

Abstract
A role for Lyn kinase as a positive regulator of immunoglobulin (Ig)E-dependent allergy has long been accepted. Contrary to this belief, Lyn kinase was found to have an important role as a negative regulator of the allergic response. This became apparent from the hyperresponsive degranulation of lyn − / − bone marrow–derived mast cells, which is driven by hyperactivation of Fyn kinase that occurs, in part, through the loss of negative regulation by COOH-terminal Src kinase (Csk) and the adaptor, Csk-binding protein. This phenotype is recapitulated in vivo as young lyn − / − mice showed an enhanced anaphylactic response. In vivo studies also demonstrated that as lyn − / − mice aged, their serum IgE increased as well as occupancy of the high affinity IgE receptor (FcεRI). This was mirrored by increased circulating histamine, increased mast cell numbers, increased cell surface expression of the high affinity IgE receptor (FcεRI), and eosinophilia. The increased IgE production was not a consequence of increased Fyn kinase activity in lyn − / − mice because both lyn − / − and lyn − / − fyn − / − mice showed high IgE levels. Thus, lyn − / − mice and mast cells thereof show multiple allergy-associated traits, causing reconsideration of the possible efficacy in therapeutic targeting of Lyn in allergic disease.
Keywords: Lyn kinase, IgE, mast cells, allergy, degranulation
Introduction
The initiation of an allergic response requires the activation of mast cells and basophils and is primarily a consequence of allergen-induced aggregation of the IgE-occupied FcεRI. This aggregation initiates Lyn kinase–dependent phosphorylation of FcεRI by transphosphorylation (1), a process that likely depends on the receptors' surrounding lipid environment (2). This initial phosphorylation step is essential for activation of the signal-amplifying Syk kinase whose activity leads to the assembly of several plasma membrane–localized macromolecular signaling complexes that govern the biochemical signals responsible for initiating mast cell degranulation and lymphokine responses (for review see references 3 and 4). Because of its central role in phosphorylating FcεRI, Lyn kinase has long been considered a positive effector in the initiation of the allergic response. However, several recent studies on B cells and monocytes suggested that Lyn kinase has a negative regulatory role because its absence resulted in the loss of phosphorylation of several inhibitory molecules and/or loss of phosphatase activity at the plasma membrane (5–7).
Bone marrow–derived mast cells (BMMCs) derived from lyn − / − mice are not inhibited in their ability to degranulate via FcεRI (8–10). This finding demonstrated Lyn kinase as dispensable for mast cell degranulation and altered the view of the role of Lyn kinase in driving this response. Our previous studies showed that degranulation of Lyn-deficient mast cells was hyperresponsive even though FcεRI phosphorylation was reduced (9, 11). On the other hand, degranulation of Lyn-deficient mast cells depended on phosphatidylinositol 3-OH kinase (PI3K) activity, and was associated with constitutive activation of PKCδ (10). Subsequently, we found that Fyn kinase was an upstream activator of PI3K and in its absence BMMCs failed to degranulate (10). Moreover, the degranulation of both wild-type and Lyn-deficient mast cells was effectively inhibited by the PI3K inhibitor, LY294002 (10), consistent with the view that downstream of FcεRI, positive regulation of mast cell degranulation is Lyn independent but Fyn and PI3K dependent.
These findings, together with genetic evidence of Lyn kinase as a negative regulator of IL-4 signaling (6), led us to explore how Lyn kinase mediates negative control on mast cell degranulation and whether its in vivo role recapitulates negative regulation of allergic responses. Among our findings, lyn − / − mice demonstrated several allergy-associated traits including increased serum IgE, increased circulating histamine, increased numbers of mast cells in the skin and peritoneum, and increased expression of mast cell FcεRI, consistent with the view that increased IgE enhances FcεRI expression, mast cell survival, and could lead to mast cell degranulation (for review see reference 12; 13, 14). BMMCs from lyn − / − mice showed increased Fyn kinase activity and were hypersensitive and hyperresponsive due in part to the loss of appropriate targeting of COOH-terminal Src kinase (Csk), which phosphorylates Src family protein tyrosine kinases (Src PTKs) at a consensus COOH-terminal tyrosine and promotes an inactive closed conformation. Collectively, our findings show that Lyn kinase plays a broad negative regulatory role in vivo that is important for control of mast cell responsiveness and allergy-associated traits.
Materials and Methods
Reagents and Antibodies.
DNP-specific mouse IgE was produced as previously described (15). DNP human serum albumin (DNP-HSA; Ag; Sigma-Aldrich) was diluted in PBS before use. Rabbit antibodies to Fyn (FYN3), Lyn (44), Hck (M28), Src (N16), Csk-binding protein (Cbp), Csk, ERK2, and p38MAPK were from Santa Cruz Biotechnology, Inc. Biotinylated antibody to phosphotyrosine (4G10) was from Upstate Biotechnology. The rabbit phospho-Src (Y418) and phospho-Lyn (Y507) antibodies, which also recognize Fyn (Y417) and (Y528), were from Biosource International and Cell Signaling Technologies, respectively. Immunoblotting antibodies to Cbp were provided by B. Schraven (Magdeburg University, Magdeburg, Germany) and T. Yamashita (Hokkaido University, Sapporo, Japan). Mouse anti–phospho-ERK2 and rabbit anti–phospho-p38MAPK were from Cell Signaling Technologies. Rabbit anti–phospho-Akt (Ser472/473/474) was from BD Biosciences. Secondary antibodies or reagents used for immunoblotting were: sheep anti–mouse IgG–horseradish peroxidase (HRP) and donkey anti–rabbit IgG-HRP (Amersham Biosciences), or Extravidin-HRP from Sigma-Aldrich. The Src-selective inhibitor, PP2, was from Calbiochem. For measurement of serum IgE levels, purified anti–mouse IgE (clone R53-72), purified mouse IgE (κ isotype control), biotin rat anti–mouse IgE (clone R35-118), and streptavidin-HRP (SAv-HRP) were purchased from BD Biosciences. ABTS peroxidase substrate and peroxidase solution B were from KPL Gaithersburg. The ELISA kit for serum histamine detection was from Beckman Coulter/Immunotech.
Mice and In Vivo Experiments, Bone Marrow Isolation, and BMMC Culture.
Mice used for these studies were as follows: fyn − / − (SV129 × C57/BL6 [N2]); lyn − / − (SV129 × C57/BL6 [N7]); lyn up/up (Y508F, SV129 × C57/BL6 [N2]; reference 7); and lyn − / − fyn − / −, wt, lyn − / −, and fyn − / − (SV129 × C57/BL6, generated from fyn − / − [N2] × lyn − / − [N2] mating). Mice were maintained and used in accordance with National Institutes of Health (NIH) guidelines and National Institute of Arthritis and Musculoskeletal and Skin Diseases–approved animal study proposal A001-04-03. Bone marrow was isolated from 5–6-wk-old wild-type and gene-disrupted mice as previously described (16). In vivo and cell responses of the gene-disrupted mice were compared with age- and sex-matched litter mates. For in vivo studies, most mice were used at an age of 4 or 7 wk. Limited numbers of mice of 5, 8, 12, and 16 wk were used to confirm the relationship of age to induction of allergic-like traits.
Peritoneal lavage was with 3 ml RPMI medium. Recovered cells were analyzed for mast cell content and FcεRI expression by staining with antibodies to KIT and FcεRI, or with an appropriate IgG isotype control as previously described (16). To determine receptor occupancy, the mean fluorescence intensity of recovered cells was compared before and after incubation with exogenous DNP-specific IgE. Cytospins of the recovered peritoneal cells were stained with Wright-Giemsa to distinguish mast cells, eosinophils, and monocytes. Passive systemic anaphylaxis (PSA) challenge was performed essentially as previously described (16). The concentration of serum IgE before challenge was determined by ELISA and plasma histamine (anaphylaxis) was measured by a competitive histamine ELISA (16). Significance was determined by an unpaired t test using a 95% confidence interval. BMMCs were grown in RPMI media supplemented with FBS, stem cell factor (SCF), and IL-3 as previously described (16, 17). For BMMC cultures, FcεRI receptor expression was monitored weekly as previously described (16) and cells were used for experiments when >95% of the population was FcεRI+. Cells were rested in the absence of serum, SCF, and IL-3 for 2–4 h before stimulation.
Ag Stimulation, Lysates, Immunoprecipitation, Immunoblots, and PP2 Experiments.
3.0 × 107 cells were sensitized with DNP-specific IgE for 2 h. Unless otherwise indicated, cells were stimulated with 300 ng/ml Ag (DNP-HSA). Preparation of cell lysates, immunoprecipitations, and immunoblotting procedures are described elsewhere (18). Relative quantitation of immunoblots was performed by densitometry. To determine the effect of PP2 on mast cell degranulation and signaling, 2.5 × 106 (degranulation) or 7.5 × 106 (MAPK and Akt phosphorylation) BMMCs were incubated with a 10-μM concentration of PP2 for 15 min at 37°C before Ag stimulation at varying doses (degranulation) or with 300 ng/ml (MAPK/Akt). After stimulation, degranulation was measured as indicated below or cells for MAPK/Akt activation were pelleted by centrifugation and lysed in 250 μl SDS sample buffer and proteins were resolved by SDS-PAGE followed by transfer to nitrocellulose membranes.
Hexosaminidase Release Assay and Northern Blots.
Degranulation was measured by assaying β hexosaminidase release as previously described (16). For control, the supernatants of IgE-sensitized mast cells not stimulated with Ag were used. Triplicate wells were used for each condition and a minimum of three individual experiments from at least two independent BMMC cultures were performed. For Northern blots, total RNA was isolated from wild-type, Lyn−/−, and Fyn−/− mast cells using TRIzol reagent (Life Technologies). Agarose/formaldehyde gel electrophoresis of RNA and transfer to the Zeta-Probe blotting membrane (Bio-Rad Laboratories) was performed according to standard procedure. The membranes were prehybridized and hybridized at 65°C in PerfectHybTM Plus hybridization buffer (Sigma-Aldrich) overnight with a Fyn cDNA probe encompassing nucleotides 15–480 or a cDNA of the entire β actin gene (Lofstrand). After three washes at room temperature in 2× SSC for 10 min, two stringent washes were performed at 65°C for 30 min in 2× SSC, 0.2% SDS. Blots were then subjected to autoradiography.
Retroviral Gene Transduction.
Bone marrow cells from femurs of Lyn−/− mice were grown in the presence of IL-3 and SCF for 10 d. The retroviral packaging cells Phoenix-E and Bosc 23 (1:9 ratio) were mixed 24 h before transfection. Cells were transfected using Fugene 6 (Roche) with an empty pMX vector (ve/ve) or with wild-type Lyn in the pMX-pIE vector (re/re) containing a green fluorescent protein (GFP) reporter. For infection, bone marrow cells were cocultured with the packaging cells for 48 h. The bone marrow cells were then harvested from the mixed culture and grown in IL-3 and SCF containing media (growth medium) for 48 h before selection of transduced cells with 1.0 μg/ml puromycin. After 14–21 d of selection, BMMCs were screened for GFP and FcεRI expression. Typical cultures used for analysis showed >77 and 95% GFP or FcεRI expression, respectively. Wild-type Lyn expression in transduced cells was ∼1.5-fold greater than in wild-type BMMCs.
In Vitro Kinase (IVK) Assays.
The activity of Src PTKs was measured by an immune complex autophosphorylation assay. 3.0 × 107 BMMCs were lysed in RIPA buffer (10 mM NaH2PO4, 50 mM NaCl, 50 mM NaF, 0.5% deoxycholic acid, 0.05% NaN3, 0.1% SDS, 1% Triton X-100, and 1 mM Na3VO4). Fyn, Hck, and Src were immunoprecipitated with 5 μg of the appropriate antibody prebound to protein A–sepharose for 3 h at 4°C. Immune-complexed proteins were recovered by centrifugation and washed twice with RIPA buffer followed by two additional washes with kinase buffer (50 mM Hepes, 0.1 mM EDTA, 0.015% Triton X-100, 0.07% 2-mercaptoethanol, 10 μM Na3VO4, pH 7.0). The IVK reaction was started by resuspension of the pellet in 30 μl kinase buffer containing 10 mM ATP and 10 mM MgCl2. Samples were incubated at 30°C for 20 min with vigorous mixing and reactions were stopped by addition of 30 μl 2× SDS sample buffer and immediate boiling for 3 min. Protein A–sepharose beads were removed by centrifugation and recovered proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Autophosphorylation was detected by antiphosphotyrosine blotting and protein loading was determined by reprobing stripped membranes with the appropriate antibody. Quantitation was by densitometry.
Online Supplemental Material.
Figs. S1 and S2 showing increased Fyn phosphorylation and protein as well as reconstitution of Cbp phosphorylation by exogenous expression of Lyn in Lyn-deficient mast cells, are available at http://www.jem.org/cgi/content/full/jem.20040382/DC1.
Results
Lyn-deficient Mast Cells Show Increased Fyn Kinase Activity.
To explore the underlying mechanism for the observation of increased degranulation in Lyn-deficient mast cells (10), we examined the activity of several Src PTKs. Fyn, Hck, and p60 Src (Src) have been demonstrated to be expressed and/or activated upon FcεRI stimulation in either the mast cell line, RBL-2H3, or in BMMCs (10, 19). Yes and Fgr are minimally detected in BMMCs as determined by RT-PCR and Western blotting (10). In IVK autophosphorylation assays, only Fyn kinase showed enhanced activity (Fig. 1 a) in Lyn-deficient mast cells. Fyn activity was constitutively enhanced but was further increased by FcεRI stimulation. This was also reflected by increased phosphorylation of Fyn on Y417 (pY417), the regulatory tyrosine found in the activation loop, relative to the COOH-terminal inhibitory tyrosine, Y528 (pY528; Fig. S1 a, available at http://www.jem.org/cgi/content/full/jem.20040382/DC1). Interestingly, the amount of Fyn protein was also slightly increased by an average of 1.3-fold in the absence of Lyn, suggesting negative regulation of Fyn protein expression by Lyn (Fig. S1 b). This was not caused by increased Fyn mRNA expression because Northern blots showed no significant difference in mRNA in Lyn-deficient and wild-type cells (Fig. S1 c). Both the autophosphorylation assay and pY417/pY528 analysis showed that the activity of Fyn kinase in Lyn-deficient mast cells was ∼2.5–4-fold greater than that seen in wild-type cells. Thus, the 30% increase in Fyn protein does not explain the total increase in Fyn activity. FcεRI-mediated activation of Src was found in Lyn-deficient mast cells but was reduced compared with wild-type mast cells (Fig. 1 b). Notably, we could not reproducibly detect Hck protein and activity in Lyn-deficient mast cells (not depicted).
Figure 1.

Fyn kinase, but not Src kinase, activity is constitutively increased in Lyn−/− mast cells. (a) Wild-type or Lyn−/− mast cells were stimulated with Ag for the indicated time. Fyn kinase was immunoprecipitated and subjected to an IVK reaction. Autophosphorylation was measured by anti-phosphotyrosine (Anti-pY) immunoblotting and normalized for protein loading (Anti-Fyn). (b) Conditions were identical to (a), however, p60 Src was immunoprecipitated and subjected to IVK. Quantitation was by densitometry. One representative experiment is shown. Fold induction is the mean of three experiments normalized to the wild-type (0 min) activity.
Loss of Cbp Phosphorylation and Csk Interaction in Lyn-deficient Mast Cells.
To investigate possible mechanisms for enhanced Fyn kinase activity, we explored the requirement for Lyn in the phosphorylation and interaction of Cbp with Csk, as this regulatory pathway has been intimately linked to control of Src PTK activity (20, 21). As seen in Fig. 2 a, immunoprecipitation of Cbp from wild-type mast cells resulted in the coprecipitation of Lyn kinase. The association of Lyn kinase with Cbp was largely unaffected by FcεRI stimulation even though a slight reduction in associated Lyn was consistently observed. Fyn kinase coprecipitation was also detected, although this interaction appeared to be weak and was not always observed. Consistent with a previous report (22), FcεRI stimulation of wild-type mast cells increased phosphorylation of Cbp and also the binding of Csk (Fig. 2 b). In contrast, Lyn-deficient mast cells showed a marked inhibition (ranging from 67 to 92%) in the phosphorylation of Cbp. This loss of phosphorylation was not restored by the possible redundancy of other Src PTKs (23). Concomitant with the loss of Cbp phosphorylation, a considerable loss (51–77%) of Csk interaction with Cbp was also observed (Fig. 2 b). Similar results were obtained by the inverse immunoprecipitation of Csk, which coimmunoprecipitated Cbp from wild-type mast cells but showed minimal Cbp coimmunoprecipitation from Lyn-deficient mast cells (Fig. 2 c). The phosphorylation of Cbp was found to be primarily Lyn dependent because Fyn-deficient mast cells showed relatively normal phosphorylation of Cbp (and Csk interaction; Fig. 2 d).
Figure 2.

Lyn kinase, but not Fyn kinase, is required for tyrosine phosphorylation of Cbp. (a) Wild-type mast cells were stimulated with Ag for the indicated time. Cbp was immunoprecipitated and recovered protein was probed for the presence of Lyn (Anti-Lyn) or Fyn (Anti-Fyn) kinases. A control immunoprecipitation with normal rabbit IgG (rIgG) is also shown. Immunoblots were reprobed with anti-Cbp for protein loading. (b) Wild-type and Lyn−/− mast cells were stimulated as described above for the indicated time. Cbp was immunoprecipitated and resolved proteins were probed for tyrosine phosphorylation (Anti-pY) or for Csk (Anti-Csk). (c) Cells were stimulated as described above and Csk was immunoprecipitated. Resolved protein was immunoblotted for Cbp phosphorylation, Csk, or Cbp protein. (d) Wild-type, Lyn−/−, or Fyn−/− mast cells were stimulated with Ag for the indicated time. Cbp was immunoprecipitated and its tyrosine phosphorylation was determined by anti-pY blotting. Stripped immunoblots were then reprobed with anti-Csk or anti-Cbp. Fold induction is the mean of three individual experiments from two BMMC cultures. For all panels, one representative of a minimum of three experiments is shown.
Expression of Lyn Kinase in Lyn-deficient Mast Cells Causes Cbp Phosphorylation, Down-regulates Fyn Kinase Activity, and Restores Normal Levels of Degranulation.
Mutation of the negative regulatory tyrosine (Y508F) in Lyn kinase results in a constitutively active form of this kinase capable of phosphorylating its substrates in vivo (7, 24). Mast cells derived from mice expressing this mutant form of Lyn (lyn up/up) were analyzed for the phosphorylation of Cbp. As shown in Fig. S2 a (available at http://www.jem.org/cgi/content/full/jem.20040382/DC1), expression of lyn up/up resulted in enhanced phosphorylation of Cbp. However, analysis of the protein levels of Fyn kinase in lyn up/up mast cells revealed that expression of this constitutively active form of Lyn caused a loss of Fyn protein (Fig. S2 b), making an analysis of the effect of Lyn on Fyn activity difficult. Therefore, Lyn-deficient mast cells were retrovirally transduced with wild-type Lyn, which did not substantially decrease the expression of Fyn kinase (Fig. 3 a). Fig. 3 b shows that the total activity of Fyn kinase in the Lyn retrovirus–transduced cells was reduced and FcεRI-dependent increases in Fyn activity were restored. Retroviral transduction alone (vector) did not cause any significant change in Fyn kinase activity. In three individual experiments, the total activity of Fyn was reduced by 37–56% when wild-type Lyn was transduced in Lyn-deficient mast cells. One might predict that if Lyn kinase is essential for negative regulation of mast cell degranulation, expression of wild-type Lyn kinase in Lyn-deficient mast cells should inhibit the hyper-degranulation restoring it to the level of wild-type cells. As shown in Fig. 3 c, expression of wild-type Lyn (lyn re/re) in Lyn-deficient mast cells restored control of degranulation and the observed response at all Ag concentrations mirrored that of wild-type cells, whereas the response of the vector-transduced cells (lyn ve/ve) was essentially identical to that of Lyn-deficient cells.
Figure 3.

Control of Fyn kinase activity and mast cell degranulation is restored by retroviral transduction of Lyn kinase in Lyn-deficient mast cells. (a) Lyn−/− mast cells were transduced (reconstituted) with wild-type Lyn kinase (Lynre/re) or with a control vector (Lynve/ve). Expression of Lyn and Fyn kinase was compared between wild-type, Lyn−/−, Lynre/re, and Lynve/ve mast cells. Expression of endogenous Akt is shown for normalization. (b) Wild-type, Lyn−/−, Lynre/re, and Lynve/ve mast cells were stimulated with Ag for the indicated time. Fyn kinase was immunoprecipitated and subjected to an IVK assay. Autophosphorylation was detected by anti-pY and stripped blots were reprobed with anti-Fyn. One representative of three experiments is shown. Fold induction is the mean of all experiments. (c) Degranulation of wild-type Lyn-containing (Lynre/re), control vector–containing (Lynve/ve), Lyn−/−, and wild-type mast cells in response to the indicated Ag concentrations. Net hexosaminidase release is reported as a percentage of total. Data is from three individual experiments.
Lyn/Fyn-deficient Mice Are Resistant to PSA and BMMCs from These Mice Are Impaired in Degranulation.
To further test the hypothesis that Fyn kinase activity was responsible for the hyperresponsive degranulation phenotype of Lyn-deficient mast cells, we generated lyn − / − fyn − / − double-deficient mice. As shown in Fig. 4 a, Lyn or Fyn protein was not detected in immunoblots of Lyn/Fyn double-deficient mast cells. As might be expected, Cbp phosphorylation in Lyn/Fyn double-deficient mast cells was impaired (Fig. 4 b) similarly to Lyn-deficient mast cells. Analysis of the FcεRI-dependent degranulation revealed that Lyn/Fyn double-deficient mast cells were also defective in this response (Fig. 4 c). The extent of degranulation was similar to that observed for Fyn-deficient mast cells (10) and was, at best, 20% of the response seen in wild-type cells (Fig. 4 c). However, Lyn/Fyn double-deficient mast cells were able to degranulate to phorbol ester (PMA) and calcium ionophore (A23187), which bypasses early signaling events, demonstrating normal development of the secretory compartment (Fig. 4 c). Interestingly, the extent of the PMA/A23187-induced degranulation mirrored that of Lyn-deficient mast cells rather than that of wild-type cells (10), suggesting that Lyn kinase may exert additional regulatory controls on mast cell degranulation that are independent of Fyn kinase.
Figure 4.

In vitro and in vivo degranulation of wild-type and Lyn-deficient mast cells is Fyn kinase dependent. (a) Doubly deficient lyn − / − fyn − / − mice were generated by initial mating of lyn − / − and fyn − / − mice. Whole cell lysates (WCL) from BMMCs derived from all genotypes were immunoblotted for Fyn, Lyn, and FcεRIγ (protein loading). (b) Wild-type and Lyn/Fyn−/− deficient mast cells were stimulated with Ag for the indicated time. Cbp was immunoprecipitated and its tyrosine phosphorylation was determined by anti-pY. Stripped blots were reprobed with anti-Cbp. (c) Degranulation of wild-type, Lyn−/−, Lyn/Fyn−/−, and Fyn−/− mast cells was determined, after stimulation with the indicated concentrations of Ag, by hexosaminidase release. Net release as a percentage of total cellular hexosaminidase is shown. PMA/A23187 stimulation of Lyn/Fyn−/− mast cells shows that these cells have no intrinsic defect in the secretory apparatus. (d) In vivo passive systemic anaphylactic challenge of 4-wk-old wt, lyn − / −, fyn − / −, and lyn − / − fyn − / − mice. Mice were passively sensitized with DNP-specific IgE (i.v.) and challenged 24 h later with DNP-HSA (Ag) or pseudochallenged with an equal volume of saline for 1.5 min (i.v.). Plasma histamine concentration was measured by competitive ELISA. Significance for all data was determined by an unpaired t test. ***, a p-value of ≤0.006.
Next, we explored whether the defective response of Lyn/Fyn double-deficient mast cells was recapitulated in vivo. PSA of lyn − / −, fyn -/-, and lyn − / − fyn − / − mice was analyzed. The initial experiments, in which mice of ∼8 wk of age were used, showed that lyn − / − mice were considerably resistant (>60% inhibited) to an anaphylactic challenge (not depicted). However, as will become evident below, the ability to sensitize these mice with Ag-specific IgE was significantly impaired. In contrast, as shown in Fig. 4 d, when mice of 4 wk of age were subjected to a PSA challenge, lyn − / − mice showed a significant augmentation (P ≤ 0.001) in their circulating histamine relative to wild-type mice. The observed mean serum histamine concentration for lyn − / − mice was 16.8 ± 0.6 μM, whereas that of wild-type mice was 12.0 ± 0.4 μM. In contrast, fyn − / − and lyn − / − fyn − / − mice (4 wk of age) were resistant. Upon PSA challenge, both fyn − / − and lyn − / − fyn − / − mice showed levels of circulating histamine that were 20% or less than in wild-type mice. These results demonstrate that the in vitro mast cell responses of each genotype are recapitulated in vivo.
Older Lyn-deficient Mice Have Increased Serum IgE, Increased FcεRI Occupancy, Increased Serum Histamine, and Increased Peritoneal Mast Cells and Eosinophils.
The decreased anaphylactic response of older lyn − / − mice led us to explore the apparent discrepancy with the observed BMMC hyperresponsiveness in our in vitro studies as well as with increased in vivo anaphylactic response of young mice. The most logical explanation is that the occupancy of FcεRI by IgE would increase with the age of the mice based on the observation that lyn − / − mice were shown to have increased levels of Igs and enhanced IL-4–dependent Ig class switching leading to IgE (5, 6, 25). Increased IgE production might preclude sensitization with Ag-specific IgE. Thus, we first explored the relationship between the serum IgE levels of lyn − / − mice and the age of the mice. As shown in Fig. 5 a, young lyn − / − mice (4 wk of age) showed no significant difference in circulating IgE levels when compared with wild-type mice of up to 16 wk of age (0.23 ± 0.04 vs. 0.31 ± 0.05 μg/ml), although a slight trend toward increased levels of IgE was noted. However, by 7 wk of age, lyn − / − mice showed as much as a 10-fold increase in serum IgE levels ranging from 0.98 to 5.8 μg/ml when compared with 7-wk-old wild-type mice (0.14–1.2 μg/ml). This suggested the possibility that as lyn − / − mice aged, FcεRI occupancy might vary from wild-type mice. Fig. 5 b shows that FcεRI occupancy of peritoneal mast cells by IgE increases with age for both wild-type and lyn − / − mice. However, by 4 wk of age, lyn − / − mice show FcεRI occupancy levels similar to that of 7-wk-old wild-type mice. By 7 wk of age, the FcεRI occupancy of lyn − / − mice by IgE is almost complete. By 12–16 wk, lyn − / − mice have no unoccupied FcεRI (not depicted). Although wild-type mice at 7 wk showed an average of 22% of their mast cell FcεRI as available for IgE binding, only 4% of these receptors are available in lyn − / − mice. Interestingly, wild-type mice of 16 wk of age showed an FcεRI occupancy of ∼86%. Nonetheless, these mice respond to a PSA challenge similarly to younger mice (not depicted), suggesting that to respond to Ag-specific IgE, >4% but as little as 14% of the receptor occupied by this IgE is required. This level of FcεRI availability for Ag-specific IgE is still seen in 4-wk-old lyn − / − mice, but not after 7 wk of age (Fig. 5 b).
Figure 5.

Lyn −/− mice show increased serum IgE, increased FcεRI occupancy, and increased circulating histamine. (a) The serum IgE of 4- or 7-wk-old age- and sex-matched unchallenged wt or lyn − / − mice was measured by ELISA. (b) FcεRI occupancy of peritoneal mast cells was determined from 4- or 7-wk-old wt and lyn − / − mice. The mean fluorescence intensity of cells before and after incubation with exogenous IgE was compared with obtained relative occupancies. Data is shown as a percentage of receptors available for binding of exogenous IgE. (c) Plasma histamine concentration of the mice in (a) was measured by competitive ELISA. (d) Correlation analysis of serum IgE and histamine concentrations from lyn − / − mice (data in a and c). Significance for all data was determined by an unpaired t test. ***, a p-value of ≤0.003; *, a p-value of ≤0.03. Pearson r2 = 0.9517 for (d). A total of five (b) or eight (a and c) mice were used.
Recent studies provide strong evidence for a role of monomeric IgE in increasing mast cell cytokines, survival, and degranulation (under conditions where a large excess of monomeric IgE is present or when negative regulatory control is lost in a mast cell; 13, 14, 26, 27). A correlation between total serum IgE levels and histamine release in vivo has also been demonstrated in several studies (28–30). Therefore, we tested whether the increased serum IgE in lyn − / − mice could translate to increased serum histamine based on the assumption that high concentrations of monomeric IgE could trigger mast cell degranulation or, alternatively, that mice housed in a clean facility could still be exposed to some environmental allergens. As seen in Fig. 5 c, we obtained a statistically significant difference of approximately twofold in the serum histamine concentrations of 7-wk-old wild-type mice (mean = 102.3 ± 19.1 nM, n = 8) and lyn − / − mice (mean = 214.1 ± 44.0 nM, n = 8). A high degree of correlation (r2 = 0.9517) was obtained for serum IgE and histamine concentrations in the lyn − / − mice (Fig. 5 d). In contrast, no correlation was found between the serum IgE and histamine concentrations in 7–12-wk-old wild-type mice (not depicted), suggesting that high concentrations of monomeric IgE might be driving mast cell degranulation in the lyn − / − mice rather than the interaction of an environmental allergen with allergen-specific IgE.
As stated above, increased concentrations of monomeric IgE were shown to increase mast cell survival, but these conditions also resulted in increased cell surface expression of mast cell FcεRI (for review see reference 12), two factors associated with increased allergic responses. Therefore, we investigated whether these factors might be observed in 7-wk-old lyn − / − mice given their increased serum IgE. Fig. 6 a shows that mast cells in a peritoneal lavage of lyn − / − mice are increased, compared with wild-type mice of the same age. Analysis on all tested mice revealed that peritoneal mast cells of wild-type mice comprised, on average, 2.2% of the total cells in the peritoneal lavage (Fig. 6 b). In contrast, lyn − / − mice showed, on average, ∼5.3% as mast cells (ranging from a low of 2.1 to a high of ∼11%). Regardless of whether the numbers of peritoneal mast cells in a given mouse were enhanced or not, all mast cells showed increased cell surface FcεRI expression (Fig. 6 b). It should be noted that lyn − / − mice also showed increased numbers of dermal mast cells, with 8.0 ± 0.4 mast cells per field (a magnification of 1,000) versus wild-type mice, which had 4.5 ± 0.3 mast cells (P ≤ 0.001; not depicted). Importantly, 4-wk-old wild-type and lyn − / − mice did not differ significantly in peritoneal mast cell numbers with 1.3 ± 0.4 and 1.7 ± 0.3% of total cells as mast cells, respectively (not depicted), even though the lyn − / − mice showed a slight trend toward increased mast cell numbers.
Figure 6.
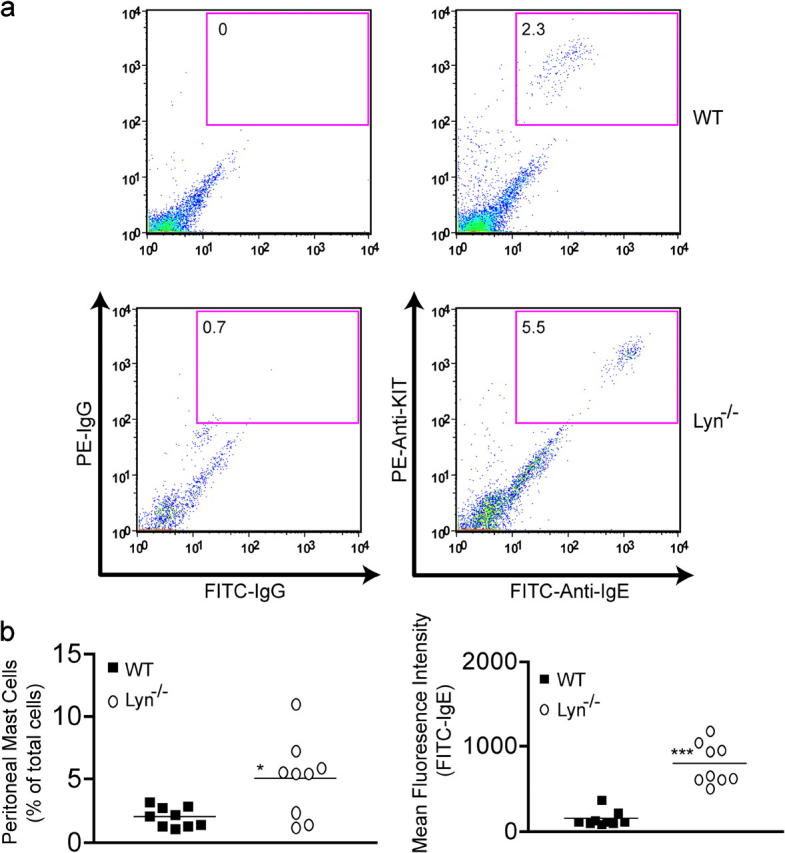
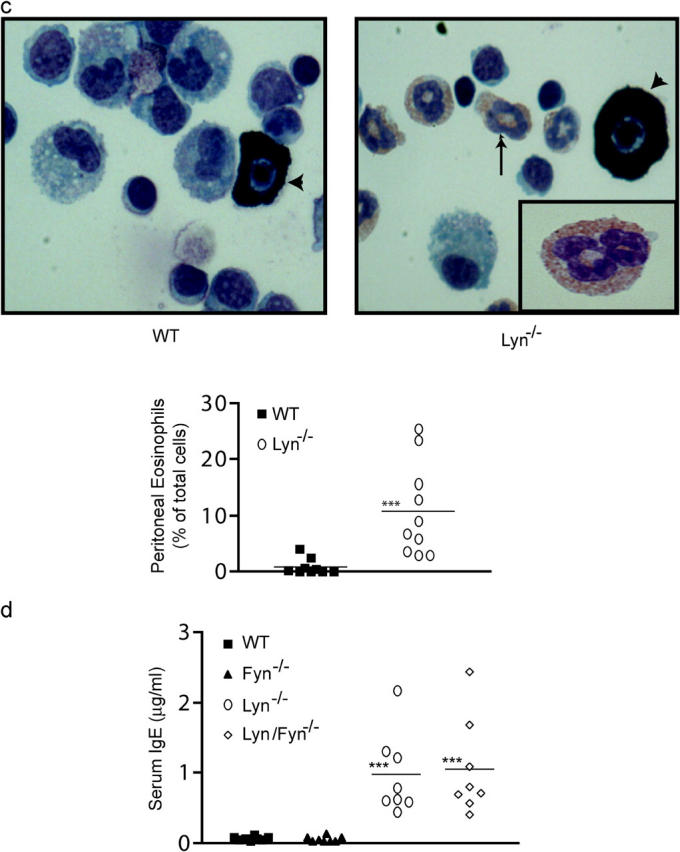
Lyn −/− mice have increased numbers of peritoneal mast cells with increased cell surface FcεRI expression and eosinophilia. Fyn is not required for increased serum IgE. (a) Cells from a peritoneal lavage of wt or lyn − / − mice were stained with antibody to KIT and IgE after incubation with IgE to ensure FcεRI saturation. As a negative control, isotype-specific IgG was also used to stain the cells. Inset numbers reflect the percentage of KIT/FcεRI+ cells. (b) The percentage of mast cells in the peritoneum and the mean fluorescence intensity of FITC-IgE staining (reflecting mast cell FcεRI expression) of all the mice analyzed (n = 10). (c) Wright-Giemsa stain of peritoneal lavage cells from wt or lyn − / − mice. Mast cells are indicated by intense granule staining (dark cells, marked with arrow head), whereas eosinophils show a light red/pink stain and multilobed/segmented nucleus (arrow, see inset in lyn − / − sample). Graph is a quantitation of the percentage of eosinophils found in the peritoneal lavage of all wt and lyn − / − mice analyzed (n = 10). (d) The serum IgE concentration of the indicated genotypes was measured by ELISA from eight sex- and age-matched mice as described in Materials and Methods. Mean of the observed IgE concentrations for lyn − / − or lyn − / − fyn − / − mice is indicated by the line. No significant difference was found between these two populations, but both were significantly different from wt or fyn − / − mice. Significance was determined by an unpaired t test. ***, a p-value of ≤0.006; *, a p-value of ≤0.03.
One of the consequences of in vivo mast cell activation is the recruitment of eosinophils (31, 32). Therefore, we investigated whether the number of eosinophils in the peritoneal lavage of 7-wk-old lyn − / − mice was significantly different from that of wild-type mice. Fig. 6 c shows that a striking eosinophilia is present in the peritoneum of lyn − / − mice. As compared with wild-type mice, where peritoneal eosinophils are ∼2% of the cell population, lyn − / − mice had an average of >10% of total peritoneal cells as eosinophils (Fig. 6 c). Preliminary experiments showed no significant difference in eosinophil numbers in the bone marrow of lyn − / − versus wild-type mice (not depicted), suggesting that enhanced eosinophil proliferation and/or differentiation in the bone marrow is not a likely explanation for the peritoneal eosinophilia.
Given the high serum IgE concentrations in older lyn − / − mice and the enhanced Fyn kinase activity in mast cells from these mice, we investigated whether Fyn might contribute to the increased IgE phenotype. We measured the serum IgE concentration of 7-wk-old wild-type, fyn − / −, lyn − / −, and lyn − / − fyn − / − mice. As shown in Fig. 6 d, fyn − / − mice showed no significant difference in serum IgE levels compared with wild-type mice. In contrast, the serum IgE levels of lyn − / − fyn − / − mice were dramatically elevated and identical to those of lyn − / − mice. Dermal mast cell numbers in fyn − / − mice (4.0 ± 0.4 per field, a magnification of 1,000) were comparable to wild-type mice (4.5 ± 0.3), whereas those of lyn − / − fyn − / − mice (7.7 ± 0.5) were comparable to lyn − / − mice (8.0 ± 0.4). This argues against the possible loss of mast cells as an explanation for the loss of anaphylactic responsiveness in the fyn − / − and lyn − / − fyn − / − mice. In contrast, it demonstrates a unique role for Lyn kinase in down-regulating B cell IgE production, which is independent of its control of Fyn kinase activity, and also in controlling mast cell numbers in vivo. The important role of Lyn in the IgE response is further supported by the in vivo data shown in Fig. 6 d. The mice in these experiments were generated from the initial mating of fyn − / − and lyn − / − mice. Thus, the lyn alleles were solely contributed by the fyn − / − strain. This inheritance of lyn alleles from one parent results in normal serum IgE levels regardless of whether fyn alleles are present or not (Fig. 6 d, wt vs. fyn − / − mice). Thus, lyn alleles, but not fyn alleles, are essential for control of B cell IgE production.
Discussion
Previous reports on the biochemical activity and responses of Lyn-deficient mast cells (8–10, 33) have provided a glimpse of negative regulation in mast cell signaling and responses. Here we explored the central mechanism through which mast cells can degranulate in the absence of Lyn kinase and investigated if Lyn exerts a broad negative influence on the allergic response. The latter question is of critical importance because of the accepted view of Lyn kinase as a positive effector in mast cell responses and thus as a possible therapeutic target in allergic disease (34). We find that Fyn kinase activity is responsible for the hyper-degranulation of Lyn-deficient mast cells. This is seemingly independent of the background genetics of the lyn − / − mice or is a dominant feature of the C57/BL6 background because mice backcrossed to C57/BL6 for seven generations or mice of mixed background (SV129 × C57/BL6, N2) showed similar in vivo responses and their mast cells showed increased Fyn kinase activity. The loss of phosphorylation of the membrane scaffold adaptor, Cbp, plays an important role in the mast cell hyperresponsiveness because membrane targeting of the Src PTK negative regulatory kinase, Csk, is severely impaired in Lyn-deficient mast cells, but this regulatory pathway is restored by expression of Lyn. The essential nature of Fyn in driving degranulation is evident in the lyn − / − fyn − / − mouse, whose mast cells (both in vitro and in vivo) showed severe impairment in degranulation. Interestingly, lyn − / − mice showed an increase in allergy-associated traits that was characterized by increased serum IgE, increased circulating histamine, and eosinophilia, supporting the view of increased mast cell responsiveness in vivo. Our findings show that inheritance of lyn alleles restores normal control of these allergy-associated traits and that fyn alleles play no apparent role in the control of these factors. These findings, together with our previous observation that in vivo expression of a Lyn gain of function mutant (Y508F) resulted in down-regulation of serum IgE levels (24), argue for the importance of Lyn kinase in controlling allergy-associated risk factors.
Monomeric IgE in the context of a hypersensitive mast cell (SHIP-1 −/−; reference 13), or incubation of wild-type mast cells with high concentrations of certain monomeric IgEs (14, 27), causes allergen-independent mast cell degranulation in vitro. High concentrations of monomeric IgE in the context of a hypersensitive mast cell is recapitulated in lyn − / − mice. This suggests that the observed increase in circulating histamine and the eosinophilia is a possible consequence of mast cell activation, although this still remains to be formally demonstrated. However, the present findings are noteworthy as they establish a clear association between the increasing age of lyn − / − mice, increasing serum IgE, and increased circulating histamine, an association not present in wild-type mice. Moreover, our finding that young (4 wk) lyn − / − mice showed an increased PSA response demonstrates the enhanced potential of mast cells to be triggered for degranulation in these mice. On the other hand, this finding was in apparent contradiction with the previous finding that lyn − / − mice were resistant to passive cutaneous anaphylaxis (25). We now know that the almost complete occupancy of FcεRI by IgE as lyn − / − mice age is a likely explanation because this would prohibit the binding of the passively transferred Ag-specific IgE, making the response to exogenous Ag difficult. Nonetheless, it should be noted that older lyn − / − mice (>7 wk) also showed an impaired anaphylactic response (20–30% inhibition in serum histamine) to an anti-IgE challenge (not depicted), albeit not to the same extent as an Ag challenge (50–80% inhibition). Because young mice (4 wk) respond well to an anti-IgE challenge, the loss of responsiveness in older mice may reflect constitutive IgE-mediated mast cell degranulation (as reflected by increased serum histamine levels before a challenge), or could also reflect that the levels of IgE in vivo are increased to the extent where an excess of fivefold of anti-IgE to serum IgE concentrations is not sufficient to elicit a full response. Our studies also show that occupancy of >4 but <14% of receptors with Ag-specific IgE was sufficient to elicit an effective anaphylactic response in lyn − / − mice that was comparable to wild-type mice, where 23–41% of the receptors were available to Ag-specific IgE. This is consistent with our previous in vitro studies showing that low occupancy of FcεRI can elicit mast cell activation and functional responses (35). However, unlike the potent lymphokine responses observed, potent degranulation was not a feature of 14% of receptor occupancy in vitro. Thus, the in vivo results likely reflect the hypersensitivity of mast cells in lyn − / − mice and the lowered threshold to elicit the signals necessary for degranulation. Alternatively, this apparent discrepancy might be explained by differences in the in vivo microenvironment in which the mast cells reside (36, 37), as it may serve to prime lyn − / − mast cells to respond more potently with less allergen-specific IgE-occupied receptors.
One mechanism by which Src PTK activity is down-regulated occurs through phosphorylation of an inhibitory (i.e., Y508/528) tyrosine residue located at the COOH terminus of the catalytic domain, which promotes an autoinhibitory interaction with its SH2 domain and a closed inactive kinase conformation (38). Csk, which is responsible for the Y508/528 phosphorylation, is a cytoplasmic kinase that is targeted to the plasma membrane by its SH2 domain interaction with the phosphorylated adaptor protein, Cbp (20, 21). Thus, loss of Cbp phosphorylation (as seen in Lyn-deficient mast cells) is likely to increase the fraction of Src PTKs in an active state. Our findings show that Fyn is highly active in the absence of Cbp phosphorylation. This is demonstrated by increased Fyn activity in an autophosphorylation assay as well as by increased phosphorylation of the activation loop Y417 relative to the inhibitory Y528 (Fig. S1). Evidence for preferential phosphorylation of the equivalent activation loop tyrosine on Lyn kinase (Y397), as reflective of an activated state, was recently provided by a study demonstrating that Lyn kinase in the lipid raft fraction of RBL-2H3 cells (39) is highly active and predominantly phosphorylated at Y397. Interestingly, active Lyn in lipid rafts is not a consequence of FcεRI engagement because active Lyn was detected in resting cells and its activity was not changed by FcεRI engagement (1, 39, 40). This is consistent with the view that Lyn phosphorylates the lipid raft resident protein Cbp in resting mast cells (Fig. 3). The increased phosphorylation of Cbp, upon FcεRI engagement, might be a consequence of additional recruitment of Lyn (41) or possibly the activation of Syk. This increase in Cbp phosphorylation causes additional recruitment of Csk, which may serve to control the amount of active Fyn and thus controls degranulation. This view is supported by the reconstitution experiments, where introduction of relatively normal levels of Lyn in Lyn-deficient mast cells restored FcεRI-dependent control of Fyn activation and reduced the overall activity of Fyn kinase. Additionally, the kinetics of pY417/pY528 phosphorylation seen in wild-type cells (Fig. S1) also support this view because peak Y417 phosphorylation was seen at 1 min after stimulation and decreased at 3 and 9 min after stimulation. Finally, support is also provided by the previous observation that overexpression of Cbp had a substantial inhibitory effect on mast cell degranulation (22).
Both positive and negative roles for Lyn kinase have been described (5–7, 25, 42-44). CD19-deficient B cells are known to be hyporesponsive, whereas the Ig hyperresponsiveness is seen in Lyn-deficient B cells (5, 25). B cells from doubly deficient mice (CD19 − / − lyn − / −) showed a suppressed Ig hyperresponsiveness (44). This correlated with Fyn kinase activity, which was defective in CD19 and CD19/Lyn double-deficient B cells but was intact in Lyn-deficient B cells. This raised the possibility that Lyn-deficient B cell Ig hyperresponsiveness is driven, at least in part, by Fyn kinase. This hypothesis was also supported by the observed reduction of IgG3 autoantibodies in fyn − / −-MRL/lpr mice, which led to a marked reduction in autoimmune disease and an increased lifespan compared with the MRL/lpr mouse (45). Therefore, we explored whether the high serum IgE levels in lyn − / − mice might be driven by Fyn kinase. Analysis of serum IgE concentrations for wild-type, fyn − / −, lyn − / −, and lyn − / − fyn − / − mice revealed that the absence of Fyn had no effect on IgE levels, regardless of whether the mice express Lyn or not. Thus, the negative regulatory role for Lyn in B cell–mediated IgE production is independent of its control of Fyn kinase activity. Recently, it was reported that Lyn-deficient mast cells have increased proliferative responses (33), raising the possibility that Fyn kinase might drive this response. We can now conclude that the increased activity of Fyn kinase in Lyn-deficient mast cells is not responsible for the increased mast cell numbers in lyn − / − mice, as our preliminary experiments also showed increased mast cell numbers in older lyn − / − fyn − / − mice.
Herein, we demonstrate that Lyn deficiency has broad consequences with respect to the allergic response. We find that Lyn negatively controls both B cell and mast cell responsiveness in a manner that directly impacts IgE-dependent activation of mast cells both in vitro and in vivo. Although the etiology of the allergic-like phenotype of Lyn deficiency is not completely clear, an increase in allergy-associated risk factors is clearly demonstrated. Given the present evidence of a negative regulatory role for Lyn in mast cell responses both in vivo and in vitro, one is led to conclude that Lyn kinase primarily functions as a negative regulator of the IgE-mediated allergic response. This brings into question the usefulness of therapeutically targeting this kinase in allergic disease.
Acknowledgments
We thank Amanda Turner and Wibke Schwarzer for technical contributions.
This work was supported by the Department of Health and Human Services, NIH, NIAMS (to J. Rivera), United States-Israel Bi-national Science Foundation grant number 2000016 (to J. Rivera), the Japan Society for Promotion of Science (to Y. Furumoto), and a Pan American Fellowship Award (to C. Gonzalez-Espinosa) from CONACyT and the Fogarty International Center (NIH). C. Gonzalez-Espinosa is currently supported by grant number 39726-Q from CONACyT.
S. Odom, G. Gomez, K.W. Harder, and J. Rivera contributed equally to this work.
The online version of this article contains supplemental material.
Abbreviations used in this paper: BMMC, bone marrow–derived mast cell; Cbp, COOH-terminal Src kinase–binding protein; Csk, COOH-terminal Src kinase; GFP, green fluorescent protein; HRP, horseradish peroxidase; HSA, human serum albumin; IVK, in vitro kinase; PI3K, phosphatidylinositol 3-OH kinase; PSA, passive systemic anaphylaxis; SCF, stem cell factor; Src PTK, Src family protein tyrosine kinase.
References
- 1.Pribluda, V.S., C. Pribluda, and H. Metzger. 1994. Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc. Natl. Acad. Sci. USA. 91:11246–11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Field, K.A., D. Holowka, and B. Baird. 1997. Compartmentalized activation of the high affinity immunoglobulin E receptor within membrane domains. J. Biol. Chem. 272:4276–4280. [DOI] [PubMed] [Google Scholar]
- 3.Rivera, J. 2002. Molecular adapters in FcɛRI signaling and the allergic response. Curr. Opin. Immunol. 14:688–693. [DOI] [PubMed] [Google Scholar]
- 4.Nadler, M.J., S.A. Matthews, H. Turner, and J.-P. Kinet. 2000. Signal transduction by the high-affinity immunoglobulin E receptor Fc epsilon RI: coupling form to function. Adv. Immunol. 76:325–355. [DOI] [PubMed] [Google Scholar]
- 5.Chan, V.W.F., F. Meng, P. Soriano, A.L. DeFranco, and C.A. Lowell. 1997. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity. 7:69–81. [DOI] [PubMed] [Google Scholar]
- 6.Janas, M.L., P. Hodgkin, M. Hibbs, and D. Tarlinton. 1999. Genetic evidence for Lyn as a negative regulator of IL-4 signaling. J. Immunol. 163:4192–4198. [PubMed] [Google Scholar]
- 7.Harder, K.W., L.M. Parsons, J. Armes, N. Evans, N. Kountouri, R. Clark, C. Quilici, D. Grail, G.S. Hodgson, A.R. Dunn, et al. 2001. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity. 15:603–615. [DOI] [PubMed] [Google Scholar]
- 8.Nishizumi, H., and T. Yamamoto. 1997. Impaired tyrosine phosphorylation and Ca2+ mobilization, but not degranulation, in Lyn-deficient bone marrow-derived mast cells. J. Immunol. 158:2350–2355. [PubMed] [Google Scholar]
- 9.Kawakami, Y., J. Kitaura, A.B. Satterthwaite, R.M. Kato, K. Asai, S.E. Hartman, M. Maeda-Yamamoto, C.A. Lowell, D.J. Rawlings, O.N. Witte, et al. 2000. Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cell activation. J. Immunol. 165:1210–1219. [DOI] [PubMed] [Google Scholar]
- 10.Parravicini, V., M. Gadina, M. Kovarova, S. Odom, C. Gonzalez-Espinosa, Y. Furumoto, S. Saitoh, L.E. Samelson, J.J. O'Shea, and J. Rivera. 2002. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat. Immunol. 3:741–748. [DOI] [PubMed] [Google Scholar]
- 11.Kovarova, M., P. Tolar, R. Arudchandran, L. Draberova, J. Rivera, and P. Draber. 2001. Structure-function analysis of Lyn kinase association with lipid rafts and initiation of early signaling events after Fc epsilon receptor I aggregation. Mol. Cell. Biol. 21:8318–8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawakami, T., and S.J. Galli. 2002. Regulation of mast-cell and basophil function and survival by IgE. Nat. Rev. Immunol. 2:773–786. [DOI] [PubMed] [Google Scholar]
- 13.Huber, M., C.D. Helgason, J.E. Damen, L. Liu, R.K. Humphries, and G. Krystal. 1998. The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc. Natl. Acad. Sci. USA. 95:11330–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitaura, J., J. Song, M. Tsai, K. Asai, M. Maeda-Yamamoto, A. Mocsai, Y. Kawakami, F.-T. Liu, C.A. Lowell, B.G. Barisas, et al. 2003. Evidence that IgE molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the FcɛRI. Proc. Natl. Acad. Sci. USA. 100:12911–12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu, F.T., J.W. Bohn, E.L. Ferry, H. Yamamoto, C.A. Molinaro, L.A. Sherman, N.R. Klinman, and D.H. Katz. 1980. Monoclonal dinitrophenyl-specific murine IgE antibody: preparation, isolation, and characterization. J. Immunol. 124:2728–2737. [PubMed] [Google Scholar]
- 16.Saitoh, S., R. Arudchandran, T.S. Manetz, W. Zhang, C.L. Sommers, P.E. Love, J. Rivera, and L.E. Samelson. 2000. LAT is essential for Fc(epsilon)RI-mediated mast cell activation. Immunity. 12:525–535. [DOI] [PubMed] [Google Scholar]
- 17.Razin, E. 1990. Culture of bone marrow-derived mast cells: a model for studying oxidative metabolism of arachidonic acid and synthesis of other molecules derived from membrane phospholipids. Methods Enzymol. 187:514–520. [DOI] [PubMed] [Google Scholar]
- 18.Arudchandran, R., M.J. Brown, M.J. Peirce, J.S. Song, J. Zhang, R.P. Siraganian, U. Blank, and J. Rivera. 2000. The Src homology 2 domain of Vav is required for its compartmentation to the plasma membrane and activation of c-jun NH2-terminal kinase 1. J. Exp. Med. 191:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eiseman, E., and J.B. Bolen. 1992. Engagement of the high-affinity IgE receptor activates src protein-related tyrosine kinases. Nature. 355:78–80. [DOI] [PubMed] [Google Scholar]
- 20.Kawabuchi, M., Y. Satomi, T. Takao, Y. Shimonishi, S. Nada, K. Nagai, A. Tarakhovsky, and M. Okada. 2000. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 404:999–1003. [DOI] [PubMed] [Google Scholar]
- 21.Brdicka, T., D. Pavlistova, A. Leo, E. Bruyns, V. Korinek, P. Angelisova, J. Scherer, A. Shevchenko, I. Hilgert, J. Cerny, et al. 2000. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J. Exp. Med. 191:1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohtake, H., N. Ichikawa, M. Okada, and T. Yamashita. 2002. Cutting edge: transmembrane phosphoprotein Csk-binding protein/phosphoprotein associated with glycosphingolipid-enriched microdomains as a negative feedback regulator of mast cell signaling through FcɛRI. J. Immunol. 168:2087–2090. [DOI] [PubMed] [Google Scholar]
- 23.Thomas, S.M., and J.S. Brugge. 1997. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13:513–609. [DOI] [PubMed] [Google Scholar]
- 24.Hibbs, M.L., K.W. Harder, J. Armes, N. Kountouri, C. Quilici, F. Casagranda, A.R. Dunn, and D.M. Tarlinton. 2002. Sustained activation of Lyn tyrosine kinase in vivo leads to autoimmunity. J. Exp. Med. 196:1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hibbs, M.L., D.M. Tarlinton, J. Armes, D. Grail, G. Hodgson, R. Maglitto, S.A. Stacker, and A.R. Dunn. 1995. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 83:301–311. [DOI] [PubMed] [Google Scholar]
- 26.Asai, K., J. Kitaura, Y. Kawakami, N. Yamagata, M. Tsai, D.P. Carbone, F.T. Liu, S.J. Galli, and T. Kawakami. 2001. Regulation of mast cell survival by IgE. Immunity. 14:791–800. [DOI] [PubMed] [Google Scholar]
- 27.Kalesnikoff, J., M. Huber, V. Lam, J.E. Damen, J. Zhang, R.P. Siraganian, and G. Krystal. 2001. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. 14:801–811. [DOI] [PubMed] [Google Scholar]
- 28.Sydbom, A., and T. Karlsson. 1979. Relationship between serum IgE levels and anaphylactic histamine release from isolated rat mast cells. Acta Physiol. Scand. 107:313–318. [DOI] [PubMed] [Google Scholar]
- 29.van Toorenenbergen, A.W., P. van Swieten, and R.C. Aalberse. 1983. Measurement of IgE on rat mast cells: relation to serum IgE and allergen-induced histamine release. Scand. J. Immunol. 17:13–18. [DOI] [PubMed] [Google Scholar]
- 30.Bergstrand, H., A. Bjornsson, I.M. Frick, B. Lundquist, R. Pauwels, and H. Bazin. 1982. Anti-IgE and con A-induced histamine release from mast cells of four rat strains: correlation with total serum IgE. Agents Actions. 12:612–618. [DOI] [PubMed] [Google Scholar]
- 31.Martins, M.A., C.P. Pasquale, P.M. e Silva, R.S. Cordeiro, and B.B. Vargaftig. 1990. Eosinophil accumulation in the rat pleural cavity after mast cell stimulation with compound 48/80 involves protein synthesis and is selectively suppressed by dexamethasone. Int. Arch. Allergy Immunol. 92:416–424. [DOI] [PubMed] [Google Scholar]
- 32.Coyle, A.J., K. Wagner, C. Bertrand, S. Tsuyuki, J. Bews, and C. Heusser. 1996. Central role of immunoglobulin (Ig) E in the induction of lung eosinophil infiltration and T helper 2 cell cytokine production: inhibition by a non-anaphylactogenic anti-IgE antibody. J. Exp. Med. 183:1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernandez-Hansen, V., G.A. Mackay, C.A. Lowell, B.S. Wilson, and J.M. Oliver. 2003. The Src kinase Lyn is a negative regulator of mast cell proliferation. J. Leukoc. Biol. 75:143–151. [DOI] [PubMed] [Google Scholar]
- 34.Metzger, H. 1999. It's spring, and thoughts turn to… allergies. Cell. 97:287–290. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Espinosa, C., S. Odom, A. Olivera, J.P. Hobson, M.E. Cid Martinez, A. Oliveira-dos-Santos, L. Barra, S. Spiegel, J.M. Penninger, and J. Rivera. 2003. Preferential signaling and induction of allergy-promoting lymphokines upon weak stimulation of the high affinity IgE receptor on mast cells. J. Exp. Med. 197:1453–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurish, M.F., and K.F. Austen. 1989. Different mast cell mediators produced by different mast cell phenotypes. IgE, Mast Cells and the Allergic Response. D. Chadwick, D. Evered, and J. Whelan, editors. John Wiley & Sons Ltd., New York. 36–45. [DOI] [PubMed]
- 37.Ochi, H., N.H. De Jesus, F.H. Hsieh, K.F. Austen, and J.A. Boyce. 2000. IL-4 and IL-5 prime human mast cells for different profiles of IgE-dependent cytokine production. Proc. Natl. Acad. Sci. USA. 97:10509–10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hubbard, S.R. 1999. Src autoinhibition: let us count the ways. Nat. Struct. Biol. 6:711–714. [DOI] [PubMed] [Google Scholar]
- 39.Young, R.M., D. Holowka, and B. Baird. 2003. A lipid raft environment enhances Lyn kinase activity by protecting the active site tyrosine from dephosphorylation. J. Biol. Chem. 278:20746–20752. [DOI] [PubMed] [Google Scholar]
- 40.Yamashita, T., S.-Y. Mao, and H. Metzger. 1994. Aggregation of the high-affinity IgE receptor and enhanced activity of p53/56lyn protein-tyrosine kinase. Proc. Natl. Acad. Sci. USA. 91:11251–11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Field, K.A., D. Holowka, and B. Baird. 1995. FcɛRI-mediated recruitment of p53/56lyn to detergent-resistant membrane domains accompanies cellular signaling. Proc. Natl. Acad. Sci. USA. 92:9201–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith, K.G.C., D.M. Tarlinton, G.M. Doody, M.L. Hibbs, and D.T. Fearon. 1998. Inhibition of the B cell by CD22: a requirement for Lyn. J. Exp. Med. 187:807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cornall, R.J., J.G. Cyster, M.L. Hibbs, A.R. Dunn, K.L. Otipoby, E.A. Clark, and C.C. Goodnow. 1998. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity. 8:497–508. [DOI] [PubMed] [Google Scholar]
- 44.Hasegawa, M., M. Fujimoto, J.C. Poe, D.A. Steeber, C.A. Lowell, and T.F. Tedder. 2001. A CD 19-dependent signaling pathway regulates autoimmunity in Lyn-deficient mice. J. Immunol. 167:2469–2478. [DOI] [PubMed] [Google Scholar]
- 45.Takahashi, T., T. Yagi, S. Kakinuma, A. Kurokawa, T. Okada, K. Takatsu, S. Aizawa, and T. Katagiri. 1997. Suppression of autoimmune disease and of massive lymphadenopathy in MRL/Mp-lpr/lpr mice lacking tyrosine kinase Fyn (p59fyn). J. Immunol. 159:2532–2541. [PubMed] [Google Scholar]