

Abstract
Cancer vaccines aim at inducing (a) tumor-specific effector T cells able to reduce/eliminate the tumor mass, and (b) long-lasting tumor-specific memory T cells able to control tumor relapse. We have shown earlier, in 18 human histocompatibility leukocyte antigen (HLA)-A*0201 patients with metastatic melanoma, that vaccination with peptide-loaded CD34–dendritic cells (DCs) leads to expansion of melanoma-specific interferon γ–producing CD8+ T cells in the blood. Here, we show in 9 out of 12 analyzed patients the expansion of cytolytic CD8+ T cell precursors specific for melanoma differentiation antigens. These precursors yield, upon single restimulation with melanoma peptide–pulsed DCs, cytotoxic T lymphocytes (CTLs) able to kill melanoma cells. Melanoma-specific CTLs can be grown in vitro and can be detected in three assays: (a) melanoma tetramer binding, (b) killing of melanoma peptide–pulsed T2 cells, and (c) killing of HLA-A*0201 melanoma cells. The cytolytic activity of expanded CTLs correlates with the frequency of melanoma tetramer binding CD8+ T cells. Thus, CD34-DC vaccines can expand melanoma-specific CTL precursors that can kill melanoma antigen–expressing targets. These results justify the design of larger follow-up studies to assess the immunological and clinical response to peptide-pulsed CD34-DC vaccines.
Keywords: tumor immunology, immunotherapy, cancer, vaccine, immunomonitoring
Introduction
Early studies in mice have demonstrated that tumor-specific T cells can control tumor growth and metastasis (1–3). The identification of T cell–defined tumor antigens in humans, particularly in melanoma (4–8), facilitated the development of tumor-specific immunotherapy (9, 10). Two independent strategies are being pursued: (a) adoptive T cell therapy where ex vivo–induced antigen-specific T cells are transferred into patients (11–13), and (b) vaccination to induce therapeutic T cell immunity and tumor-specific immune memory in vivo (14–17).
Considerable clinical responses have been observed after adoptive transfer of melanoma antigen–specific CD8+ T cell clones and tumor-infiltrating T cell lines (18–20), demonstrating the value of tumor-specific T cells in treatment of cancer. Yet, passive transfer of T cells is not expected to yield the long-lived tumor-specific immunity that might be required to prevent tumor progression/relapse.
Cancer vaccines aim at inducing (a) tumor-specific effector T cells able to reduce/eliminate the tumor mass, and (b) long-lasting tumor-specific memory T cells able to control tumor relapse. Owing to their capacity to induce and regulate T cell immunity, DCs are increasingly used as adjuvants for vaccination in cancer (21–23). The immunogenicity of DCs charged with antigens ex vivo has now been demonstrated in healthy volunteers (24) and in patients with cancer (21, 25–27). Indeed, a number of pilot clinical trials have used tumor antigen–loaded DCs as vaccines and demonstrated safety as well as some clinical and immune responses (28–35). We vaccinated 18 HLA-A*0201 patients with metastatic melanoma with peptide-pulsed CD34-DCs. We have recently reported that vaccination with DCs led to enhancement of melanoma-specific CD8+ T cell immunity as measured by IFN-γ production (ELISPOT) upon in vitro exposure to melanoma antigen–derived peptides (28, 36). Here, we further analyzed 12 patients to determine whether vaccination with peptide-pulsed CD34-DCs permits expansion of melanoma-specific CD8+ T cells that can yield functional CTLs able to kill melanoma antigen–expressing cells. Melanoma-specific CD8+ T cells were measured after single restimulation with peptide-pulsed DCs in three independent assays: (a) killing of melanoma peptide–pulsed T2 cells, (b) killing of HLA-A*0201 allogenic melanoma cell lines, and (c) binding of melanoma tetramers. Each of the assays indicates that CD34-DCs vaccines can enhance CTL precursors specific for melanoma differentiation antigens.
Materials and Methods
Study Design, Patient Characteristics, and Eligibility Criteria
All of the details have been previously described (21, 28). In brief, 18 HLA-A*0201 patients with metastatic melanoma were injected with melanoma peptide–pulsed CD34-DCs. Inclusion criteria were the following: biopsy-proven American Joint Committee on Cancer metastatic melanoma; age >18 yr; Karnofsky performance status >80%; HLA-A*0201 phenotype; intradermal skin test positive to recall antigen; normal blood CD4 and CD8 T cell numbers; and normal quantitative immunoglobulin levels. Exclusion criteria included the following: prior chemotherapy or biologicals <4 wk before trial entry; untreated central nervous system lesions; bulky hepatic metastatic lesions; pregnancy; or concurrent corticosteroids/immunosuppressive therapy. All patients were presented with several treatment alternatives before they gave a written informed consent. The study was approved by the Food and Drug Administration, the National Cancer Institute, and the Institutional Review Board. Patients received a 6-wk outpatient vaccination course with antigen-loaded CD34-DCs given s.c. every 14 d for a total of four vaccinations. DCs were administered in a dose-escalation design at the dose level per cohort of 0.1, 0.25, 0.5, and 106 DCs/kg/vaccination. 12 out of 18 patients subsequently received additional vaccines as described elsewhere (unpublished data).
The trial was initiated in 1999 and the majority of patients completed the four vaccinations by mid 2000. Progressive disease means the appearance of ≥1 new lesions or ≥20% increase in the longest diameter of specified lesions, taking as reference the longest diameter recorded at the treatment onset according to RECIST criteria (37). Nonprogressive disease means the absence of disease progression as defined above. Patients underwent computed tomographic (CT) scanning, magnetic resonance imaging, or positron emission tomography scanning, and physical examination within 6 wk of the first vaccination and within 30 d after the fourth vaccination (unless otherwise indicated). Details are provided in Table I. All charts were reviewed by an independent outside clinical monitor. Scans were reviewed by two independent radiologists.
Table I.
Patient Characteristics
| Patient number | Previous therapy | Disease sites on entry | Status after four injections |
|---|---|---|---|
| 4 | Chemo/IL-2 IFN-α | CT scan: subcarinal LN mass, pulmonary nodule | CT scan: NPD |
| 5 | Chemo/IL-2 IFN-α | CT and PET scan: spleen, liver, and lung nodules | CT scan: NPD |
| 6 | Surgery/IL-2 | CT scan and PE: skin nodules and LN | PE: PD, new lesions |
| 9 | Surgery/RT cell vaccine | PE: axillary LN mass | PE: NPD |
| 10 | Surgery | PE: femoral LN next to biopsy proven LN mass | PE: NPD |
| 12 | Surgery/RT | MRI and PE: skin nodule, CNS | MRI and PE: NPD |
| 13 | Surgery/RT | MRI and CT scan: axillary LN | MRI and CT scan: PD in CNS |
| 16 | Chemo | CT scan: retroperitoneal LN, liver | CT scan: PD new lesions |
| 17 | Surgery | CT and PET scan: lung nodules | CT scan: NPD |
| 18 | Surgery | CT/PET scan and PE: pericardial nodule and vaginal wall | CT scan and PE: NPD |
| 19 | RT | PET scan and PE: parotid nodule | PE and CT scan: NPD |
| 21 | Surgery | MRI and PE: three liver lesions | MRI and PE: NPD |
NPD, nonprogressive disease; PD, progressive disease; PE, physical exam; PET, positron emission tomography; MRI, magnetic resonance imaging; CNS, central nervous system. Assessment of disease sites at baseline (pre) and after four DC vaccinations (post). Patient number 9: cell vaccine in 1995 while in stage III disease. CT scan at baseline negative. Patient number 10: CT scan of chest, abdomen, and pelvis negative at baseline. Patient number 12: received whole brain radiation therapy within 4 wk before first DC vaccination. Patient number 13: altered vaccination schedule due to appearance of a central nervous system lesion after two vaccinations; the lesion was resected. Because of steroid therapy, the patient received two additional vaccinations 3 mo later. Patient number 18: baseline scans performed 8 wk before first vaccination. Patient number 19: CT scan 2 mo after fourth DC vaccination.
Preparation and Administration of the DC Vaccine
The procedure has been described in detail (21, 28). In brief, DCs were generated by culture of CD34+ hematopoietic progenitor cells with 50 ng/ml GM-CSF (Immunex), 100 ng/ml Flt3 liter (Immunex), and 10 ng/ml TNF (CellPro, Inc.). DC vaccine was pulsed with 2 μg/ml KLH (Intracell), 2.5 μg/ml HLA-A*0201–restricted flu-matrix peptide (Flu-MP) GILGFVFTL58-66, and a mix of four HLA-A*0201–restricted peptides derived from melanoma antigens (Melan A/MART-127–35: AAGIGILTV; gp100g209–2M: IMDQVPFSV; Tyrosinase368–376: YMDGTMSQV; MAGE-3271–279: FLWGPRALV; 2.5 μg/ml). Vaccine was administered s.c. in three injection sites.
Assessment of CTL Precursors
Media and Reagents.
Complete culture medium (CM) consisted of RPMI 1640, 1% l-glutamine, 1% penicillin/streptomycin, 1% sodium pyruvate, 1% nonessential amino acids, 25 mM Hepes, and 10% heat-inactivated FCS (GIBCO BRL). For T cell cultures, FCS was replaced by 10% human AB serum (Gemini Bio-Products). 100 ng/ml GM-CSF (Leukine; Immunex), 25 ng/ml IL-4 (R&D Systems), 10 UI/ml IL-7 (R&D Systems), and 10 UI/ ml IL-2 (R&D Systems) were used.
Synthetic Peptides.
MelanA/MART-127–35 (AAGIGILTV), gp100g209–2M (IMDQVPFSV), TYR368–376 (YMDGTMSQV), Flu-MP58–66 (GILGFVFTL), CMV pp65495–563 (NLVPMVATV), MAGE-3271–279 (FLWGPRALV), and PSA1141–150 (FLTPKKLQCV) were used.
Cell Lines.
K562, A375 melanoma cell line, MCF7 breast cancer cell line, and T2 were from the American Type Culture Collection. The Me275 and Me290 melanoma cell lines, established at the Ludwig Cancer Institute in Lausanne, were provided by J.-C. Cerottini and D. Rimoldi (Ludwig Institute for Cancer Research, Lausanne, Switzerland). All cell lines were maintained in CM.
Monocyte-derived DCs.
Monocyte-derived DCs were generated from G-CSF (Amgen)–mobilized PBMCs from either HLA-A*0201 healthy volunteers (Institutional Review Board 097–053) or from patients. Adherent monocytes were cultured for 6 d in CM and GM-CSF plus IL-4. Maturation was induced by 30 h of culture with LPS. At day 6, the DCs were loaded with 10 μg/ml HLA-A*0201–restricted peptides overnight.
CD8+ T Cell Purification.
PBMCs were depleted of NK cells using CD56 and CD16 microbeads (Miltenyi Biotec) and CD8+ T cells were positively selected using microbeads (>90% CD8+).
Recall Assay.
104 peptide-loaded DCs were plated with 105 T cells in a final volume of 200 μl CM plus 10% AB serum, 10 UI/ml IL-7 for 3 d, and 10 UI/ml IL-2 for 7 d.
Tetramer Analysis.
Streptavidin-PE–labeled tetramers were purchased from Beckman Coulter. The peptides used for the tetramers were NLVPMVATV (HLA-A*0201 CMV pp65495–563) and the same peptides used for the vaccine: GILGFVFTL (HLA-A*0201 Flu-MP58–66), ELAGIGILTV (HLA-A*0201 MelanA/MART-127–35), IMDQVPFSV (HLA-A*0201 gp100g209–2M), YMDGTMSQV (HLA-A*0201 TYR368–376), and FLWGPRALV (HLA-A*0201 MAGE-3271–279). After culture, the T cells were divided into aliquots of 106 cells/well in a 96-well plate. 5 μl of each tetramer and 3 μl CD8 FITC or CD3 FITC were added to pelleted cells. Pellets were resuspended and incubated at room temperature for 30 min in the dark. After two washes with PBS, the cells were resuspended in 1% paraformaldehyde in PBS and analyzed by flow cytometry.
Cytotoxicity Assay and Class I MHC Blocking.
Cytotoxicity was measured in a standard 4-h 51Cr release assay. In brief, T2 cells were pulsed overnight with 10 μg/ml of indicated peptides. Targets (T2 cells and allogenic cell lines) were labeled with 51Cr (NEN Life Science Products), washed, and cocultured (103 cells) at 37°C for 4 h with CTLs. Percentage of specific lysis was calculated as (cpmexperiment − cpmspontaneous release)/(cpmmaximum release − cpmspontaneous release). 30 μg/ml anti-HLA ABC mAb (clone W6/32; DakoCytomation) or 30 μg/ml isotype control–purified mouse IgG2a (Becton Dickinson) was added at the onset of the cytotoxicity assay.
Statistics
As the form of the relationships between our measurements and the immunologic effectiveness is unknown, we used nonparametric tests based on u-scores, i.e., Spearman correlation and Mann-Whitney test, as indicated. To score activity profiles measured by the overall response to several melanoma antigens, we used a novel extension of u-statistics to multivariate ordinal data (38). In short, one first determines the partial ordering among all pairs of patients based on their response profiles. If one patient has a higher response than the other for some antigens and a lower response in none, the former and the latter patient have a “higher” and “lower” profile, respectively. If one patient has a higher response in some antigens, but a lower response in others, the pair-wise order among their profiles is undetermined, because the relative importance of the antigens is unknown. For each patient, the u-score is then computed as the number of patients whose profiles are lower, minus the number of patients whose profiles are higher. Pairs with undetermined order do not contribute to the score. These scores can then be used with standard nonparametric methods including linear rank procedures such as Spearman correlation (39, 40).
Online Supplemental Material
Table S1 shows CTL activity against peptide-pulsed T2 cells and HLA-A*0201 tumor cell lines. Table S2 shows the percentage of CD8+ T cells that bind tetramer in the recall assay, and Table S3 shows tetramer profiles. Tables S1–S3 are available at http://www.jem.org/cgi/content/full/jem.20032118/DC1.
Results
Expansion of Melanoma-specific CTL Precursors in the Recall Assay.
We investigated whether vaccination of patients with metastatic melanoma with peptide-pulsed CD34-DCs leads to expansion of melanoma-specific CD8+ T cells, which can mature into cytotoxic effectors upon single in vitro restimulation with melanoma peptide–pulsed DCs. CD8+ T cells, isolated from apheresis samples collected either at baseline or 2–4 mo after four vaccinations, were depleted of CD16+ CD56+ cells and restimulated with autologous peptide-pulsed DCs with 10 U/ml IL-7 and IL-2. We found robust proliferation of CD8+ T cells (2.5–11-fold) in cultures with both viral peptide– or melanoma peptide–pulsed DCs. There was no difference in T cell expansion between pre- and post-DC vaccination samples (unpublished data) or between the patients. Cultured CD8+ T cells were assessed in three assays: (a) killing of peptide-pulsed T2 cells, (b) killing of HLA-A*0201 melanoma cells, and (c) binding of melanoma tetramers.
Expanded CTLs Are Able to Kill Melanoma Peptide–pulsed T2 Cells.
Maturation of CD8+ T cells into CTLs was first assessed in a standard 4-h 51Cr release assay using as targets T2 cells pulsed with either the four melanoma peptides used in the vaccine, i.e., MelanA/MART-1, gp100, tyrosinase, and MAGE-3, or with a control peptide. Fig. 1, a and b, shows the CTL activity of cultured CD8+ T cells isolated from post-DC vaccination blood of two patients. Patient number 9 restimulated CD8+ T cells killed melanoma peptide–pulsed T2 cells with 40% lysis at the E/T ratio of 50:1 (Fig. 1 a). In contrast, restimulated CD8+ T cells from patient number 13 were not able to kill melanoma peptide–loaded T2 cells (Fig. 1 b). Killing was specific as no lysis of PSA peptide–pulsed T2 cells could be detected. Control cultures, in which CD8+ T cells were restimulated with CMV peptide–pulsed DCs (Fig. 1 a, Pt#9) or Flu-MP peptide–loaded DCs (Fig. 1 b, Pt#13), yielded highly efficient specific CTLs with ∼80% lysis at the E/T ratio of 50:1. These results suggested that some of the vaccinated patients do not display circulating melanoma-specific T cells able to mature into melanoma-specific CTLs, whereas they show functional virus-specific CD8+ T cells.
Figure 1.
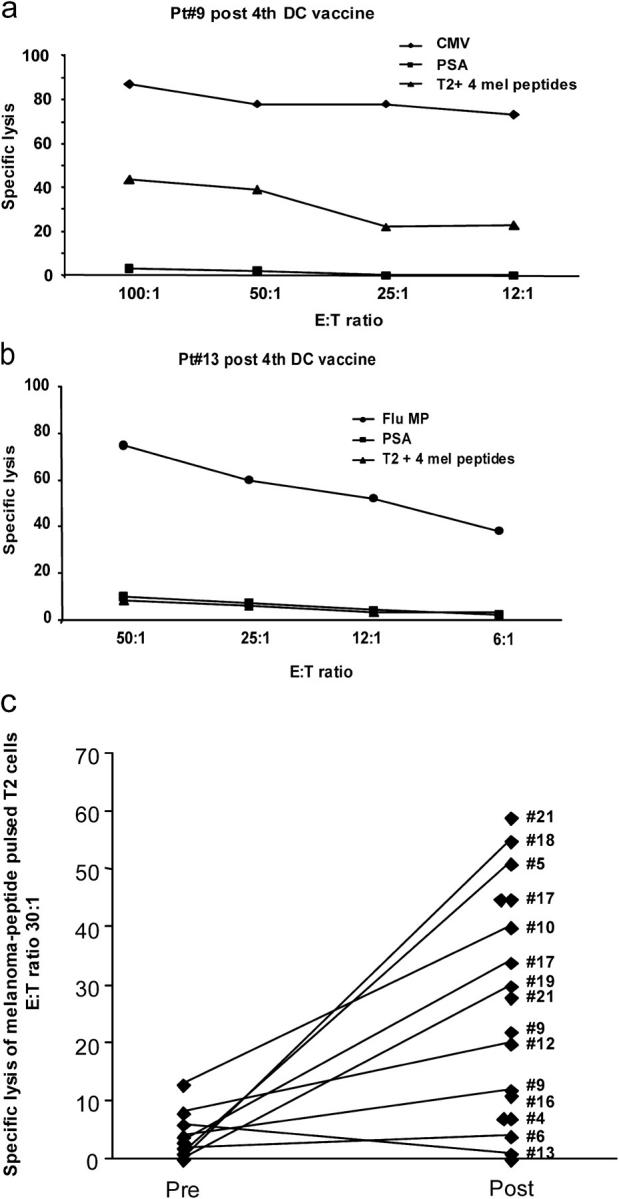
CTL activity against melanoma peptide–pulsed T2 cells before and after vaccination. Purified CD8+ T cells after a single stimulation with peptide-pulsed DCs are used as effectors in a standard 4-h 51Cr assay with T2 cells pulsed with either a control PSA peptide, or with viral peptides (Flu-MP, CMV), or with a mix of the four melanoma peptides (GP100, MART-1/Melan A, tyrosinase, and MAGE-3) at indicated E/T ratios. (a) CD8+ T cells from patient number 9 (see Table I, after fourth DC vaccine) are capable of specific lysis (ordinate) of T2 cells pulsed with viral (CMV) or melanoma peptides at several E/T ratios (abscissa). (b) CD8+ T cells from patient number 13 (see Table I, after fourth DC vaccine) are capable of specific lysis (ordinate) of T2 cells pulsed with Flu-MP peptide at several E/T ratios (abscissa), but not of melanoma peptide–pulsed T2 cells. (c) Killing of melanoma peptide–pulsed T2 cells (a mix of four melanoma peptides) by cultured CD8+ T cells from all tested patients at baseline (Pre) and after fourth DC vaccination (Post). Ordinate/specific lysis at the E/T ratio of 30–25:1, after subtraction of values obtained with PSA peptide–loaded T2 (see Table S1), patient number (see Table I). All experiments are shown.
Sufficient numbers of post-DC vaccination PBMCs were available to assess the CTL function of CD8+ T cells from 12 of the 18 vaccinated patients (Table S1, available at http://www.jem.org/cgi/content/full/jem.20032118/DC1). The median specific lysis was 21% after subtraction of values obtained with control PSA peptide–pulsed T2 cells (range: 0–55%; Fig. 1 c). In 10 of these patients (nos. 5, 6, 9, 10, 12, 13, 16, 17, 18, and 19) the killing of melanoma peptide–pulsed T2 cells could be compared prevaccination and after the fourth vaccination. After vaccination, restimulated CD8+ T cells from seven patients (nos. 5, 9, 10, 12, 17, 18, and 19) showed more than twofold higher killing of melanoma peptide–pulsed T2 cells than before vaccination. Killing of T2 cells pulsed with control PSA peptide was not increased, consistent with specificity (Table S1).
Thus, vaccination with peptide-pulsed CD34-DCs leads to expansion of circulating melanoma-specific CD8+ T cells that can mature into melanoma-specific CTLs able to kill model target cells expressing melanoma antigen.
Expanded CTLs Are Able to Kill Melanoma Cells.
Although T2 cells facilitate determination of antigen specificity, they do not yield conclusion as to whether a given cytotoxic cell can actually kill tumor cells. The cultured CD8+ T cells were analyzed for their capacity to kill the HLA-A*0201 melanoma cell lines Me275 and Me290. The use of allogenic melanoma cell lines expressing endogenous antigens to measure the immune response to DC vaccination has an advantage over the autologous tumor, available only in a few of our patients, as it allows comparing the T cell functions of different patients and avoids the tumor-dependent variability.
Fig. 2 shows the CTL activity of restimulated CD8+ T cells from patient number 12, which killed two melanoma cell lines, Me290 and Me275 (25% specific lysis), even at a relatively low E/T ratio of 25:1 (Fig. 2 a). No killing of control targets, the HLA-A*0201 MCF7 breast cancer cell line, and the NK-sensitive K562, was observed. Killing of melanoma cells was restricted by the expression of HLA class I, as the pretreatment of target cells with the HLA class I–blocking mAb W6/32 resulted in >90% inhibition of Me275 killing at different E/T ratios (Fig. 2 b, Pt#12). In line with the results observed with melanoma peptide–pulsed T2 cells, patient number 13's CD8+ T cells did not kill melanoma cell lines (Table S1).
Figure 2.

CTL activity against allogenic tumor cell lines. Purified CD8+ T cells after single stimulation are used as effectors in a standard 4-h 51Cr assay with control (K562 and MCF-7) and HLA-A*0201 melanoma (Me275 and Me290) cell lines at the indicated E/T ratios. (a) Purified CD8+ T cells from patient number 12 (see Table I, after fourth DC vaccine) are capable of specific lysis (ordinate) of two melanoma cells lines, but do not kill control targets at several E/T ratios (abscissa). (b) Killing of Me275 cells is restricted by MHC class I expression on targets cells. Me275 cells are used without pretreatment or are preincubated with either an isotype control or anti–HLA class I W6/32 mAb and used as targets. Specific lysis (ordinate) at two E/T ratios (abscissa) representative of three independent experiments with T cells from three patients is shown.
The capacity of cultured CD8+ T cells from post-DC vaccination blood to kill melanoma cell lines was then analyzed in 12 patients (Fig. 3 and Table S1). The median-specific lysis of Me275 cells was 12% (range: 0–39). The lysis of control MCF7 and K562 cells was 8 (range: 0–17) and 9% (range: 0–46), respectively. Independent experiments measuring CD8+ T cells from the same patient yielded reproducible results (Table S1). No induction of CTL activity against Me275 melanoma cells could be found when CD8+ T cells from three healthy HLA-A*0201 volunteers were used (median 2%).
Figure 3.

CTL activity against allogenic tumor cell lines before and after vaccination. Killing of Me275 melanoma cells, and of control MCF7 breast cancer and K562 cells, by cultured CD8+ T cells from all patients tested at baseline (Pre) and after fourth DC vaccine (Post). Ordinate, nonsubtracted specific lysis at the E/T ratio of 30–25:1; abscissa, patient number (see Table I). Average of all experiments for each patient (see Table S1) is shown. Prevaccination PBMCs were analyzed only once due to the limited availability of cells. Wilcoxon paired test.
In 10 of these 12 patients, the killing of melanoma cells could be compared prevaccination and after the fourth DC vaccination. As shown in Fig. 3, there was a significant increase in the lysis of Me275 melanoma cells post-DC vaccination, but not of control K562 and MCF7 cells. CD8+ T cells from seven patients showed a more than twofold increase in Me275 melanoma cell killing after DC vaccinations (Fig. 3). The patient with the highest prevaccination CTL activity also had IFN-γ–making T cells in the ELISPOT assay using pre-DC vaccination PBMCs (28). These results suggest that peptide-pulsed CD34-DC vaccination can enhance melanoma-specific CTLs able to kill melanoma cells.
Expanded CTLs Bind Melanoma Tetramers.
The increased ability of cultured CD8+ T cells to kill tumor targets could reflect an increased function of specific T cells and/or an increased frequency. Therefore, we measured the frequency of melanoma-specific CD8+ T cells using tetramers loaded with the four melanoma peptides used for vaccination, i.e., gp100, MART-1/Melan A, tyrosinase, and MAGE-3. The binding of Flu-MP tetramer was also analyzed as the Flu-MP peptide was a component of the DC vaccine. The frequency of melanoma-specific T cells was determined as the fraction of CD8+ T cells showing high intensity staining (41).
Fig. 4 shows examples of tetramer staining in cultured CD8+ T cells, which efficiently killed Me275 melanoma cells (Fig. 3, #17 and #21). CD8+ T cells from patient number 17 (Fig. 4 a) displayed predominant specificity for one antigen, i.e., gp100, with 16% of total tetramer-binding T cells, even higher than for Flu-MP–specific T cells (Fig. 4 a, 13.5%). MART-1–specific CD8+ T cells, and tyrosinase- and MAGE-3–specific T cells, were barely detectable. CD8+ T cells from patient number 21 contained cells specific for three of the four melanoma peptides used for immunization, i.e., gp100 (13.5%), MART-1 (1.6%), and MAGE-3 (2.2%; Fig. 4 b). CD8+ T cells specific for gp100 could be detected in this patient (patient no. 21) in the cultured prevaccination T cells, albeit at a lower frequency (5 vs. 13.5%), a finding consistent with the CTL data shown above (Fig. 3) and the earlier ELISPOT data from this patient (28).
Figure 4.

Flow cytometry analysis of tetramer binding by expanded CD8+ T cells. Restimulated CD8+T cells are labeled with anti–CD8-FITC (abscissa) and PE tetramers of a given specificity (ordinate). The analysis is performed on T cells gated on CD8 expression and the high affinity T cells are distinguished based on the intensity of tetramer fluorescence (square). (a) Patient number 17 and (b) patient number 21. For details, see Table S2. (c) Comparison of the percentage of high intensity tetramer binding CD8+ T cells (ordinate, log scale) at baseline and after fourth DC vaccination. Wilcoxon paired test.
The proportion of melanoma tetramer–binding T cells among CD8+ T cell cultures was measured in 10 patients before vaccination and after 4 vaccinations (Fig. 4 c and Table S2). The majority of the patients demonstrated considerable increase in the proportion of gp100-specific CD8+ T cells and three patients (nos. 5, 10, and 12) showed an increase in MART-1/Melan A–specific CD8+ T cells. The proportion of CD8+ T cells binding tyrosinase and/or MAGE-3 tetramer was much lower than that of gp100 and/or MART-1, the mechanism of which remains to be determined. Nevertheless, in some patients, considerable increase in tyrosinase- and/or MAGE-3–specific CD8+ T cells post-DC vaccinations could be observed (Fig. 4 c and Table S2). Thus, tetramer binding analysis indicates that vaccination with peptide-pulsed CD34-DCs leads to the expansion of melanoma-specific CD8+ T cells.
Killing of Melanoma Antigen–expressing Targets Correlates with Expansion of Tetramer-binding CD8+ T Cells.
Lastly, we determined whether the increased killing of cells expressing melanoma antigens (either T2 or melanoma cells) in response to DC vaccination correlates with the increased frequency of melanoma-specific CD8+ T cells, using u-scores for the profiles of the counts of CD8+ T cells binding each of the melanoma tetramers, i.e., gp100, MART-1/Melan A, tyrosinase, and MAGE-3 (Table S3). As shown in Fig. 5 a, the killing of melanoma peptide–pulsed T2 targets was strongly correlated (r = 0.75, P = 0.006) with the overall frequency of melanoma-specific CD8+ T cells. In contrast, there was no correlation between the frequency of melanoma-specific CD8+ T cells and the killing of control PSA peptide–pulsed T2 targets (r = 0.12, P = 0.73; Fig. 5 a). In line with the T2 assay, the killing of Me275 melanoma cells was strongly correlated (r = 0.85, P = 0.0008) with the overall frequency of melanoma-specific CD8+ T cells (Fig. 5 b). The killing of control cell lines MCF7 or K562, did not correlate with the frequency of melanoma-specific CD8+ T cells (P > 0.1), which again indicates specificity.
Figure 5.

CTL function correlates with the frequency of tetramer-binding CD8+ T cells. (a) The specific lysis, by CD8+ T cells restimulated with melanoma peptide–pulsed DCs, of melanoma peptide–pulsed T2 cells (ordinate), but not of PSA peptide–pulsed T2 cells, correlates with the frequency of all high affinity/intensity melanoma tetramers binding CD8+ T cells determined used u-scores (abscissa). Nonparametric Spearman correlation. (b) The specific lysis of Me275 melanoma cells by CD8+T cells restimulated with melanoma peptide–pulsed DCs (ordinate) correlates with the frequency of all high affinity/intensity melanoma tetramers binding CD8+ T cells determined used u-scores (abscissa). There was no correlation between tetramer scores and the killing of control MCF7 cells. Nonparametric Spearman correlation.
Thus, vaccination with antigen-loaded CD34-DC vaccines expands melanoma-specific CD8+ T cell precursors that promptly yield melanoma-specific CTLs.
Discussion
To the best of our knowledge, this represents the first study in which the quality of melanoma-specific CTLs elicited by vaccination with antigen-pulsed CD34-DCs was systematically measured in a cohort of 12 patients in three independent assays. The results show that vaccination with CD34-DCs can elicit CD8+ T cell precursors specific for melanoma differentiation antigens. These precursors can expand and mature into specific CTLs in a recall assay, i.e., upon single restimulation with melanoma peptide–pulsed DCs. These CTLs can be detected by three independent measurements: (a) binding of melanoma tetramers, (b) killing of melanoma peptide–pulsed T2 cells, and (c) killing of HLA-A*0201 melanoma cells.
Most clinical DC vaccination studies reported thus far measured CD8+ T cell responses using antigen-specific IFN-γ ELISPOT as a parameter of T cell function and specificity (for review see references 25 and 26). Only a few studies demonstrated the induction of tumor-specific cytotoxic T cells using as targets either peptide-pulsed T2 cells (29, 35, 42) or tumor RNA–loaded DCs (43). Except for two studies (44, 45), the induction of CTLs active against tumor cells could be analyzed only in a limited number of patients (33, 46, 47). In one study (44), 2 out of 10 patients with breast/ovarian cancer vaccinated with peptide-pulsed monocyte-derived DCs displayed killing of HLA-A*0201 tumor cell lines. In a second study (45), four out of nine patients with glioblastoma vaccinated with peptide-pulsed monocyte-derived DCs demonstrated the induction of effector cells capable to kill autologous tumors.
We found that vaccination with peptide-pulsed CD34-DCs leads to expansion of melanoma-specific cytolytic CD8+ T cell precursors in several patients. Enhancement of melanoma-specific CD8+ T cell immunity was assessed by measuring their CTL function against multiple targets expressing melanoma antigens in a standard 4-h 51Cr release assay. The release of 51Cr from labeled melanoma cells was significantly higher after culture with T cells isolated from post-DC vaccination blood as compared with T cells isolated at baseline. Yet, in some patients the expanded CD8+ T cells induced low 51Cr release from labeled melanoma cells (∼10% of specific lysis, patients 9, 10, 18, and 19). Because loading Me275 melanoma cells with a mix of the four melanoma peptides did not significantly increased the killing (unpublished data), the recognition of endogenous antigen was not a limiting factor. Determination of specific lysis is particularly complex when analyzing the killing of tumor cells. Two issues present themselves in the data interpretation, i.e., the lysis of control tumor cell lines and low 51Cr release. We interpret the observed lysis of K562 and MCF7 cells as the reflection of lytic activity of NK cells and therefore, we have chosen to present all the data without subtractions or thresholds. Indeed, the lysis of MCF7 or K562 cells did not correlate with the frequency of melanoma tetramer–specific T cells present in T cell cultures. Furthermore, although the killing of Me275 cells is restricted by HLA class I expression and can be nearly abolished by pretreatment of targets with respective mAb, this was not the case with MCF7 cells. To resolve low 51Cr release, we have exploited alternative measurements of CTL function. Preliminary data suggest that T cells from patients 9, 10, 18, and 19 were indeed able to reduce the number of viable Me275 cells in cocultures, suggesting that 10% of specific lysis at 4 h might indicate the presence of specific CTLs (unpublished data). Thus, T cells may use different mechanisms to control tumor survival/growth and a 51Cr release assay may not be sufficient for their evaluation.
One patient (no. 21) displayed, before vaccination, gp100-specific CD8+ T cells able to kill melanoma cells. As the only previous therapy was surgery, this suggests the existence of naturally occurring melanoma-specific immunity (6). This observation is consistent with earlier observations that melanoma-infiltrated LNs resected from patients may contain MART-1/Melan A tetramer-binding CD8+ T cells that display memory phenotype and can be expanded in vitro (48). Furthermore, previously untreated melanoma patients may display melanoma tetramer–binding T cells in the blood, particularly those specific for MART-1/Melan A (49, 50). Yet, in the majority of patients these T cells do not seem functional (28, 36, 49, 50).
We could measure CD8+ T cells specific for gp100 and MART-1, however, the frequency of T cells specific for the two other antigens presented on the vaccine, i.e., tyrosinase and MAGE-3, was considerably lower. Although this observation requires further studies, possible explanations may arise from (a) frequency of precursors, with MART-1 being higher than other specificities, (b) peptide used for immunization, with mutated gp100 having a high affinity for MHC class I (51), and (c) experimental conditions that do not allow sufficient in vitro expansion of T cells with lower frequency. For example, we cannot exclude that culturing CD8+ T cells with IL-2 leads to selection of certain specificities, whereas using other cytokines, for example IL-15 (52, 53), or culturing in the presence of CD4 T cells, might permit detection of a larger repertoire of CD8+ T cells.
Measuring the quality of melanoma-specific CD8+ T cell immunity in vaccinated patients revealed two groups of patients, i.e., those who did mount melanoma-specific CTLs able to kill the tumor and those who did not. Such inability to mount CD8+ T cell immunity seems to be restricted to melanoma antigens because highly efficient CMV- or Flu-MP–specific CTLs can be generated from these patients' blood. Thus, lack of melanoma-specific CTL induction is not a consequence of a global alteration of the immune system. The lack of melanoma-specific T cells by all three measurements was particularly apparent in three patients (nos. 6, 13, and 16), all of which experienced early disease progression with appearance of new lesions. Studies in larger patient cohorts are necessary to determine whether disease progression is indeed linked with the absence of melanoma-specific immunity in response to vaccination.
In conclusion, concurring with earlier clinical studies (29, 35), our study demonstrates that vaccination with DCs can induce functional CD8+ T cell immunity in cancer patients. The present results justify the design of larger follow-up studies to assess the immunological and clinical response to peptide-pulsed CD34-DC vaccines.
Acknowledgments
We thank our patients for volunteering to participate in this study. We are grateful to BiJue Chang and Susan Hicks for excellent help with accrual of patients and healthy volunteers, follow-up, and regulatory issues. We thank Susan Burkeholder, Jennifer Finholt-Perry, Nicolas Taquet, and Leena John from the Baylor Institute for Immunology Research Manufacturing Practice Lab for excellent work and commitment; John Kohl, Sebastien Coquery, and Elizabeth T. Kraus for processing and analyzing blood samples; and Kathy Brooks and Cindy Samuelsen for continuous help. We thank Dr. Franco Marincola for clinical monitoring. We thank Dr. Michael Ramsay for continuous support.
This work is supported by grants from Baylor Health Care Systems Foundation, Falk Foundation, Cancer Research Institute (to J. Fay), the National Institutes of Health (CA78846 and CA085540 to J. Banchereau, PO-1 CA84512 to J. Banchereau and A.K. Palucka, CA89440 to A.K. Palucka, and RR00102 to K.M. Wittkowski), and Association pour la Recherche sur le Cancer (to S. Paczesny). J. Banchereau is the recipient of the Max & Gayle Clampitt Chair for Immunology Research.
The online version of this article contains supplemental material.
Abbreviations used in this paper: CM, culture medium; CT, computed tomography; Flu-MP, flu-matrix peptide.
References
- 1.Greenberg, P.D., J.P. Klarnet, D.E. Kern, and M.A. Cheever. 1988. Therapy of disseminated tumors by adoptive transfer of specifically immune T cells. Prog. Exp. Tumor Res. 32:104–127. [DOI] [PubMed] [Google Scholar]
- 2.Herlyn, D., A. Linnenbach, H. Koprowski, and M. Herlyn. 1991. Epitope- and antigen-specific cancer vaccines. Int. Rev. Immunol. 7:245–257. [DOI] [PubMed] [Google Scholar]
- 3.North, R.J. 1984. The murine antitumor immune response and its therapeutic manipulation. Adv. Immunol. 35:89–155. [DOI] [PubMed] [Google Scholar]
- 4.Boon, T., J.C. Cerottini, B. Van den Eynde, P. van der Bruggen, and A. Van Pel. 1994. Tumor antigens recognized by T lymphocytes. Annu. Rev. Immunol. 12:337–365. [DOI] [PubMed] [Google Scholar]
- 5.Houghton, A.N. 1994. Cancer antigens: immune recognition of self and altered self. J. Exp. Med. 180:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagorsen, D., C. Scheibenbogen, F.M. Marincola, A. Letsch, and U. Keilholz. 2003. Natural T cell immunity against cancer. Clin. Cancer Res. 9:4296–4303. [PubMed] [Google Scholar]
- 7.Rosenberg, S.A. 1997. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol. Today. 18:175–182. [DOI] [PubMed] [Google Scholar]
- 8.Urban, J.L., and H. Schreiber. 1992. Tumor antigens. Annu. Rev. Immunol. 10:617–644. [DOI] [PubMed] [Google Scholar]
- 9.Kiessling, R., K. Wasserman, S. Horiguchi, K. Kono, J. Sjoberg, P. Pisa, and M. Petersson. 1999. Tumor-induced immune dysfunction. Cancer Immunol. Immunother. 48:353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein, G., and T. Boon. 1993. Tumor immunology: present perspectives. Curr. Opin. Immunol. 5:687–692. [DOI] [PubMed] [Google Scholar]
- 11.Dudley, M.E., and S.A. Rosenberg. 2003. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer. 3:666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirshaut, Y., and S.F. Slovin. 1985. Harnessing T-lymphocytes for human cancer immunotherapy. Cancer. 56:1366–1373. [DOI] [PubMed] [Google Scholar]
- 13.Melief, C.J., and W.M. Kast. 1991. T-cell immunotherapy of cancer. Res. Immunol. 142:425–429. [DOI] [PubMed] [Google Scholar]
- 14.Finn, O.J. 2003. Cancer vaccines: between the idea and the reality. Nat. Rev. Immunol. 3:630–641. [DOI] [PubMed] [Google Scholar]
- 15.Gilboa, E. 1999. The makings of a tumor rejection antigen. Immunity. 11:263–270. [DOI] [PubMed] [Google Scholar]
- 16.Pardoll, D.M. 1998. Cancer vaccines. Nat. Med. 4:525–531. [DOI] [PubMed] [Google Scholar]
- 17.Ribas, A., L.H. Butterfield, J.A. Glaspy, and J.S. Economou. 2003. Current developments in cancer vaccines and cellular immunotherapy. J. Clin. Oncol. 21:2415–2432. [DOI] [PubMed] [Google Scholar]
- 18.Dudley, M.E., J.R. Wunderlich, P.F. Robbins, J.C. Yang, P. Hwu, D.J. Schwartzentruber, S.L. Topalian, R. Sherry, N.P. Restifo, A.M. Hubicki, et al. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 298:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meidenbauer, N., J. Marienhagen, M. Laumer, S. Vogl, J. Heymann, R. Andreesen, and A. Mackensen. 2003. Survival and tumor localization of adoptively transferred Melan-A-specific T cells in melanoma patients. J. Immunol. 170:2161–2169. [DOI] [PubMed] [Google Scholar]
- 20.Yee, C., J.A. Thompson, D. Byrd, S.R. Riddell, P. Roche, E. Celis, and P.D. Greenberg. 2002. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci. USA. 99:16168–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banchereau, J., B. Schuler-Thurner, A.K. Palucka, and G. Schuler. 2001. Dendritic cells as vectors for therapy. Cell. 106:271–274. [DOI] [PubMed] [Google Scholar]
- 22.Gilboa, E., S.K. Nair, and H.K. Lyerly. 1998. Immunotherapy of cancer with dendritic-cell-based vaccines. Cancer Immunol. Immunother. 46:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinman, R.M., and M. Dhodapkar. 2001. Active immunization against cancer with dendritic cells: the near future. Int. J. Cancer. 94:459–473. [DOI] [PubMed] [Google Scholar]
- 24.Dhodapkar, M.V., J. Krasovsky, R.M. Steinman, and N. Bhardwaj. 2000. Mature dendritic cells boost functionally superior CD8(+) T-cell in humans without foreign helper epitopes. J. Clin. Invest. 105:R9–R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis, I.D., M. Jefford, P. Parente, and J. Cebon. 2003. Rational approaches to human cancer immunotherapy. J. Leukoc. Biol. 73:3–29. [DOI] [PubMed] [Google Scholar]
- 26.Schuler, G., B. Schuler-Thurner, and R.M. Steinman. 2003. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 15:138–147. [DOI] [PubMed] [Google Scholar]
- 27.Timmerman, J.M., and R. Levy. 1999. Dendritic cell vaccines for cancer immunotherapy. Annu. Rev. Med. 50:507–529. [DOI] [PubMed] [Google Scholar]
- 28.Banchereau, J., A.K. Palucka, M. Dhodapkar, S. Burkeholder, N. Taquet, A. Rolland, S. Taquet, S. Coquery, K.M. Wittkowski, N. Bhardwaj, et al. 2001. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 61:6451–6458. [PubMed] [Google Scholar]
- 29.Fong, L., Y. Hou, A. Rivas, C. Benike, A. Yuen, G.A. Fisher, M.M. Davis, and E.G. Engleman. 2001. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc. Natl. Acad. Sci. USA. 98:8809–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geiger, J.D., R.J. Hutchinson, L.F. Hohenkirk, E.A. McKenna, G.A. Yanik, J.E. Levine, A.E. Chang, T.M. Braun, and J.J. Mule. 2001. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 61:8513–8519. [PubMed] [Google Scholar]
- 31.Hsu, F.J., C. Benike, F. Fagnoni, T.M. Liles, D. Czerwinski, B. Taidi, E.G. Engleman, and R. Levy. 1996. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat. Med. 2:52–58. [DOI] [PubMed] [Google Scholar]
- 32.Mackensen, A., B. Herbst, J.L. Chen, G. Kohler, C. Noppen, W. Herr, G.C. Spagnoli, V. Cerundolo, and A. Lindemann. 2000. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoietic progenitor cells. Int. J. Cancer. 86:385–392. [DOI] [PubMed] [Google Scholar]
- 33.Nair, S.K., M. Morse, D. Boczkowski, R.I. Cumming, L. Vasovic, E. Gilboa, and H.K. Lyerly. 2002. Induction of tumor-specific cytotoxic T lymphocytes in cancer patients by autologous tumor RNA-transfected dendritic cells. Ann. Surg. 235:540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nestle, F.O., S. Alijagic, M. Gilliet, Y. Sun, S. Grabbe, R. Dummer, G. Burg, and D. Schadendorf. 1998. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 4:328–332. [DOI] [PubMed] [Google Scholar]
- 35.Thurner, B., I. Haendle, C. Roder, D. Dieckmann, P. Keikavoussi, H. Jonuleit, A. Bender, C. Maczek, D. Schreiner, P. von den Driesch, et al. 1999. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J. Exp. Med. 190:1669–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palucka, A.K., M.V. Dhodapkar, S. Paczesny, S. Burkeholder, K.M. Wittkowski, R.M. Steinman, J. Fay, and J. Banchereau. 2003. Single injection of CD34+ progenitor-derived dendritic cell vaccine can lead to induction of T-cell immunity in patients with stage IV melanoma. J. Immunother. 26:432–439. [DOI] [PubMed] [Google Scholar]
- 37.Therasse, P., S.G. Arbuck, E.A. Eisenhauer, J. Wanders, R.S. Kaplan, L. Rubinstein, J. Verweij, M. Van Glabbeke, A.T. van Oosterom, M.C. Christian, et al. 2000. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 92:205–216. [DOI] [PubMed] [Google Scholar]
- 38.Wittkowski, K., E. Lee, R. Nussbaum, F. Chamian, and J.G. Krueger. 2004. Combining several ordinal measures in clinical studies. Stat. Med. 23:1579–1592. [DOI] [PubMed] [Google Scholar]
- 39.Hajek, J., and Z. Sidak. 1967. Theory of Rank Tests. Elsevier, New York. 250 pp.
- 40.Wittkowski, K.M., E. Susser, and K. Dietz. 1998. The protective effect of condoms and nonoxynol-9 against HIV infection. Am. J. Public Health. 88:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yee, C., P.A. Savage, P.P. Lee, M.M. Davis, and P.D. Greenberg. 1999. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J. Immunol. 162:2227–2234. [PubMed] [Google Scholar]
- 42.Godelaine, D., J. Carrasco, S. Lucas, V. Karanikas, B. Schuler-Thurner, P.G. Coulie, G. Schuler, T. Boon, and A. Van Pel. 2003. Polyclonal CTL responses observed in melanoma patients vaccinated with dendritic cells pulsed with a MAGE-3.A1 peptide. J. Immunol. 171:4893–4897. [DOI] [PubMed] [Google Scholar]
- 43.Heiser, A., D. Coleman, J. Dannull, D. Yancey, M.A. Maurice, C.D. Lallas, P. Dahm, D. Niedzwiecki, E. Gilboa, and J. Vieweg. 2002. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J. Clin. Invest. 109:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brossart, P., S. Wirths, G. Stuhler, V.L. Reichardt, L. Kanz, and W. Brugger. 2000. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood. 96:3102–3108. [PubMed] [Google Scholar]
- 45.Yu, J.S., C.J. Wheeler, P.M. Zeltzer, H. Ying, D.N. Finger, P.K. Lee, W.H. Yong, F. Incardona, R.C. Thompson, M.S. Riedinger, et al. 2001. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 61:842–847. [PubMed] [Google Scholar]
- 46.Su, Z., J. Dannull, A. Heiser, D. Yancey, S. Pruitt, J. Madden, D. Coleman, D. Niedzwiecki, E. Gilboa, and J. Vieweg. 2003. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res. 63:2127–2133. [PubMed] [Google Scholar]
- 47.Timmerman, J.M., D.K. Czerwinski, T.A. Davis, F.J. Hsu, C. Benike, Z.M. Hao, B. Taidi, R. Rajapaksa, C.B. Caspar, C.Y. Okada, et al. 2002. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 99:1517–1526. [DOI] [PubMed] [Google Scholar]
- 48.Romero, P., P.R. Dunbar, D. Valmori, M. Pittet, G.S. Ogg, D. Rimoldi, J.L. Chen, D. Lienard, J.C. Cerottini, and V. Cerundolo. 1998. Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J. Exp. Med. 188:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee, P.P., C. Yee, P.A. Savage, L. Fong, D. Brockstedt, J.S. Weber, D. Johnson, S. Swetter, J. Thompson, P.D. Greenberg, et al. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5:677–685. [DOI] [PubMed] [Google Scholar]
- 50.Pittet, M.J., D. Valmori, P.R. Dunbar, D.E. Speiser, D. Lienard, F. Lejeune, K. Fleischhauer, V. Cerundolo, J.C. Cerottini, and P. Romero. 1999. High frequencies of naive Melan-A/MART-1–specific CD8+ T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med. 190:705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenberg, S.A., J.C. Yang, D.J. Schwartzentruber, P. Hwu, F.M. Marincola, S.L. Topalian, N.P. Restifo, M.E. Dudley, S.L. Schwarz, P.J. Spiess, et al. 1998. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 4:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waldmann, T.A., S. Dubois, and Y. Tagaya. 2001. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity. 14:105–110. [PubMed] [Google Scholar]
- 53.Waldmann, T.A., and Y. Tagaya. 1999. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu. Rev. Immunol. 17:19–49. [DOI] [PubMed] [Google Scholar]