach, R.L. Coffman, and F. Powrie. 1999. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 190:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kingsley, C.I., M. Karim, A.R. Bushell, and K.J. Wood. 2002. CD25+ CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J. Immunol. 168:1080–1086. [DOI] [PubMed] [Google Scholar]
- 29.Belkaid, Y., C.A. Piccirillo, S. Mendez, E.M. Shevach, and D.L. Sacks. 2002. CD4+ CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 420:502–507. [DOI] [PubMed] [Google Scholar]
- 30.Thornton, A.M., and E.M. Shevach. 1998. CD4+ CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piccirillo, C.A., J.J. Letterio, A.M. Thornton, R.S. McHugh, M. Mamura, H. Mizuhara, and E.M. Shevach. 2002. CD4+ CD25+ regulatory T cells can mediate suppressor function in the absence of transforming growth factor β1 production and responsiveness. J. Exp. Med. 196:237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dieckmann, D., H. Plottner, S. Berchtold, T. Berger, and G. Schuler. 2001. Ex vivo isolation and characterization of CD4+ CD25+ T cells with regulatory properties from human blood. J. Exp. Med. 193:1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apostolou, I., A. Sarukhan, L. Klein, and H. von Boehmer. 2002. Origin of regulatory T cells with known specificity for antigen. Nat. Immunol. 3:756–763. [DOI] [PubMed] [Google Scholar]
- 34.Levings, M.K., R. Sangregorio, and M.G. Roncarolo. 2001. Human CD25+ CD4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J. Exp. Med. 193:1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jonuleit, H., E. Schmitt, M. Stassen, A. Tuettenberg, J. Knop, and A.H. Enk. 2001. Identification and functional characterization of human CD4+ CD25+ T cells with regulatory properties isolated from peripheral blood. J. Exp. Med. 193:1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levings, M.K., R. Sangregorio, C. Sartirana, A.L. Moschin, M. Battaglia, P.C. Orban, and M.G. Roncarolo. 2002. Human CD25+ CD4+ T suppressor cell clones produce transforming growth factor β, but not interleukin 10, and are distinct from type 1 T regulatory cells. J. Exp. Med. 196:1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barthlott, T., G. Kassiotis, and B. Stockinger. 2003. T cell regulation as a side effect of homeostasis and competition. J. Exp. Med. 197:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stockinger, B., T. Barthlott, and G. Kassiotis. 2001. T cell regulation: a special job or everyone's responsibility? Nat. Immunol. 2:757–758. [DOI] [PubMed] [Google Scholar]
- 39.Kukreja, A., G. Cost, J. Marker, C. Zhang, Z. Sun, K. Lin-Su, S. Ten, M. Sanz, M. Exley, B. Wilson, et al. 2002. Multiple immuno-regulatory defects in type-1 diabetes. J. Clin. Invest. 109:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gregori, S., N. Giarratana, S. Smiroldo, and L. Adorini. 2003. Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J. Immunol. 171:4040–4047. [DOI] [PubMed] [Google Scholar]
- 41.Yamazaki, S., T. Iyoda, K. Tarbell, K. Olson, K. Velinzon, K. Inaba, and R.M. Steinman. 2003. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 198:235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarbell, K.V., M. Lee, E. Ranheim, C.C. Chao, M. Sanna, S.K. Kim, P. Dickie, L. Teyton, M. Davis, and H. McDevitt. 2002. CD4+ T cells from glutamic acid decarboxylase (GAD)65-specific T cell receptor transgenic mice are not diabetogenic and can delay diabetes transfer. J. Exp. Med. 196:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masteller, E.L., M.R. Warner, W. Ferlin, V. Judkowski, D. Wilson, N. Glaichenhaus, and J.A. Bluestone. 2003. Peptide-MHC class II dimers as therapeutics to modulate antigen-specific T cell responses in autoimmune diabetes. J. Immunol. 171:5587–5595. [DOI] [PubMed] [Google Scholar]
- 44.Lenschow, D.J., Y. Zeng, K.S. Hathcock, L.A. Zuckerman, G. Freeman, J.R. Thistlethwaite, G.S. Gray, R.J. Hodes, and J.A. Bluestone. 1995. Inhibition of transplant rejection following treatment with anti-B7-2 and anti-B7-1 antibodies. Transplantation. 60:1171–1178. [DOI] [PubMed] [Google Scholar]
- 45.Gavin, M.A., S.R. Clarke, E. Negrou, A. Gallegos, and A. Rudensky. 2002. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol. 3:33–41. [DOI] [PubMed] [Google Scholar]
- 46.Tang, Q., K.J. Henriksen, E.K. Boden, A.J. Tooley, J. Ye, S.K. Subudhi, X.X. Zheng, T.B. Strom, and J.A. Bluestone. 2003. Cutting edge: CD28 controls peripheral homeostasis of CD4+ CD25+ regulatory T cells. J. Immunol. 171:3348–3352. [DOI] [PubMed] [Google Scholar]
- 47.Kanagawa, O., A. Militech, and B.A. Vaupel. 2002. Regulation of diabetes development by regulatory T cells in pancreatic islet antigen-specific TCR transgenic nonobese diabetic mice. J. Immunol. 168:6159–6164. [DOI] [PubMed] [Google Scholar]
- 48.Qin, S., S.P. Cobbold, H. Pope, J. Elliott, D. Kioussis, J. Davies, and H. Waldmann. 1993. “Infectious” transplantation tolerance. Science. 259:974–977. [DOI] [PubMed] [Google Scholar]
- 49.Chen, Z.K., S.P. Cobbold, H. Waldmann, and S. Metcalfe. 1996. Amplification of natural regulatory immune mechanisms for transplantation tolerance. Transplantation. 62:1200–1206. [DOI] [PubMed] [Google Scholar]
- 50.van Maurik, A., M. Herber, K.J. Wood, and N.D. Jones. 2002. Cutting edge: CD4+ CD25+ alloantigen-specific immunoregulatory cells that can prevent CD8+ T cell-mediated graft rejection: implications for anti-CD154 immunotherapy. J. Immunol. 169:5401–5404. [DOI] [PubMed] [Google Scholar]
- 51.Reijonen, H., W.W. Kwok, and G.T. Nepom. 2003. Detection of CD4+ autoreactive T cells in T1D using HLA class II tetramers. Ann. NY Acad. Sci. 1005:82–87. [DOI] [PubMed] [Google Scholar]
- 52.Eisenbarth, G.S., and G.T. Nepom. 2002. Class II peptide multimers: promise for type 1A diabetes? Nat. Immunol. 3:344–345. [DOI] [PubMed] [Google Scholar]
- 53.Maus, M.V., J.L. Riley, W.W. Kwok, G.T. Nepom, and C.H. June. 2003. HLA tetramer-based artificial antigen-presenting cells for stimulation of CD4+ T cells. Clin. Immunol. 106:16–22. [DOI] [PubMed] [Google Scholar]