

Abstract
Early B cell development is characterized by stepwise, ordered rearrangement of the immunoglobulin (Ig) heavy (HC) and light (LC) chain genes. Only one of the two alleles of these genes is used to produce a receptor, a phenomenon referred to as allelic exclusion. It has been suggested that pre–B cell receptor (pre-BCR) signals are responsible for down-regulation of the VDJH-recombinase machinery (Rag1, Rag2, and terminal deoxynucleotidyl transferase [TdT]), thereby preventing further rearrangement on the second HC allele. Using a mouse model, we show that expression of an inducible μHC transgene in Rag2−/− pro–B cells induces down-regulation of the following: (a) TdT protein, (b) a transgenic green fluorescent protein reporter reflecting endogenous Rag2 expression, and (c) Rag1 primary transcripts. Similar effects were also observed in the absence of surrogate LC (SLC) components, but not in the absence of the signaling subunit Ig-α. Furthermore, in wild-type mice and in mice lacking either λ5, VpreB1/2, or the entire SLC, the TdT protein is down-regulated in μHC+LC− pre–B cells. Surprisingly, μHC without LC is expressed on the surface of pro–/pre–B cells from λ5−/−, VpreB1−/−VpreB2−/−, and SLC−/− mice. Thus, SLC or LC is not required for μHC cell surface expression and signaling in these cells. Therefore, these findings offer an explanation for the occurrence of HC allelic exclusion in mice lacking SLC components.
Keywords: B cell development, allelic exclusion, Rag, TdT, pre-BCR
Introduction
B lymphocytes develop from hematopoietic progenitors, a process that follows a sequence of cellular stages during which Ig variable regions of the heavy chain (HC) and light chain (LC) loci are rearranged from a pool of DNA segments (1). Checkpoints along this developmental line ensure that only cells expressing functionally rearranged Ig genes proceed in differentiation (2). At the transition from the pro–B to the pre–B stage, cells are screened and selected for surface expression of a signaling-competent pre–B cell receptor (pre-BCR). The pre-BCR consists of a μHC pairing with the surrogate LC (SLC) components λ5 and VpreB1/2 in association with the signaling module Ig-α/-β (3). Cell surface expression of the pre-BCR induces clonal expansion (4). However, this is abolished in gene-targeted mice with either mutated or lacking pre-BCR components (for review see reference 3).
Furthermore, signals transduced through the pre-BCR are also implicated in maintaining allelic exclusion at the Ig HC locus by preventing further rearrangement of the second, DJH-rearranged allele. This is presumably achieved by shutting down the recombination machinery (e.g., the Rag genes; reference 5) and closing of the remaining Ig HC allele during subsequent LC rearrangement. Interestingly, Ig HC allelic inclusion is observed in mice in which the transmembrane region of μHC has been mutated (μmT), demonstrating that the membrane-form of the Ig HC is crucial for allelic exclusion to occur (6). In contrast, all mouse models in which components of the SLC have been deleted still display allelic exclusion at the level of surface Ig expression (7–9).
Several hypotheses have been put forward to explain this inconsistency. It was suggested that premature rearrangement and expression of κLC in a small number of cells could “rescue” B cell development and Ig HC allelic exclusion (10). The observation that some μHCs can associate with VpreB alone (11) led to the hypothesis that this pre-BCR–like complex could signal allelic exclusion in the absence of λ5. However, the observation that allelic exclusion is still maintained in VpreB1−/−VpreB2−/− double-deficient and VpreB1−/−VpreB2−/−λ5−/− triple-deficient mice (8, 9) suggested that in the absence of λ5 and/or VpreB1/2 μHC pairs with other, yet undefined partners resulting in a signaling complex could substitute for some, but not all, pre-BCR–specific functions.
To achieve a greater understanding of the mechanisms contributing to allelic exclusion, especially in SLC-deficient mice, we asked how the de novo synthesis of a tetracycline-controlled transgenic μHC affects the expression of the cellular recombination machinery (Rag1/2 and terminal deoxynucleotidyl transferase [TdT]) in the presence or absence of either λ5 or VpreB1/2. The same question was also investigated in mice lacking the entire SLC (SLC−/−). In addition, we have analyzed the cellular location and composition of putative signaling complexes in these same mice.
Materials and Methods
Animals.
Double transgenic Ig-tTA/tet-μ mice on a Rag2−/− (tet-μHC), Rag2−/−λ5−/− (tet-μHC λ5−/−), or Rag2−/− VpreB1−/−VpreB2−/− (tet-μHC VpreB1−/−VpreB2−/−) genetic background, as well as VpreB1−/−VpreB2−/− and VpreB1−/−VpreB2−/−λ5−/− (SLC−/−) mice have been described previously (4, 8, 9). tet-μHC, Rag2−/−λ5−/− mice were bred with mice deficient for Ig-α (12) to create mice deficient for all these genes. NG-BAC transgenic mice were provided by M. Nussenzweig (The Rockefeller University, New York, NY; reference 13). All mice were bred and maintained under pathogen-free conditions in the animal facility of the Nikolaus-Fiebiger-Center or the Babraham Institute. Genotypes were confirmed by PCR and/or flow cytometric analysis (NG-BAC). Transgenic mice were treated with 100 μg/ml tetracycline hydrochloride (Sigma-Aldrich) diluted in drinking water for a minimum of 7 d before BM preparation.
Sorting and Culturing of BM-derived B Lineage Cells.
Pro–B cells were isolated from BM single cell suspension using anti-CD19–coated magnetic beads (Miltenyi Biotec) following the manufacturer's instructions or by sorting green fluorescent protein+ (GFP) cells (NG-BAC) using a MoFlo™ high-speed sorter (DakoCytomation). Pro–B cells were cultured on γ-irradiated ST2 cells in medium with or without IL-7 (14). IMDM medium supplemented with 2% FCS, 1 mM glutamine, 50 μM β-mercaptoethanol (GIBCO™; Invitrogen), and 0.3 mg/ml Primatone® RL (Sheffield Products) was used for all cell culture experiments unless stated otherwise. Tetracycline hydrochloride was added to the culture medium at a concentration of 100 ng/ml to block transgenic HC expression.
For enzymatic amplification staining of surface pre-BCR components, MACS-sorted tet-μHC CD19+ BM cells were cultured for 2 d in the presence of Tet, splitted, and recultured for an additional 18 h in the presence or absence of Tet on γ-irradiated stromal cells in the presence of IL-7. For SLC−/− mice, MACS-sorted CD19+ cells were cultured for 4 d on γ-irradiated stromal cells in the presence of IL-7 in RPMI 1640 and supplemented with 50 μM β-mercaptoethanol, antibiotics, and 10% FCS.
Antibodies and Flow Cytometric Analysis.
Antibodies for surface stainings include: PE-conjugated anti-CD19 mAb, FITC- or PerCP™-conjugated anti-B220 mAbs, PE-conjugated anti–c-kit, and anti-CD25 mAbs (BD Biosciences).
For intracellular stainings, cells were fixed and permeabilized using the FIX & PERM® kit (Caltag Laboratories) and the following antisera: FITC- or Cy™5-conjugated polyclonal goat anti–mouse IgM (μH chain specific) and FITC- or Cy™5-conjugated polyclonal donkey anti–rabbit IgG (Dianova); FITC-conjugated anti–κL-chain and anti–λ1+λ2L-chain mAbs (BD Biosciences); polyclonal rabbit anti-TdT (Supertechs); and polyclonal rabbit anti-Ku70 (DPC Biermann).
RNA Fluorescent In Situ Hybridization (FISH).
The Rag1 cDNA fragment was amplified by PCR using the following primers: 5′-AGTGAGGTCTTCTCCTAGCACCTA-3′; and 5′-ATGATTTTCTGAACCTCTCTTGG-3′. The PCR product was cloned into the pCR2.1 vector (GIBCO™; Invitrogen). To prepare probes for RNA FISH, the plasmid was first linearized by restriction enzyme digest, either 5′ or 3′ of the insert for preparation of antisense and sense probes. Thereafter, RNA was synthesized using T7 or T3 RNA polymerase as instructed using the MEGAscript™ High Yield Transcription Kit (Ambion), followed by RT-PCR amplification using p(dN)6 random primers and digoxygenin-dUTP.
RNA FISH was performed as described by Gribnau et al. (15) with the exception that the hybridization mix contained 50% formamide. Before application to the slides, the hybridization mix containing the probes was denatured at 85°C. Antibody detection of labeled probe was preceded by a 45-min incubation of the slides with TSB (0.1 M Tris, 0.15 M NaCl, and blocking reagent; Roche Diagnostics Ltd.) at room temperature. Probe was detected using the following antibodies that were diluted in TSB and each incubated with the slides for 45 min at room temperature: sheep anti-DIG (Roche Diagnostics Ltd.), FITC-labeled rabbit anti–sheep, and goat anti–rabbit (Calbiochem-Novabiochem). Images were examined by microscope under oil with a 100× objective.
Enzymatic Amplification Staining.
Membrane pre-BCR components were detected by enzyme amplification staining (EAS) using the Flow-Amp™ kit for cell surface molecules stained by biotinylated primary antibodies (Flow-Amp Systems, Ltd.). EAS staining achieves signal enhancement with enzymes that catalyze the deposition of labeled molecules onto the cell surface. Although the label binds covalently to any cell surface protein, the deposition is specific because it is proximity controlled by specific antibody–dependent binding to a targeted cell surface molecule. Abs used for this method include: biotin-conjugated polyclonal goat anti–mouse IgM (μH chain specific; Sigma-Aldrich), biotin-conjugated rat mAbs R6-60.2 (anti–mouse IgM), SL156 (anti–mouse pre–B cell receptor), and LM34 (anti–mouse λ5; BD Biosciences). Hybridomas producing rat mAbs VP245 and R5 (anti–mouse VpreB) were provided by F. Melchers (University of Basel, Basel, Switzerland; reference 16) and M.D. Cooper (University of Alabama, Birmingham, AL; reference 17). These two mAbs were purified from hybridoma supernatants on protein G–Sepharose, biotinylated using standard procedures, and tested by flow cytometry on the pre–B cell line NFS5. Biotin was revealed using PE-streptavidin purchased from BD Biosciences. After EAS surface staining, cells were incubated with propidium iodide (Sigma-Aldrich) to exclude dead cells from analysis. Fluorescence of cells in the lymphocyte gate was determined on a four-color FACSCalibur™ instrument (BD Biosciences), and data files were analyzed using FlowJo™ software (Tree Star, Inc.) or CellQuest™ (BD Biosciences).
Results
TdT Is Down-regulated in Pre–B Cells after De Novo Expression of μHC in the Absence of either λ5 or VpreB1/2.
During V(D)J recombination, nontemplate-derived nucleotides (N regions) are added at the segment joints through the activity of the enzyme TdT (18). It is highly expressed in pro–B cells where Ig HC genes are rearranged, but is sharply down-regulated at the pre–B cell stage when cytoplasmic μHC protein is expressed (19). Thus, although N regions are found in the majority of Ig HC rearrangements, they are uncommon in Ig LC joints of adult murine B cells (20). Expression of transgenic μHCs in Rag1−/− pro–B cells and μHC constructs transfected into pro–B cell lines both resulted in a similar decrease in TdT expression, suggesting that its down-regulation is induced by the pre-BCR (21).
To confirm a causal connection, we took advantage of a tetracycline-controlled μHC transgene that can be expressed in the absence of endogenous V(D)J rearrangements, called tet-μHC (4). In these mice, μHC expression is completely suppressed, and B cell development is blocked at the pro–B cell stage after treatment with tetracycline in the drinking water for 7 d (4). CD19+ BM pro–B cells from these mice were cultured on stromal cells in the presence of IL-7. Expression of the transgenic μHC was induced by omitting tetracycline. After 24 h, the cells were harvested, stained for intracellular μHC and TDT expression, and analyzed by FACS®. Fig. 1 A demonstrates that μHC positive cells (−Tet) showed a marked decrease in TdT protein level compared with μHC negative cells (−Tet). Surprisingly, after μHC induction (−Tet), a similar, though less pronounced down-regulation of TdT was observed in cells that lack the SLC components λ5 or VpreB1/2, and, hence, cannot express a complete pre-BCR complex. Parallel staining for Ku70, an ubiquitously expressed protein involved in V(D)J recombination, as well as general DNA repair (22), revealed no decrease in μHC positive cells, confirming the specificity of μHC-mediated TdT down-regulation in the absence of λ5 or VpreB1/2. Comparable results were obtained with cells cultured in the absence of IL-7 (unpublished data). Thus, de novo expression of μHC is causally involved in the down-regulation of TdT and, unexpectedly, de novo expression of a μHC can signal the down-regulation of TdT in the absence of the pre-BCR.
Figure 1.

TdT is down-regulated in pre–B cells after de novo synthesis of transgenic μHC in the absence of λ5 or VpreB1/2. (A) CD19+ BM cells were isolated by MACS from tet-μHC, tet-μHC λ5−/−, and tet-μHC VpreB1−/−VpreB2−/− mice that had received Tet in the drinking water for 7 d and cultured on stromal cells in medium containing IL-7 in the presence (top) or absence (bottom) of Tet. After 24 h, cells were fixed, permeabilized, and stained for cytoplasmatic μHC expression in combination with TdT, Ku70, or βGal (isotype control). Fluorescence was determined by flow cytometry. Numbers within the gates represent mean fluorescence intensities for βGal, TdT, and Ku70, respectively. (B) CD19+ BM cells were isolated by MACS from tet-μHC λ5−/− Ig-α−/− mice that had received Tet in the drinking water for 7 d and cultured on stromal cells in medium containing IL-7 in the absence of Tet. After 48 h, cells were fixed, permeabilized, and stained for cytoplasmatic μHC expression in combination with TdT or βGal (isotype control). Numbers within the gates represent mean fluorescence intensities for βGal and TdT, respectively.
To further analyze whether the signal transduction molecule Ig-α is involved in the unexpected down-regulation of TdT after expression of a μHC transgene in the absence of λ5, tet-μHCλ5−/−Ig-α−/− mice were generated. Comparable levels of TdT were found in μHC+ and μHC− cells 11, 24, and 48 h after transgene induction (Fig. 1 B and not depicted). We conclude that the signaling subunit Ig-α is necessary for the μHC-induced down-regulation of TdT in the absence of a functional pre-BCR.
TdT Is Down-regulated in μHC-expressing BM Pre–B Cells in the Absence of SLC Components and Conventional LC.
To further investigate the unexpected down-regulation of TdT expression in the absence of the pre-BCR, we analyzed wild-type mice that express a polyclonal μHC repertoire as well as mice deficient in all SL chain components. We compared TdT levels of pro–B and pre–B cells in recombination competent wild-type, VpreB1−/−VpreB2−/−, and SLC−/− mice. For this purpose, four-color flow cytometric analyses were performed on CD19+ BM cells. B lineage cells were stained for surface c-kit or CD25, followed by fixation and permeabilization to detect cytoplasmic μHC and nuclear TdT. Antibodies specific for κ and λLC were also included to exclude cells expressing surface or intracellular LC.
In wild-type mice, as expected, most c-kit+μHC− pro–B cells showed high levels of TdT expression, whereas most c-kit+μHC+ cells had down-regulated TdT (Fig. 2 A, top), presumably because they already express the pre-BCR (23). As expected, in VpreB1−/− VpreB2−/− and SLC−/− mice, most c-kit+μHC− cells were positive for TdT (Fig. 2 A, middle and bottom). However, in contrast with wild-type cells, TdT levels stay elevated in most c-kit+μHC+ cells from both VpreB1−/−VpreB2−/− and SLC−/− mice.
Figure 2.

TdT is down-regulated in μHC-expressing BM pre–B cells in the absence of SLC components and conventional LC. CD19+ BM cells isolated from C57BL/6, VpreB1/2−/−, or VpreB1/2−/− λ5−/− (SLC−/−) mice were stained for surface c-kit or CD25, followed by fixation and permeabilization to detect intracellular κ/λLC and μHC in combination with TdT or βGal (isotype control). Cells positive for conventional LC were excluded from the analysis. (A) Cells positive for c-kit (i.e., pro–B cells) and negative (G1) or positive (G2) for cytoplasmic μHC were analyzed separately for TdT expression (unshaded). Staining for βGal was used as isotype control (shaded). (B) Cells positive for surface CD25 and cytoplasmic μHC (i.e., pre–B cells) were analyzed as in A. Numbers in the dot plots indicate the percentage of cells within the gates. Numbers in the histograms represent mean fluorescence intensities for TdT or βGal (shaded). The purity of CD19+ enriched cells was 96 (C57BL/6), 91 (VpreB1/2−/−), and 89% (SLC−/−) in this experiment. Representative results of two FACS® analyses are shown.
In mouse BM, CD25 is a marker for pre–B cells (24) and, in wild-type mice, it has been shown that these cells express μHC, but are negative for TdT mRNA (5). The data shown in Fig. 2 B (top) confirm these findings at the protein level. As described previously (8, 9), a small but distinct population of CD25+ BM cells is detected in both VpreB1−/−VpreB2−/− and SLC−/− mice. As shown in Fig. 2 B (middle and bottom), these cells are μHC positive and, most importantly, negative for TdT. This population of μHC+LC−TdT− cells can also be detected in λ5−/− mice (unpublished data). Together, these data show that μHC expression mediates down-regulation of TdT also in the absence of λ5, VpreB1/2, or the entire SLC and conventional LC.
Induction of μHC in Pro–B Cells Mediates Down-Regulation of Rag Expression in the Absence of λ5.
Although TdT is dispensable for Ig rearrangements, Rag1/2 complexes are crucial for execution of the initial steps of V(D)J recombination. Rag1/2 gene expression is down-regulated in large pre–B cells that display the pre-BCR on the cell surface (5). Surface expression of the μHC in complex with SLC (pre-BCR) is believed to be mandatory for this down-regulation and, thereby, might be implicated in allelic exclusion by preventing further rearrangement on the DJ-rearranged allele. Because allelic exclusion is still maintained in all SLC knockout mice (7–9), we analyzed the effect of transgenic μHC induction on Rag1/2 expression in the presence or absence of λ5.
For this purpose, we used tet-μHC and tet-μHCλ5−/− mice carrying a bacterial artificial chromosome that contains the Rag2 gene regulatory sequences, but encodes a GFP reporter instead of Rag2 (NG-BAC). The GFP reporter was shown to reflect the documented patterns of Rag expression (13). GFP+ BM cells from tetracycline-treated tet-μHC mice (pro–B cells) were isolated by flow cytometry cell sorting and cultured on stromal cells in medium containing IL-7 without Tet (Fig. 3). Cells were fixed at 24, 48, and 72 h; permeabilized; stained for expression of μHC; and analyzed by FACS®. The de novo expression of μHC resulted in a more pronounced down-regulation of GFP, clearly measurable after 48 h (Fig. 3, left; mean fluorescence intensities: 78 in μ negative versus 56 in μ positive cells). Because GFP degradation was shown to lag behind Rag mRNA (GFP in vivo half-life: 52 h; reference 25), more rapid effects could not be expected. We also observed a decrease in the GFP level in μHC negative cells during the cultivation (Fig. 3, mean fluorescence intensities: 189 for 24 h to 55 for 72 h). This effect presumably is induced by IL-7 in the medium as described previously (26). Surprisingly, comparison of GFP levels in μHC positive and μHC negative cells 48 and 72 h after the conditional expression of the μHC in cells lacking λ5 shows that Rag2 reporter gene expression is down-regulated to a greater extent in μHC positive cells (Fig. 3, right). This result indicates that Rag2 down-regulation might be mediated by μHC, but does not depend on formation of a complete pre-BCR.
Figure 3.
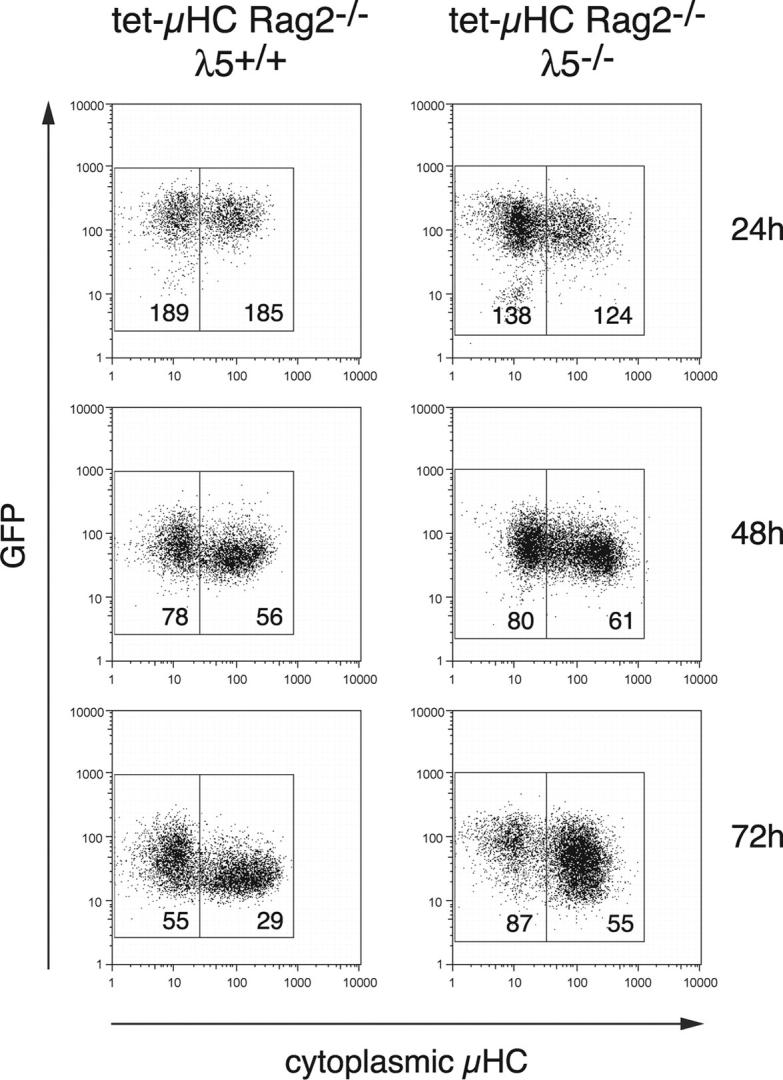
Expression of μHC in pro–B cells mediates down-regulation of a transgenic GFP reporter reflecting endogenous Rag2 expression in the absence of λ5. GFP+ BM cells were isolated from tet-μHC and tet-μHC λ5−/− mice carrying a bacterial artificial chromosome that encodes a green fluorescent protein (GFP) reporter instead of Rag2 (NG-BAC) and cultured on stromal cells in medium containing IL-7 without Tet. At 24, 48, and 72 h, cells were fixed and stained for cytoplasmic μHC expression. Fluorescence intensities for μHC versus GFP were analyzed by FACS®. Numbers printed within the gates indicate mean fluorescence intensity for GFP.
Transgenic μHC expression is already detectable 12 h after withdrawal of Tet (4), but Rag-driven GFP down-regulation was not observed until 48 h. Therefore, to determine directly whether μHC mediates termination of Rag transcription and whether this has already taken place after 12 h, RNA FISH was performed (15). The advantage is that this technique allows the detection of RNA produced at the site of transcription (i.e., at individual alleles, in single cells [nuclei]). For this, we made a probe that specifically detects Rag1 primary transcripts (Fig. 4 and not depicted). As a control, we used a probe that detects CD45, which is expressed on both pro–B and pre–B cells and, hence, is independent of μHC. The CD45 gene is biallelically transcribed (27). Numbers of nuclei showing 0, 1, or 2 foci of transcription were counted (>150 nuclei per slide). Nuclei with one CD45 probe signal may reflect an oscillating transcriptional status of this gene (28, 29). We assume that this is also the case for the Rag gene because a comparable number of nuclei (5–10%) were observed with only one detectable CD45 or Rag1 signal. Therefore, percentages of nuclei with one or two signals were combined.
Figure 4.
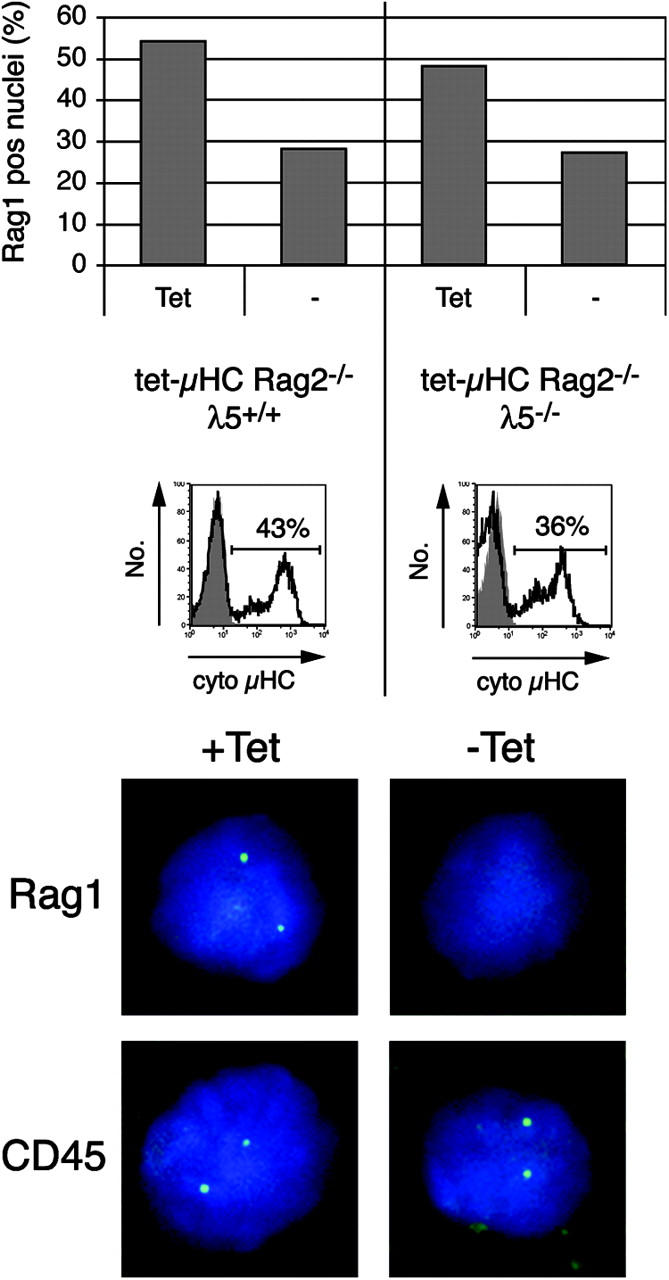
Induction of μHC in pro–B cells induces down-regulation of Rag1 primary transcripts in the absence of λ5. CD19+ BM cells from tet-μHC and tet-μHC λ5−/− mice were isolated by MACS and cultured on stromal cells with or without Tet for 12 h. Cells were fixed onto slides and subjected to RNA FISH to detect primary transcripts on individual alleles in single nuclei using ssDNA probes specific for Rag1 and CD45 (control). Hybridized probes were detected and visualized by fluorochrome-labeled Abs, and cells displaying signals were counted (top, >150 nuclei per slide). Induction of cytoplasmic μHC expression was verified by intracellular FACS® (middle, unshaded). Nuclei were counterstained with 4,6-diamino-2-phenylindole. Microscopical images (bottom) show representative nuclei hybridized for Rag1 or CD45, respectively. The experiment was repeated with comparable results.
As shown in Fig. 4, in tet-μHC cell cultures in the absence of μHC expression (+Tet), 54% of pro–B cells contained signals for Rag1 primary transcripts. In contrast, 12 h after induction of μHC (−Tet), this percentage was reduced to 28%. This decrease of 48% corresponded well with the proportion of cytoplasmic μHC+ cells present in the culture (43%). In tet-μHCλ5−/− cell cultures, a similar decrease of cells with Rag1 signals (from 48 to 27%) was observed after μHC induction (−Tet). As with the tet-μHC cell cultures, the 44% decrease in the absence of λ5 was similar to the proportion of cytoplasmic μHC+ cells (36%). As expected, the expression of μHC had no effect on the percentage of nuclei with CD45 primary transcripts, independent of genotype (unpublished data). Thus, the percentage of cells that express μHC is similar to the percentage of nuclei in which Rag1 transcription is terminated. Therefore, we conclude that μHC expression causes down-regulation of Rag1 transcription. Furthermore, this is independent of λ5 and, hence, a complete pre-BCR.
Transgenic μHC Detected on the Surface of Pre–B Cells in the Absence of LC and λ5 or VpreB1/VpreB2.
Our findings that μHC mediates down-regulation of TdT and Rag1/2 in the absence of SLC components raised the question of how the responsible signals are generated. It is thought that incomplete pre-BCRs are retained in the ER through binding to the Ig HC binding protein BIP (30). Because the various SLC-deficient mice still show HC allelic exclusion (7–9), it has been suggested that an ER-resident μHC complex may still be able to signal. However, a pre-BCR apparently has to be released from the ER to efficiently bind Src-related protein tyrosine kinases and gain signaling capacity (31). Therefore, we favor the hypothesis that a signaling competent pre-BCR–like complex is expressed on the cell surface in the absence of λ5 and/or VpreB1/2.
To allow detection of low levels of surface μHC complexes, we used biotinylated Abs in combination with EAS. We analyzed surface expression of transgenic μHC and SLC on CD19+ BM cells from tet-μHC, tet-μHC λ5−/−, and tet-μHC VpreB1−/−VpreB2−/− mice. Cells were isolated and expanded on stromal cells in medium containing IL-7 and Tet for 48 h, washed, and recultured for 18 h in the presence (Fig. 5 A, shaded) or absence (Fig. 5 A, unshaded) of Tet. Expression of the transgenic μHC in the absence of Tet was confirmed by parallel cytoplasmic staining for μHC (unpublished data).
Figure 5.

μH chain is detectable on the surface of λ5−/−, VpreB1−/−VpreB2−/−, and SLC−/− pre–B cells. (A) CD19+ BM cells were isolated by MACS from tet-μHC, tet-μHC λ5−/−, and tet-μHC VpreB1−/−VpreB2−/− mice that had received Tet in drinking water for 7 d. Cells were expanded on stromal cells in medium containing IL-7 and Tet for 48 h, washed, and recultured for an additional 18 h in the presence of IL-7 with (shaded) or without Tet (unshaded). Cells were harvested and stained with biotinylated Abs against μH chain (goat polyclonal), pre-BCR (SL156), or SLC components (LM34, VP245, and R5). Surface-bound biotin was amplified and revealed by EAS and streptavidin-PE. Propidium iodide negative cells were analyzed by flow cytometry. (B) CD19+ BM cells were isolated by MACS from SLC−/− or Rag1−/− (control) mice and expanded on stromal cells in medium containing IL-7 for 4 d. Cells were harvested and stained with FITC-labeled Abs against κ/λLC and biotinylated Abs against μHC (goat polyclonal) or λ5 (LM34). Surface-bound biotin was amplified and revealed aforementioned. Numbers represent the percentage of cells within the quadrants.
As expected, λ5 and VpreB was detectable on cells from tet-μHC mice cultured in the presence of Tet (μHC−; reference 16), whereas surface μHC and pre-BCR was only detected on cells cultured in the absence of tetracycline (Fig. 5 A, top, unshaded). Remarkably, after induction of its expression, μHC was also detected on the surface of cells lacking either λ5 or VpreB1/2 (Fig. 5 A, middle and bottom, unshaded). Comparable results were obtained with a different mAb (R6-60.2)-recognizing μH chain (unpublished data). It has been suggested that VpreB can associate with μHC in the absence of λ5 (11). However, using two different mAbs (17), we were unable to detect VpreB on the surface of μHC+ cells from λ5−/− mice. Interestingly, cells from VpreB1−/−VpreB2−/− mice express low levels of λ5 on the surface of μHC− cells, which increased after μHC induction (Fig. 5 A, bottom). Therefore, we assume that λ5 can be transported to the cell surface in the absence of VpreB1/2. Although association of λ5 with μHC is not essential, it apparently facilitates surface expression. The possibility that a μHC-λ5 complex might be formed in the absence of VpreB1/2 is supported by the detection of a complex on the cell surface using the mAb SL156 only after μHC expression. This observation also indicates that the epitope recognized by the mAb SL156 is formed by μHC and λ5 and is not dependent on VpreB1/2. Together, our data show that μHC can be transported to the cell surface in the absence of λ5 or VpreB1/2. In cells lacking λ5, μHC does not associate with VpreB but in the absence of VpreB1/2, it possibly forms a complex with λ5.
μHC Detected on the Surface of Pre–B Cells from SLC−/− Mice.
To exclude the possibility that only the μHC transgene used in these experiments can give rise to surface expression and to exclude the possibility that both λ5 and VpreB are able to support surface μHC expression independently, we analyzed BM pre–B cells from SLC−/− mice for cell surface expression of μHC using EAS. BM pro–B cells from Rag−/− mice were used as negative control. As shown in Fig. 5 B, after culturing sorted CD19+ cells for 4 d in vitro, a distinct μHC positive population was detected in cultures from SLC−/− but not Rag−/− mice. As expected, the SLC−/−-cultured cells did not express λ5. Furthermore, the μHC positive cells did not express either κ or λLCs. Thus, a variety of μHCs can be transported to the cell surface in the absence of the entire SLC.
Discussion
The pre-BCR is the central signaling complex during early B cell development. It grants survival, induces clonal expansion, and permits further differentiation of precursor cells (3, 4). However, the most important function attributed to μHC in complex with the SLC is to repress further rearrangement, thereby preventing the production of a second functional μHC within a developing B cell. In the first instance, this is probably accomplished by shutting off the V(D)J recombination machinery, primarily Rag1/2 (5). Simultaneously, TdT is down-regulated, which results in the absence of N-nucleotide addition in VJL recombinations of the LC genes of murine B lymphocytes (21). Here, we show that not only a complete pre-BCR but also μHC in the absence of SLC can mediate down-regulation of TdT and Rag1, providing an explanation as to why allelic exclusion is still operating in SLC-deficient mice (7–9), but not in μmT mice (6).
It has been suggested that premature rearrangement and expression of κLC in a small number of cells can “rescue” B cell development and HC allelic exclusion in the absence of SLC (10, 32). However, this mechanism would predict N-nucleotide additions in LC genes of peripheral B cells in λ5−/− and VpreB1/2−/− mice, which is not the case (33). Herein, our results clearly demonstrate that the down-regulation of TdT and Rag1 does not depend on LC expression. First, our experiments using a tetracycline-inducible μHC transgene were performed in Rag2-deficient mice, which are unable to express conventional LCs. Second, FACS® analyses of BM cells from SLC-deficient mice revealed a readily detectable population of cells that expresses μHC and has down-regulated TdT; these cells were negative for cytoplasmic or surface conventional LC. Therefore, we conclude that at least some μHCs are able to deliver a signal for down-regulation of TdT and Rag1 gene expression in the absence of SLC and conventional LC.
TdT protein was found to be down-regulated in the majority of c-kit+/μHC+ cells from wild-type mice. In contrast, TdT was not down-regulated in these cells in SLC-deficient mice. In wild-type mice, a significant proportion of cells expressing the pre-BCR are still c-kit+ and presumably already cycling (5, 23) Therefore, the down-regulation of TdT mRNA levels (5) might rapidly lead to loss of TdT protein during proliferative expansion. However, the μHC does not induce proliferative expansion in SLC-deficient mice (4). Therefore, it is conceivable that in the absence of SLC, TdT protein remains detectable for longer periods of time. Importantly, TdT is clearly down-regulated at the CD25+ stage in SLC-deficient BM cells.
The analysis of Rag1 nuclear transcripts after the de novo expression of a μHC transgene demonstrates that down-regulation of the recombinase machinery in both wild-type and SLC-deficient cells is comparable and fairly rapid. This, together with asynchronous replication (34) and nonequivalent nuclear location (27) of the IgH alleles may provide sufficient time for a productive μHC to affect feedback inhibition of the rearrangement machinery. Such a situation could also occur in mice lacking SLC. It is assumed that, either at the time when the recombinase machinery is down-regulated or later during pre–B cell differentiation, the second IgH allele is rendered inaccessible, thus preventing further rearrangement (35). However, the molecular mechanisms responsible for this process are currently unknown. Thus, we can only speculate as to whether this process is affected by the absence of the SLC. As immature B cells in SLC−/− mice show allelic exclusion, we propose that, as in wild-type mice, the second IgH allele is already inaccessible for the recombinase machinery at the time of LC gene rearrangement.
In the BM of SLC-deficient mice, a small population of cells has up-regulated CD25, a marker associated with differentiation into pre–B cells (8, 9, 24). We found that these cells, representing ∼5–10% of all CD19+ surface Ig− BM cells in SLC-deficient mice, were negative for TdT as well as conventional LC and, therefore, are comparable in phenotype to pre–B cells from wild-type mice (5, 19, 24). Presumably, in a fraction of these cells, the Rag genes are reexpressed, and the LC gene loci are undergoing recombination. In all probability, these pre–B cells can give rise to the few allelically excluded immature B cells that are detected in SLC−/− mice (7–9).
Where do the signals for the down-regulation of Rag1 and TdT and the up-regulation of CD25 originate? It has been discussed that ER-resident μHCs without obligate surface expression could induce signaling cascades (3), but it is doubtful whether elements of the appropriate signaling pathways (e.g., the Src family kinases) are efficiently recruited to the ER (31). Therefore, we investigated whether some μHCs can be transported to the surface and acquire signaling competence by forming pre-BCR–like complexes in the absence of SLC. Indeed, using an enzymatic amplification staining technique, we were able to detect μHC surface expression on pre–B cells from λ5−/−, VpreB1−/−VpreB2−/−, and SLC−/− mice, which may be complexed with Ig-α/-β and, thus, mediate the effects of μHC in SLC-deficient mice. These data confirm and significantly extend recent data obtained from SLP-65−/− mice (36). In SLP-65−/− mice, there is increased μHC expression on the surface of pre–B cells and a novel pre-BCR complex was detected in the absence of λ5. Importantly, our results provide evidence for the presence of this incomplete pre-BCR on SLP-65–containing cells as well as on cells that lack all SLC components. We propose that this receptor, as has been shown for the receptor in SLP-65–deficient cells (36), is signaling competent and responsible for the observed down-regulation of the recombinase machinery. The absence of a μHC-mediated down-regulation of TdT in λ5/Ig-α–deficient cells provides further evidence for signaling competence.
It can be speculated that the incomplete pre-BCR–like receptor represents an alternative, possibly rudimentary mechanism to select functional μHCs and facilitate surface expression. Reports of human μHCs that do not require association with SLC for surface transport support this scenario (37). In the absence of SLC, μHCs are believed to be retained in the ER through binding to the Ig HC binding protein BIP (30). Consequently, there is a requirement for a partner to facilitate exit from the ER. A possible candidate is VpreB3, a protein resident in the ER and able to associate with μHC (38). Alternatively, other hitherto unknown proteins may be essential for this process.
It remains unclear at present whether the incomplete pre-BCR described here has a physiological role in normal pre–B cell development. The SLC genes are highly expressed in pro–B cells and rapidly down-regulated in pre–B cells during clonal expansion (5, 39, 40). Therefore, the degree of proliferation might be controlled, not only by the dilution of SLC with successive cell divisions but also by the half-life of SLC proteins. It is conceivable that an incomplete pre-BCR has a physiological function in normal pre–B cell development. Possibly, this takes place at the stage when conventional pre-BCR is down-regulated, which is observed during clonal expansion of pre–B cells and before LC gene recombination (i.e., at the transition from large to small pre–B cells). This is supported by the observation that the tetracycline-inducible μHC transgene is stably expressed in pre–B cell cultures over a period of at least 4 d, whereas λ5, VpreB, and the conformational epitope, recognized by the pre-BCR–specific antibody SL156, were undetectable even by surface EAS staining after only 2–3 d (unpublished data). Interestingly, it has been observed that prolonged survival of pre–B cells in μHC-transgenic, Bcl-2-transgenic Rag2−/− mice allows accumulation of novel precursor B cells in the spleen that showed considerable μHC expression on the cell surface (41). Most of these cells are expected to have down-regulated SLCs (42) and, therefore, the μHC might form an incomplete pre-BCR similar to the one described in our studies.
In summary, we provide evidence that in the absence of SLC, μHC can induce down-regulation of the recombinase machinery, and that this effect is dependent on Ig-α. Induction of a μHC transgene in Rag2−/− pro–B cells can mediate down-regulation of TdT, Rag1, and probably Rag2, providing a model that could explain how the mechanisms involved in the establishment of allelic exclusion are still functional in the absence of SLC and, therefore, a conventional pre-BCR.
Acknowledgments
We thank M. Nussenzweig for providing the NG-BAC mice and F. Melchers and M. Cooper for providing mAbs against SLC components. We thank H. Welzel for technical support as well as R. Voll, R. Slany, and S. Licence for critical reading of the manuscript.
G. Galler and T. Winkler are supported by the Deutsche Forschungsgemeinschaft (grant nos. DFG WI 1183/3 and GK592). C. Mundt, M. Parker, and I.-L. Mårtensson are supported by the Biotechnology and Biological Sciences Research Council.
Abbreviations used in this paper: EAS, enzyme amplification staining; FISH, fluorescent in situ hybridization; GFP, green fluorescent protein; HC, heavy chain; LC, light chain; pre-BCR, pre–B cell receptor; SLC, surrogate LC; TdT, terminal deoxynucleotidyl transferase.
References
- 1.Alt, F.W., G.D. Yancopoulos, T.K. Blackwell, C. Wood, E. Thomas, M. Boss, R. Coffman, N. Rosenberg, S. Tonegawa, and D. Baltimore. 1984. Ordered rearrangement of immunoglobulin heavy chain variable region segments. EMBO J. 3:1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melchers, F., D. Haasner, U. Grawunder, C. Kalberer, H. Karasuyama, T. Winkler, and A.G. Rolink. 1994. Roles of IgH and L chains and of surrogate H and L chains in the development of cells of the B lymphocyte lineage. Annu. Rev. Immunol. 12:209–225. [DOI] [PubMed] [Google Scholar]
- 3.Martensson, I.L., A. Rolink, F. Melchers, C. Mundt, S. Licence, and T. Shimizu. 2002. The pre-B cell receptor and its role in proliferation and Ig heavy chain allelic exclusion. Semin. Immunol. 14:335–342. [DOI] [PubMed] [Google Scholar]
- 4.Hess, J., A. Werner, T. Wirth, F. Melchers, H.M. Jack, and T.H. Winkler. 2001. Induction of pre-B cell proliferation after de novo synthesis of the pre-B cell receptor. Proc. Natl. Acad. Sci. USA. 98:1745–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grawunder, U., T.M. Leu, D.G. Schatz, A. Werner, A.G. Rolink, F. Melchers, and T.H. Winkler. 1995. Down-regulation of RAG1 and RAG2 gene expression in preB cells after functional immunoglobulin heavy chain rearrangement. Immunity. 3:601–608. [DOI] [PubMed] [Google Scholar]
- 6.Kitamura, D., and K. Rajewsky. 1992. Targeted disruption of mu chain membrane exon causes loss of heavy-chain allelic exclusion. Nature. 356:154–156. [DOI] [PubMed] [Google Scholar]
- 7.Kitamura, D., A. Kudo, S. Schaal, W. Muller, F. Melchers, and K. Rajewsky. 1992. A critical role of lambda 5 protein in B cell development. Cell. 69:823–831. [DOI] [PubMed] [Google Scholar]
- 8.Mundt, C., S. Licence, T. Shimizu, F. Melchers, and I.L. Martensson. 2001. Loss of precursor B cell expansion but not allelic exclusion in VpreB1/VpreB2 double-deficient mice. J. Exp. Med. 193:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimizu, T., C. Mundt, S. Licence, F. Melchers, and I.L. Martensson. 2002. VpreB1/VpreB2/lambda 5 triple-deficient mice show impaired B cell development but functional allelic exclusion of the IgH locus. J. Immunol. 168:6286–6293. [DOI] [PubMed] [Google Scholar]
- 10.Novobrantseva, T.I., V.M. Martin, R. Pelanda, W. Muller, K. Rajewsky, and A. Ehlich. 1999. Rearrangement and expression of immunoglobulin light chain genes can precede heavy chain expression during normal B cell development in mice. J. Exp. Med. 189:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seidl, T., A. Rolink, and F. Melchers. 2001. The VpreB protein of the surrogate light-chain can pair with some mu heavy-chains in the absence of the lambda 5 protein. Eur. J. Immunol. 31:1999–2006. [DOI] [PubMed] [Google Scholar]
- 12.Pelanda, R., U. Braun, E. Hobeika, M.C. Nussenzweig, and M. Reth. 2002. B cell progenitors are arrested in maturation but have intact VDJ recombination in the absence of Ig-alpha and Ig-beta. J. Immunol. 169:865–872. [DOI] [PubMed] [Google Scholar]
- 13.Yu, W., H. Nagaoka, M. Jankovic, Z. Misulovin, H. Suh, A. Rolink, F. Melchers, E. Meffre, and M.C. Nussenzweig. 1999. Continued RAG expression in late stages of B cell development and no apparent re-induction after immunization. Nature. 400:682–687. [DOI] [PubMed] [Google Scholar]
- 14.Rolink, A., A. Kudo, H. Karasuyama, Y. Kikuchi, and F. Melchers. 1991. Long-term proliferating early pre B cell lines and clones with the potential to develop to surface Ig-positive, mitogen reactive B cells in vitro and in vivo. EMBO J. 10:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gribnau, J., E. de Boer, T. Trimborn, M. Wijgerde, E. Milot, F. Grosveld, and P. Fraser. 1998. Chromatin interaction mechanism of transcriptional control in vivo. EMBO J. 17:6020–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karasuyama, H., A. Rolink, and F. Melchers. 1993. A complex of glycoproteins is associated with VpreB/λ 5 surrogate light chain on the surface of μ heavy chain-negative early precursor B cell lines. J. Exp. Med. 178:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephan, R.P., E. Elgavish, H. Karasuyama, H. Kubagawa, and M.D. Cooper. 2001. Analysis of VpreB expression during B lineage differentiation in lambda5-deficient mice. J. Immunol. 167:3734–3739. [DOI] [PubMed] [Google Scholar]
- 18.Komori, T., A. Okada, V. Stewart, and F.W. Alt. 1993. Lack of N regions in antigen receptor variable region genes of TdT-deficient lymphocytes. Science. 261:1171–1175. [DOI] [PubMed] [Google Scholar]
- 19.Li, Y.S., K. Hayakawa, and R.R. Hardy. 1993. The regulated expression of B lineage–associated genes during B cell differentiation in bone marrow and fetal liver. J. Exp. Med. 178:951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bangs, L.A., I.E. Sanz, and J.M. Teale. 1991. Comparison of D, JH, and junctional diversity in the fetal, adult, and aged B cell repertoires. J. Immunol. 146:1996–2004. [PubMed] [Google Scholar]
- 21.Wasserman, R., Y.S. Li, and R.R. Hardy. 1997. Down-regulation of terminal deoxynucleotidyl transferase by Ig heavy chain in B lineage cells. J. Immunol. 158:1133–1138. [PubMed] [Google Scholar]
- 22.Gu, Y., S. Jin, Y. Gao, D.T. Weaver, and F.W. Alt. 1997. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc. Natl. Acad. Sci. USA. 94:8076–8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winkler, T.H., A. Rolink, F. Melchers, and H. Karasuyama. 1995. Precursor B cells of mouse bone marrow express two different complexes with the surrogate light chain on the surface. Eur. J. Immunol. 25:446–450. [DOI] [PubMed] [Google Scholar]
- 24.Rolink, A., U. Grawunder, T.H. Winkler, H. Karasuyama, and F. Melchers. 1994. IL-2 receptor α chain (CD25, TAC) expression defines a crucial stage in pre-B cell development. Intern. Immunol. 6:1257–1264. [DOI] [PubMed] [Google Scholar]
- 25.Nagaoka, H., G. Gonzalez-Aseguinolaza, M. Tsuji, and M.C. Nussenzweig. 2000. Immunization and infection change the number of recombination activating gene (RAG)-expressing B cells in the periphery by altering immature lymphocyte production. J. Exp. Med. 191:2113–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Billips, L.G., C.A. Nunez, F.E. Bertrand III, A.K. Stankovic, G.L. Gartland, P.D. Burrows, and M.D. Cooper. 1995. Immunoglobulin recombinase gene activity is modulated reciprocally by interleukin 7 and CD19 in B cell progenitors. J. Exp. Med. 182:973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skok, J.A., K.E. Brown, V. Azuara, M.L. Caparros, J. Baxter, K. Takacs, N. Dillon, D. Gray, R.P. Perry, M. Merkenschlager, and A.G. Fisher. 2001. Nonequivalent nuclear location of immunoglobulin alleles in B lymphocytes. Nat. Immunol. 2:848–854. [DOI] [PubMed] [Google Scholar]
- 28.Levsky, J.M., S.M. Shenoy, R.C. Pezo, and R.H. Singer. 2002. Single-cell gene expression profiling. Science. 297:836–840. [DOI] [PubMed] [Google Scholar]
- 29.Kimura, H., K. Sugaya, and P.R. Cook. 2002. The transcription cycle of RNA polymerase II in living cells. J. Cell Biol. 159:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hendershot, L.M. 1990. Immunoglobulin heavy chain and binding protein complexes are dissociated in vivo by light chain addition. J. Cell Biol. 111:829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brouns, G.S., E. de Vries, J.J. Neefjes, and J. Borst. 1996. Assembled pre-B cell receptor complexes are retained in the endoplasmic reticulum by a mechanism that is not selective for the pseudo-light chain. J. Biol. Chem. 271:19272–19278. [DOI] [PubMed] [Google Scholar]
- 32.Pelanda, R., S. Schaal, R.M. Torres, and K. Rajewsky. 1996. A prematurely expressed Ig(kappa) transgene, but not V (kappa)J(kappa) gene segment targeted into the Ig(kappa) locus, can rescue B cell development in lambda5-deficient mice. Immunity. 5:229–239. [DOI] [PubMed] [Google Scholar]
- 33.Rolink, A., D. Haasner, F. Melchers, and J. Andersson. 1996. The surrogate light chain in mouse B-cell development. Int. Rev. Immunol. 13:341–356. [DOI] [PubMed] [Google Scholar]
- 34.Mostoslavsky, R., N. Singh, T. Tenzen, M. Goldmit, C. Gabay, S. Elizur, P. Qi, B.E. Reubinoff, A. Chess, H. Cedar, and Y. Bergman. 2001. Asynchronous replication and allelic exclusion in the immune system. Nature. 414:221–225. [DOI] [PubMed] [Google Scholar]
- 35.Bassing, C.H., W. Swat, and F.W. Alt. 2002. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 109:S45–S55. [DOI] [PubMed] [Google Scholar]
- 36.Su, Y.W., A. Flemming, T. Wossning, E. Hobeika, M. Reth, and H. Jumaa. 2003. Identification of a pre-BCR lacking surrogate light chain. J. Exp. Med. 198:1699–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minegishi, Y., M.E. Conley, and S. Mizutani. 2003. In vivo functional characterization of light chain-independent μ heavy chain. Keystone Symposia, B Cells and Antibodies: Laboratory to Clinic. M.C. Nussenzweig, F.W. Alt, and L.M. Staudt, editors. Keystone Resort, Keystone, Colorado. 75.
- 38.Shirasawa, T., K. Ohnishi, S. Hagiwara, K. Shigemoto, Y. Takebe, K. Rajewsky, and T. Takemori. 1993. A novel gene product associated with mu chains in immature B cells. EMBO J. 12:1827–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karasuyama, H., A. Rolink, Y. Shinkai, F. Young, F.W. Alt, and F. Melchers. 1994. The expression of Vpre-B/lambda 5 surrogate light chain in early bone marrow precursor B cells of normal and B cell-deficient mutant mice. Cell. 77:133–143. [DOI] [PubMed] [Google Scholar]
- 40.Dul, J.L., Y. Argon, T. Winkler, E. ten Boekel, F. Melchers, and I.L. Martensson. 1996. The murine VpreB1 and VpreB2 genes both encode a protein of the surrogate light chain and are co-expressed during B cell development. Eur. J. Immunol. 26:906–913. [DOI] [PubMed] [Google Scholar]
- 41.Young, F., E. Mizoguchi, A.K. Bhan, and F.W. Alt. 1997. Constitutive Bcl-2 expression during immunoglobulin heavy chain-promoted B cell differentiation expands novel precursor B cells. Immunity. 6:23–33. [DOI] [PubMed] [Google Scholar]
- 42.Martensson, I.L., F. Melchers, and T.H. Winkler. 1997. A transgenic marker for mouse B lymphoid precursors. J. Exp. Med. 185:653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]