

Abstract
DNAX accessory molecule 1 (DNAM-1; CD226) is a transmembrane glycoprotein involved in T cell and natural killer (NK) cell cytotoxicity. We demonstrated recently that DNAM-1 triggers NK cell–mediated killing of tumor cells upon engagement by its two ligands, poliovirus receptor (PVR; CD155) and Nectin-2 (CD112). In the present paper, we show that PVR and Nectin-2 are expressed at cell junctions on primary vascular endothelial cells. Moreover, the specific binding of a soluble DNAM-1–Fc molecule was detected at endothelial junctions. This binding was almost completely abrogated by anti-PVR monoclonal antibodies (mAbs), but not modified by anti–Nectin-2 mAbs, which demonstrates that PVR is the major DNAM-1 ligand on endothelial cells. Because DNAM-1 is highly expressed on leukocytes, we investigated the role of the DNAM-1–PVR interaction during the monocyte transendothelial migration process. In vitro, both anti–DNAM-1 and anti-PVR mAbs strongly blocked the transmigration of monocytes through the endothelium. Moreover, after anti–DNAM-1 or anti-PVR mAb treatment, monocytes were arrested at the apical surface of the endothelium over intercellular junctions, which strongly suggests that the DNAM-1–PVR interaction occurs during the diapedesis step. Altogether, our results demonstrate that DNAM-1 regulates monocyte extravasation via its interaction with PVR expressed at endothelial junctions on normal cells.
Keywords: CD226, CD155, Nectin, endothelium, diapedesis
Introduction
DNAX accessory molecule 1 (DNAM-1; CD226) is a transmembrane glycoprotein member of the immunoglobulin superfamily. In humans, it is constitutively expressed on a subset of B lymphocytes and on all T lymphocytes, NK cells, monocytes, and platelets (1). mAb-mediated cross-linking of DNAM-1 enhances the triggering of both T and NK cell cytotoxicity (1). We demonstrated recently that DNAM-1 specifically recognizes two cellular ligands named poliovirus receptor (PVR; CD155) and Nectin-2 (CD112). Tumor target cells frequently coexpress both PVR and Nectin-2, whereas they display low levels of HLA class I molecules. Upon engagement by these ligands, DNAM-1 was shown to induce NK cell–mediated cytotoxicity (2), and its involvement in the NK cell–mediated lysis strictly correlated with the expression of PVR and Nectin-2 on tumor cells (2). PVR and Nectin-2 are members of the Nectin family that also belongs to the immunoglobulin superfamily. In humans, this family comprises five structurally related molecules as follows: PVR (3), Nectin-1, -2, -3, and -4 (4–7). Both PVR and Nectin-2 are accessory receptors for some α herpesviruses (3, 8, 9). Nectins have been shown to play a fundamental role during the establishment of cell polarity and in the regulation of intercellular junctions in epithelial and neuronal cells (10). They are involved in the formation of both adherens and tight junctions in epithelial cells (10). Nectins recruit, regulate, and interact with E-cadherin at adherens junctions (AJs) via their cytoplasmic associated molecules (i.e., AF-6/afadin and catenins, respectively; reference 11). Also, they recruit and target junctional adhesion molecule (JAM)-A during the formation of tight junctions (10). They assume their functions by mediating homophilic and heterophilic transinteractions following defined combinations; among them, PVR and Nectin-2 both interact with Nectin-3 (2, 7) and DNAM-1 (2). Nectin-2 was also shown to display homophilic adhesion on endothelial cells (12).
PVR and Nectin-2 are not tumor-specific antigens because they are widely expressed on normal cells including neuronal, epithelial, endothelial, and fibroblastic cells (13). Thus, in an autologous setting, DNAM-1 interaction with PVR and Nectin-2 does not result in NK cell–mediated lysis of normal cells. Indeed, these latter express normal levels of self-HLA class I molecules that block the NK cell function upon interaction with inhibitory NK receptors (14). Recently, a new DNAM-1 function has been proposed during platelet aggregation (15). From all these data, we speculated that DNAM-1 interaction with PVR and/or Nectin-2 expressed on endothelial cells may take part in cellular transmigration through endothelial cells. Leukocytes escape the blood circulation by interacting with the endothelial cells of the vessel walls from blood to secondary lymphoid organs or underlying tissues through a well-characterized multistep adhesion molecule cascade (16–21). Although actors involved in the first steps of tethering and rolling are well established, less is known about the late phase of diapedesis. Herein, we show that both PVR and Nectin-2 are expressed at vascular interendothelial junctions and demonstrate that PVR is the major DNAM-1 ligand on endothelial cells. Finally, by using human monocytes that highly expressed DNAM-1, we provide evidence that DNAM-1 interacts directly with PVR at cell junctions and promotes monocyte transendothelial migration (TEM) by acting specifically during the diapedesis step.
Materials and Methods
Endothelial Cells.
Human umbilical vein endothelial cells (HUVECs) were harvested from umbilical cord veins, pooled, and established as primary cultures in EBM2 medium (Cambrex). Primary HUVEC cultures were serially passed, maintained in EBM2 medium, and used before the fifth passage. The Nalm6 cell line was cultivated in RPMI 1640 medium, supplemented with 10% FCS, 2 mM glutamine, 100 U/ml penicillin, and 50 μg/ml streptomycin. All cells were cultivated at 37°C in an air 5% CO2 atmosphere at constant humidity.
Isolation of PBMCs.
PBMCs were purified from healthy blood donors or from cytaphereses. Samples were diluted 1:2 or 1:10 in PBS, respectively, layered over Ficoll separation medium (Lymphoprep; Axis-Shield), and centrifuged at 2,000 revolutions/min for 20 min at room temperature to remove erythrocytes and PMNs. PBMCs were collected at the interface and washed twice with PBS.
Antibodies.
mAbs against human DNAM-1 (FS123 and KRA236) were described previously (2). Anti–Nectin-1 (R1.302 [IgG1]), Nectin-2 (R2.477 [IgG1] and L14 [IgG2a]), Nectin-4 (N4.40 [IgG1]), PVR (PV.404, L95, M5A10, and M2C24 [IgG1]) were described previously (2, 7, 12, 22, 23). Anti-PVR D171 (IgG1) mAb was purchased from Neomarkers. Anti-CD146 mAb (clone F4-5H7) was provided by F. Dignat-George (Faculté de Pharmacie, Marseille, France).
Murine monoclonal anti–VE-cadherin (clone 75; Transduction Laboratories), anti–platelet–endothelial cell adhesion molecule 1 (PECAM-1)/CD31 (clone JC/70A; DakoCytomation), rabbit polyclonal anti–β-catenin (Zymed Laboratories) were purchased from commercial sources. Anti-CD99 (clone 12E7) mAb was provided by A. Bernard (Institut National de la Santé et de la Recherche Medicale, Nice, France). Anti-CD11a mAb (clone 25.3.1 [IgG1]) was provided by D. Olive (Institut National de la Santé et de la Recherche Medicale, Marseille, France). Anti-CD34 (clone Immu133 [IgG1]), anti–CD14–PE, anti–CD14-FITC (clone RMO52), and isotypic controls were purchased from Beckman Coulter and Immunotech. Alexa-488–labeled goat anti–mouse antibody, and TRITC-labeled goat anti–rabbit antibody were obtained from Molecular Probes. Anti-CD146 and anti–VE-cadherin mAbs were labeled with a Zenon mouse Ig labeling kit (Alexa Fluor 546) following the instructions of the manufacturer (Molecular Probes). PE-labeled goat anti–human Fc fragment antibody were obtained from Beckman Coulter and Immunotech. Purification of Fab fragments was achieved by papain treatment as recommended by the manufacturer (Pierce Chemical Co.). Purity, assessed on Coomassie blue–stained SDS-PAGE, was >95%. Fab fragments were validated by flow cytometry.
Production and Purification of Soluble Forms of Nectins and DNAM-1.
Nectins–Fc constructions and productions have been described previously (7). All plasmids were purified with an endotoxin-free kit (QIAGEN), sterilized through a 0.22-μm filter, aliquoted, and stored at −20°C.
The sequence coding for the extracellular portion of DNAM-1 receptor (nucleotide 181-951; GenBank/EMBL/DDBJ accession no. U56102) was amplified starting from the pcDNA3.1 TOPO DNAM-1 plasmid using the following primers: 5′-ACGCGTCGACAACCAGCCTTTCAAACAG-3′ (DNAM-1 SalI up) and 5′-CGGGATCCTGGTTATCGGTTTTACCC-3′ (DNAM-1 BamHI dw). Amplification was performed with Vent DNA Polymerase (New England Biolabs, Inc.) for 20 cycles (30 s at 95°C, 30 s at 58°C, and 30 s at 72°C) followed by a 7-min elongation step at 72°C. The 771-bp PCR product was digested with SalI and BamHI restriction enzymes and subcloned in the SalI-BamHI–digested pRB1-2B4Fcmut vector (provided by M. Falco, Istituto Giannina Gaslini, Genova, Italy) in frame with the sequence coding for the human IgG1 portion, which was mutagenized to obtain a mutated Fc that does not bind to Fc receptors. To ensure that the correct DNAM-1 sequence was in frame with the human IgG1, the nucleotidic sequence was performed. The pRB1-DNAM-1Fcmut construct was stably transfected into the HEK293 cell line (human embryonic fibroblast) using Fugene 6 (Roche). Supernatants were collected from the cell transfectant cultured in DMEM/10% ultra-low IgG FCS (Invitrogen), and the DNAM-1–Fc molecule was purified by affinity chromatography using protein A–Sepharose 4 Fast Flow (Amersham Biosciences). Purified protein was checked by SDS-PAGE followed by silver staining and ELISA using DNAM-1–specific mAbs.
ELISA.
A sandwich ELISA was used to define the region of PVR recognized by the anti-PVR mAbs PV.404, D171, M5A10, L95, and M2C24. 96-well trays were coated with an antibody against the human Fc fragment (Sigma-Aldrich) at 10 μg/ml. After saturation of wells with PBS containing 1% BSA, 10−9 M of PVR-vcc-Fc or PVR-v-Fc was reacted with 2.5 μg/ml of the different mAbs followed by an incubation with a 1/2,000 dilution of a peroxidase-labeled goat anti–mouse and revealed with the One Step ABTS (Pierce Chemical Co.). Optical density was read at 405 nm.
In Vitro Binding Studies.
In vitro physical interaction studies between PVR–DNAM-1 and Nectin-2–DNAM-1 were performed as follows. Ultrasorb 96-well trays (Nunc) were incubated overnight at 4°C with 1 μg/ml of goat anti–human Fc affinity purified serum (Sigma-Aldrich). After three washes with PBS containing 0.5% of Tween 20, wells were incubated with PBS containing 1% BSA. After three washes, 10−7 M of different chimeric-Fc proteins (BT3-Fc, Nec3-Fc, and DNAM-1–Fc) were incubated for 2 h at 37°C. After washes, free anti–human-Fc antibodies were blocked with PBS containing 100 μg/ml of human immunoglobulin (Novartis) for 1 h at 25°C. Biotinylated PVR-Fc or Nec2-Fc (10−7M) was incubated for 2 h at 37°C in the absence of Ca2+. After three washes, 2 μg/ml streptavidin peroxidase (Sigma-Aldrich) was incubated for 1 h at 37°C. After five washes, binding was assessed by incubation with the One-Step ABTS substrate (Pierce Chemical Co.). Optical density was read at 405 nm.
Immunohistochemistry.
Immunodetection of Nectin-2, PVR, and PECAM-1–CD31 were performed on 5-μm frozen sections on human placenta or human normal skin using different concentrations of mAbs. Specimens were processed with the Universal Kit ChemMate according to the manufacturer's recommendations, counterstained for 5 min in Harris hematoxylin, and mounted in glycergel mounting medium (DakoCytomation).
Immunofluorescence Microscopy.
HUVECs were grown on 13-mm round glass coverslips as a confluent monolayer to reach optimal cell-to-cell contact. Cells were fixed with 3.7% paraformaldehyde in PBS for 20 min at room temperature. After blocking aldehydes with 50 mM NH4Cl for 10 min at room temperature, fixed cells were incubated with either 0.2% gelatin in PBS, or with 0.2% gelatin and 0.075% saponin (all obtained from Sigma-Aldrich) in PBS for 20 min. Fixed samples were first labeled with the appropriate antibodies for 60 min at room temperature and incubated with secondary antibodies in the same conditions. Alternatively, immunofluorescence analysis on filters by conjugating mAbs with the 546-nm–labeled IgG2b isotype-specific Zenon Fab (Molecular Probes). Finally, samples were washed, mounted onto slides, embedded with mounting medium (DakoCytomation), and visualized using a confocal microscope (Leica). Images were processed using the Adobe Photoshop software. In some experiments, staining was performed on live cells followed by paraformaldehyde fixation.
Flow Cytometry.
HUVECs were trypsinized and resuspended after centrifugation in 5% FCS containing PBS. In some experiments, HUVECs were detached with 0.02% (wt/vol) disodium EDTA in PBS. Cells were incubated with 2–10 μg/ml of the appropriate antibody and with a PE-labeled goat anti–mouse antibody (Immunotech).
TEM Assay.
HUVEC subcultures were removed with trypsin/EDTA and plated onto 0.1% gelatin (Sigma-Aldrich)-coated Costar transwells (5-μm pore size and 6.5-mm diameter), at 3.5 × 104 cells/well. Endothelial cell monolayers were grown to confluency for 4 d at 37°C. 5 × 105 PBMCs were loaded on each transwell. We assessed the permeability of HUVEC monolayers using 38,900 D of FITC dextran at 1 mg/ml. Fluorescence was monitored at 520 nm using a Wallac Victor fluorometer. Cell were allowed to transmigrate for 1 h at 37°C. Migrated monocytes were recovered from the bottom of the well and mixed with 105 calcein-labeled Nalm6 cells. Cells were centrifuged and incubated with an anti–CD14-PE antibody for 15 min. Finally, cells were analyzed with flow cytometry. Each condition was performed in quadruplicate. In some experiments, assays were performed in the presence of 10 μM polymyxin B.
Results
DNAM-1 Is Highly Expressed On Monocytes, Whereas Its Ligands PVR and Nectin-2 Are Expressed at Intercellular Junctions in Vascular Endothelial Cells.
We demonstrated recently that PVR and Nectin-2 are cell surface ligands of DNAM-1 that specifically induce NK cell–mediated cytolytic activity against tumor cell targets (2). In the present paper, we investigated the role of the interactions between DNAM-1 and its ligands on normal cells. Most leukocytes were shown to express DNAM-1 (1). PVR and Nectin-2 are cell adhesion molecules specifically expressed at E-cadherin–based AJs in epithelial cells. AJs are structurally and functionally similar in endothelial cells because VE-cadherin is the major component of endothelial AJ-mediating homophilic interaction between adjacent cells. In a first set of experiments, we analyzed the expression of DNAM-1, PVR, and Nectin-2 on leukocytes and endothelial cells. As shown in Fig. 1 a, low surface densities of PVR and Nectin-2 were detected on monocytes. Nectin-3 and Nectin-4 were not detected on monocytes (unpublished data). According to previous data, different leukocyte subsets, including monocytes (Fig. 1 a), highly express DNAM-1 (1). The expression of DNAM-1, PVR, and Nectin-2 was assessed on primary endothelial cells by both flow cytometry and immunofluorescence microscopy. DNAM-1 is not or just faintly expressed on HUVECs, whereas PVR and Nectin-2 are highly expressed at levels similar to PECAM-1 (Fig. 1 b). Moreover, both PVR and Nectin-2 are mostly expressed at endothelial cell junctions on HUVECs, similar to PECAM-1 (Fig. 1 c). They show an extremely similar distribution at cell borders compared with the VE-cadherin cytoplasmic partner β-catenin (Fig. 1 d). Finally, PVR and Nectin-2 as well as PECAM-1 are expressed on endothelial cells of placental blood vessels (Fig. 2 a, black arrows). In particular, in some sections and in accordance with their intercellular localization on HUVECs (Fig. 1 c), staining is observed at cell-to-cell contacts (Fig. 2 a, white arrows). PVR and Nectin-2 are also expressed in vessels from normal skin (Fig. 2 b). These data show that PVR and Nectin-2 are two new junctional molecules expressed at vascular endothelial cell junctions, whereas their counter receptor, DNAM-1, is expressed on monocytes.
Figure 1.

Analysis of the cell surface expression of DNAM-1, PVR, and Nectin-2 on monocytes and primary vascular endothelial cells. (a) Fresh PBMCs were gated on monocytes on the basis of both size and granularity. Cells were analyzed by two-color immunofluorescence and FACS® analysis with anti-CD14 mAb in combination with anti–DNAM-1 (FS.123), anti-PVR (PV.404), or anti–Nectin-2 (R2.477) mAbs followed by FITC- or PE-conjugated goat anti–mouse second reagents. (b) HUVECs were analyzed by one-color immunofluorescence and FACS® analysis with anti–DNAM-1 (FS123), anti-PVR (PV.404), anti–Nectin-2 (R2.477), or anti–PECAM-1 (JC/70A) mAbs followed by FITC-conjugated goat anti–mouse second reagents. Gray profiles indicate cells incubated with the second reagent only. We previously controlled that trypsin treatment does not affect cell surface expression of Nectins. Similar results were obtained when HUVECs were detached with 0.02% (wt/vol) disodium EDTA in PBS. (c) Localization of Nectin-2 and PVR on primary endothelial cells. HUVECs were fixed; incubated with 2 μg/ml of anti–Nectin-2 (R2.477), anti-PVR (PV.404), and anti–PECAM-1 (JC/70), followed by Alexa-488–labeled goat anti–mouse second reagent; and analyzed by immunofluorescence microscopy. Staining was similar on live cells (not depicted). (d) HUVECs were fixed, permeabilized, and stained with the anti–Nectin-2 (R2.477) mAb and the anti–β-catenin rabbit antiserum followed by Alexa-488–labeled goat anti–mouse antibody and TRITC-labeled goat anti–rabbit second reagents, respectively. Cells were analyzed by immunofluorescence microscopy.
Figure 2.

Expression of Nectin-2 and PVR on human placenta and skin. Frozen sections of placenta (a) and normal skin (b) were analyzed by immunohistochemistry with 5 μg/ml anti–Nectin-2 (R2.477), 5 μg/ml anti-PVR (D171), or 2 μg/ml anti–PECAM-1 (JC/70) mAbs as described in Materials and Methods. mAb-specific staining was observed on endothelial cells of the placenta and skin blood vessels (black arrows) and at cell-to-cell junctions (white arrow). Staining specificity was assessed by an irrelevant isotype match control mAb and also by the fact that other cell types in the section were negative for Nectin-2 and PVR. Bars, 20 μm.
PVR Is the Major DNAM-1 Counter-Receptor on Endothelial Cells.
Our data showed that DNAM-1, PVR, and Nectin-2 are expressed on monocytes. Potential interactions between these molecules with PVR and Nectin-2 on endothelial cells were assessed. To this end, chimeric molecules formed with the whole ectodomain of these molecules fused to the Fc fragment of human IgG1; namely NAM-1–Fc, PVR-Fc, and Nec2-Fc were analyzed for their binding on endothelial cells. Nec2-Fc and PVR-Fc do not significantly bind to HUVECs when compared with an irrelevant control (Fig. 3 a, N4vtr-Fc). Alternately, DNAM-1–Fc strongly binds to HUVECs. Confocal microscopy shows that DNAM-1–Fc, but not Nec2-Fc, specifically stains and delineates endothelial cell junctions, which suggests that DNAM-1 ligands are junctional molecules (Fig. 3 b). This is in agreement with the localization of PVR and Nectin-2 at cell junctions (Fig. 1).
Figure 3.

DNAM-1 recognizes junctional ligands expressed on endothelial cells. (a) HUVECs were analyzed by one-color immunofluorescence and FACS® analysis with Nec2-Fc, PVR-Fc, DNAM-1–Fc, and N4vtr-Fc (negative control) followed by FITC-conjugated goat anti–human second reagents. Gray profiles indicate cells incubated with the second reagent only. All soluble proteins were used at 20 μg/ml. Similar results were obtained when HUVECs were detached with 0.02% (wt/vol) disodium EDTA in PBS. (b) Immunofluorescence microscopy analysis of HUVECs using 20 μg/ml DNAM-1–Fc followed by FITC-conjugated goat anti–human second reagents. No staining was revealed with Nec2-Fc. The DNAM-1–Fc staining delineates the junctional systems between adjacent cells suggesting that DNAM-1 interacts with ligands localized at endothelial cell junctions. Similar results were obtained on live confluent HUVECs (not depicted).
We analyzed the binding of DNAM-1–Fc relative to the expression of PVR or Nectin-2 molecules ectopically expressed in CHO cells (Fig. 4 a). We observed that 1.5 μM DNAM-1–Fc strongly binds to PVR-expressing cells, but binds less efficiently to Nectin-2–expressing CHO cells, whereas it does not bind to control CHO cells. These data suggest that, under these conditions, DNAM-1 preferentially binds to PVR. Of note, DNAM-1–Fc binding was never detected on other Nectin-expressing CHO cells (i.e., Nectin-1, -3, and -4; unpublished data). Even though these data strongly suggest a direct interaction, binding was analyzed by means of recombinant molecules. Both PVR-Fc and Nec2-Fc strongly bind to coated DNAM-Fc and not to the irrelevant control (Fig. 4 b). Of note, both PVR-Fc and Nec2-Fc also bind to coated Nec3-Fc as described previously (Fig. 4 b and reference 7). Interestingly, PVR-Fc binding to DNAM-Fc is stronger than Nec2-Fc, which confirms our FACS® analysis. Altogether, these data highlight for the first time the direct DNAM-1–PVR and DNAM-1–Nectin-2 interactions.
Figure 4.

PVR and Nectin-2 directly bind to DNAM-1. (a) CHO/K cells either nontransfected or transfected with PVR or Nectin-2 cDNAs were analyzed by FACS® analysis with anti-PVR (L95) or anti–Nectin-2 (L14) mAbs, followed by a PE-conjugated goat anti–mouse second reagents (left). CHO/K: gray line, isotype matched mAb; black line, L95 mAb; and dashed line, L14 mAbs. CHO/K transfectants: gray line, isotype matched mAb and black histogram, L95 and L14 mAbs. Binding of DNAM-1–Fc at 1.5 μM on CHO/K cells was revealed by PE-conjugated goat anti–human second reagents (right): (gray line) incubation with the second reagent.; (black histogram) incubation with 1.5 μM DNAM-1–Fc. (b, left) PVR and Nectin-2 directly bind to Nectin-3. (right) To analyze PVR and Nectin-2 interactions with DNAM-1, biotinylated PVR-Fc and Nectin-2–Fc binding was measured by ELISA on wells coated with BT3-Fc (unrelated protein), Nectin-3–Fc (positive control), or DNAM-1–Fc. PVR-Fc and Nectin-2–Fc directly bind to DNAM-1–Fc and as expected with Nectin-3–Fc.
DNAM-1–Fc binding was performed on endothelial cells. To discriminate between PVR, Nectin-2, or still-unidentified junctional ligands expressed by these cells, anti-PVR and anti–Nectin-2 mAbs were tested for their ability to block DNAM-1–Fc binding on HUVECs. Thus, HUVECs, either preincubated with anti-PVR or anti–Nectin-2 mAbs and used alone, in combination, or with control mAbs, were analyzed by double fluorescence for DNAM-1–Fc binding used at 1.5 μM (Fig. 5 a). DNAM-1–Fc binding to HUVECs is not significantly reduced by anti–Nectin-2 mAb preincubation, and is similar to controls without mAb or with the irrelevant anti–Nectin-1 mAb. On the contrary, anti-PVR mAb alone nearly induces a full inhibition (97%) of DNAM-1–Fc binding. Using immunofluorescence microscopy, we show that DNAM-1–Fc binding to endothelial cell junctions was not modified by anti–Nectin-1 and anti–Nectin-2 mAbs, but was strongly inhibited by anti-PVR mAbs. This strengthens the specificity of DNAM-1 binding to PVR, expressed at interendothelial junctions (Fig. 5 b). To extend and confirm these observations, five different anti-PVR mAbs were tested for their ability to interfere with DNAM-1–Fc binding on HUVECs. As shown in Fig. 5 c, all of them efficiently inhibit DNAM-1–Fc binding. The various anti-PVR mAbs were mapped by ELISA assay (Fig. 5 d). PV.404, M5A10, and M2C24 react with the IgC-like domain of PVR, whereas D171 and L95 are directed against the IgV-like domain of the molecule. These results suggest that both V and C Ig-like domains of PVR are involved in the interaction with DNAM-1. Altogether, our results demonstrate that DNAM-1 interacts directly and specifically with PVR on endothelial cells, and that this interaction occurs at endothelial cell junctions.
Figure 5.

PVR is the major DNAM-1 ligand on endothelial cells. (a) Inhibition of DNAM-1–Fc binding by different anti-Nectin mAbs. HUVECs, either untreated or preincubated with anti-PVR (L95) and anti–Nectin-2 (L14) mAbs used alone or in combination, were analyzed by two-color immunofluorescence and FACS® analysis for DNAM-1–Fc binding used at 1.5 μM. FITC-conjugated goat anti–mouse or PE-conjugated goat anti–human were used as second reagents. (b) Similar experiments were performed by immunofluorescence microscopy on confluent HUVECs. 1.5 μM DNAM-1–Fc binding at endothelial junctions was not affected by preincubation with anti–Nectin-1 (R1.302) or anti–Nectin-2 (L14) mAbs. Anti-PVR (L95) mAb preincubation strongly affects the DNAM-1–Fc binding at endothelial junctions. mAbs were used at 20 μg/ml. (c) HUVECs, preincubated with the different anti–Nectin-2 (gray bars) or anti-PVR (black bars) mAbs, were analyzed by FACS® analysis for DNAM-1–Fc binding used at 0.15 μM followed by FITC-conjugated goat anti–human second reagents. Untreated HUVECs stained with N4vtr-Fc were used as negative control (white bars). The value 100% corresponds to DNAM-1–Fc binding in the presence of the irrelevant anti–Nectin-1 (R1.302) mAb. (d) The different anti-PVR mAbs were mapped by ELISA using PVR-Fc molecules. PVR-vcc-Fc is constituted by the full-length ectodomain of PVR, whereas PVR-v-Fc is only constituted by the V domain of PVR.
PVR and DNAM-1 Are Involved in Monocyte Transmigration.
Endothelial junctions have been described as key regulators of blood vessel permeability. Consistent with their distribution at endothelial cell junctions, PVR and Nectin-2 may thus participate in the events that regulate leukocyte extravasation through the endothelium. Having demonstrated that PVR is the major ligand of DNAM-1 on endothelial cells, we investigated the potential role of the PVR–DNAM-1 interaction during the TEM of monocytes. We controlled that anti-PVR mAbs readily access to the junctions of live HUVECs (not depicted), and we showed that anti-PVR mAbs do not modify the endothelial permeability (not depicted) and the endothelial integrity (see Fig. 7). Their ability to block monocyte transmigration in an in vitro TEM model was explored. Anti-PVR mAbs induced a significant blocking in monocyte transmigration when compared with isotype-matched irrelevant antibodies (Fig. 6 a, anti-CD34 and anti–Nectin-4 mAbs). Inhibition of monocyte transmigration was observed with five different anti-PVR mAbs (blockade between 60 and 80%) and was similar to that obtained with anti-CD99 mAb (Fig. 6 a, 12E7) and anti–LFA-1/CD11a (Fig. 6 b, 25.3.1), taken as positive controls (24, 25). Targeting of DNAM-1 with two different mAbs also strongly inhibits monocyte transmigration to 80%. Fab fragments of anti-PVR (mAbs) and anti–DNAM-1 (mAbs) efficiently inhibited TEM, as they blocked monocyte transmigration to the same extent as mAbs (Fig. 6 a). This rules out the possibility that inhibition may be due to Fc binding of mAbs to monocyte Fc receptors. The inhibition of monocyte TEM upon anti-PVR or anti–DNAM-1 mAb treatment is due to a blockade and not a slow down, as the same blockade was observed after a 3-h transmigration run. When incubating anti-PVR mAbs only on HUVECs, monocyte TEM was strongly blocked, which was not the case with anti–DNAM-1 mAb (Fig. 6 b). Because DNAM-1 is not or just faintly expressed on HUVECs, these results strongly suggest that DNAM-1 on monocytes is involved in this process. These results also suggest that anti-PVR mAbs block monocyte at the level of TEM and not by interfering with chemotaxis. To definitively demonstrate that mAbs did not inhibit chemotaxis, anti-PVR and anti–DNAM-1 Fabs were tested in a chemotaxis assay. No inhibition was noted when monocytes were incubated with both Fabs (Fig. 6 c). Altogether, our results demonstrate that endothelial PVR interacts with DNAM-1 on monocyte to ensure monocyte TEM.
Figure 7.
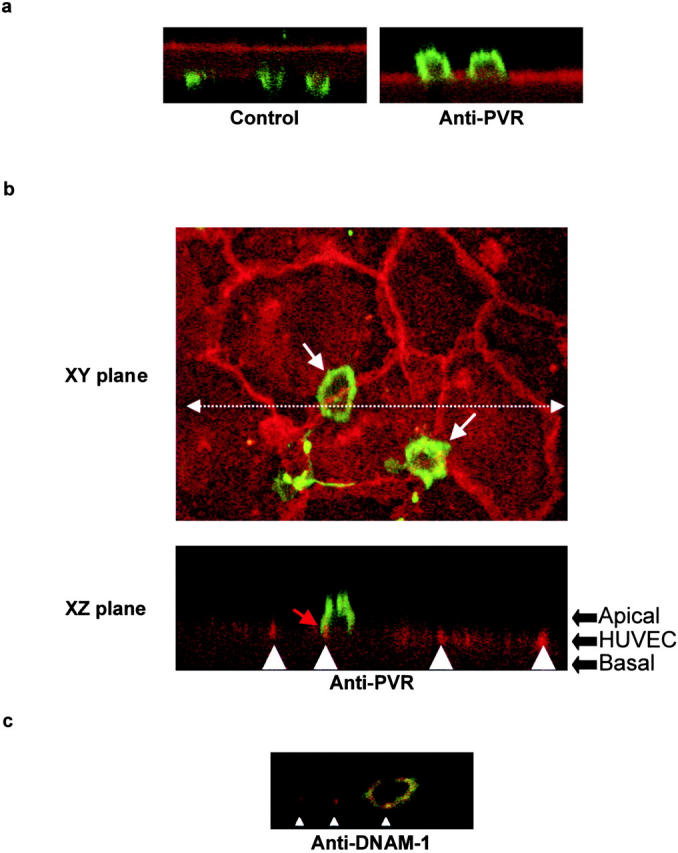
Blocking PVR–DNAM-1 transinteractions arrest monocytes over intercellular junctions at the apical surface of endothelial cells. Staining was performed after a 1-h migration run on transwell filters. (a) Monocyte migration was run in the absence (control) or the presence of anti-PVR mAb (PV.404) at 20 μg/ml. Cells were fixed, stained with 10 μg/ml anti-CD146 (previously conjugated to the 564-nm–labeled IgG1 isotype-specific Zenon Fab) and 2 μg/ml anti-CD14 (FITC-conjugated) mAbs to homogeneously stain endothelial cells and visualize monocytes, respectively. A series of images was recorded simultaneously in the XY and XZ planes. The results are representative of three independent experiments. (left) Monocytes untreated (control) are ending their transmigration and can be seen through the filter. (right) Upon anti-PVR treatment, monocytes are blocked over the endothelial cell monolayer. (b) Migration was run in the presence of 20 μg/ml of anti-PVR mAb (PV.404). Cells were fixed and stained with 10 μg/ml anti–VE-cadherin (previously conjugated to the 564-nm–labeled IgG1 isotype-specific Zenon Fab) and 2 μg/ml anti-CD14 (FITC-conjugated) mAbs to visualize endothelial junctions and monocytes, respectively. (top) White arrows indicate the position of blocked monocytes over the endothelial cell junctions in the XY plane. (bottom) XZ plane cross-section view according to the dotted line arrow in the XY plane. White arrowheads indicate the junctional staining of VE-cadherin. Red arrow marks the protruding monocyte membrane close to junctional VE-cadherin. (c) Monocyte transmigration through the HUVEC monolayer was performed as in b. Anti–DNAM-1 mAb (FS123) was used at 20 μg/ml. XZ plane cross-section view of monocytes arrest over the endothelial cell junctions as described in panel a. The results are representative of three independent experiments.
Figure 6.

PVR and DNAM-1 contribute to monocyte transmigration. Monocyte transmigration through the HUVEC monolayer was performed in the presence of 20 μg/ml of the indicated anti-PVR or anti–DNAM-1 (sodium azide free) mAbs, and 250 ng/ml MCP-1 was added to the lower chamber. Each measurement was done in quadruplicate. The results are representative of three independent experiments. (a) The N4.40 mAb that does not bind monocytes and HUVECs and Immu-133 mAb that strongly binds to HUVECs were used as isotype-matched irrelevant antibodies. Anti-CD99 mAb 12E7 was used as an inhibitory control. The value 100% corresponds to the number of monocytes that migrate in the presence of the Immu-133 mAb; FACS® analysis confirmed that almost every monocyte of the PBMC suspension transmigrates under this condition. Fab fragments of PV.404 or FS123 mAbs were used at 20 μg/ml. (b) Anti-CD11a mAb was used as a positive control. In this experiment, anti-PVR and anti–DNAM-1 mAbs were incubated on both monocytes and HUVECs (M+E) or only on endothelial cells (E). In the latter condition (E), mAbs were incubated for 30 min with HUVECs and washed with fresh medium. Monocytes were incubated, and TEM was processed as in a. In this experiment, the different antibodies used were treated with 10 μM polymyxin B. (c) Chemotaxis assay. Chemotaxis, in response to 250 ng/ml MCP-1, was assessed for 1 h on costar transwells (5-μm pore size and 6.5-mm diameter). Experimental conditions were similar to the TEM assay, except that filters were not layered with endothelial cells. Fabs were incubated through the process of migration.
PVR and DNAM-1 Take Part in the Diapedesis Process.
Transmigration is a multistep process and interfering at any step usually leads to blocking of the whole process. Thus, the precise step of action of PVR and DNAM-1 during monocyte transmigration was investigated. To distinguish between a blockade at the step of adhesion or during diapedesis itself, in vitro adhesion experiments were performed under static conditions with monocytes on HUVECs. No blockade of monocyte adhesion with anti-PVR mAbs was observed (not depicted), which was consistent with PVR localization at cell junctions (Fig. 1 a). The precise step at which monocytes were blocked during the TEM process was examined by the simultaneous staining of monocytes and endothelial cells. Cross-sections of the endothelial monolayer, analyzed in the z-plane, showed that, in the absence of mAbs, monocytes transmigrate through the HUVEC monolayer. After 1 h, some of them were still observed finishing their transmigration through the lowest part of the filter below the endothelial cell monolayer (Fig. 7 a, left). When incubating HUVECs and monocytes with anti-PVR mAbs, monocytes were blocked at the apical surface of the endothelium (Fig. 7 a, right). Moreover, monocytes were arrested over intercellular junctions as demonstrated by the staining with an anti–VE-cadherin mAb (Fig. 7 b, XY plane, arrow). Interestingly, monocyte membranes slightly protrude inside the intercellular junctions as shown by the tight localization between CD14 and VE-cadherin staining (Fig. 7 b, XZ plane, arrow). These data indicate that PVR is involved in the process of monocyte diapedesis rather than in the step of adhesion, as reported previously for PECAM-1 (24).
The site of monocyte blockade was also analyzed after anti–DNAM-1 treatment of monocytes. In this case, monocytes were also blocked at the surface of endothelial cells and in particular over intercellular junctions, at the same site as PVR (Fig. 7 c). Altogether, our results strongly suggest that DNAM-1–PVR interactions occur during the diapedesis phase to promote monocyte TEM.
Discussion
We described recently that DNAM-1 is involved in NK cell–mediated target cell killing of tumor cells by interacting specifically with PVR and Nectin-2 (2). Although PVR and Nectin-2 are expressed on tumor cell lines, they are also widely expressed on normal cells that are protected from NK cell–mediated lysis by their expression of HLA class I molecules. The aim of our analysis was to clarify the role of DNAM-1 interactions with its ligands in a normal setting. Nectins are homophilic and heterophilic cell adhesion molecules involved in several cell recognition processes, including the formation of cell junctions between epithelial cells and between neurons (10). For the first time, we demonstrate that PVR is the major ligand of DNAM-1 on endothelial cells. Our results highlight a role for DNAM-1 and PVR during leukocyte extravasation because they appear to be crucial for monocyte migration through endothelial junctions during the diapedesis step. This hypothesis is supported by the following observations. First, DNAM-1 binds specifically to the endothelium at cell junctions where PVR is expressed with a distribution similar to Nectin-2 and β-catenin. Second, targeting endothelial PVR, but not Nectin-2, inhibits DNAM-1 binding to endothelial cells. Third, mAb-mediated masking of DNAM-1 or PVR almost abrogates monocyte TEM. Last, during the TEM process, anti–DNAM-1 or anti-PVR mAb treatment blocks monocytes at the surface of endothelial cells specifically over intercellular junctions, similar to treatment with a blocking mAb to PECAM-1 (26).
DNAM-1 has been described previously to be cis-associated with LFA-1 in T cells (27). For the first time, we show that PVR and also Nectin-2 can bind directly to DNAM-1 and that this binding is independent of LFA-1. We found that Nectin-2–DNAM-1 interaction was reproducibly lower than PVR–DNAM-1 interaction according to our previous results (2). This observation is probably related to a difference of affinity. Our results show that DNAM-1 does not interact with endothelial Nectin-2 despite its high level of expression on HUVECs. This suggests that the interaction between DNAM-1 and PVR and/or Nectin-2 may be regulated by complex mechanisms in endothelial cells. We described previously two isoforms of Nectin-2 (α, the short isoform, and δ, the long isoform) that differ by their transmembrane and cytosolic regions and can homo- or heterodimerize at the cell surface (12). Taken individually, we found that these two isoforms equally bind DNAM-1–Fc (unpublished data) and are both expressed in HUVECs (unpublished data). Thus, the lack of interaction between DNAM-1 and endothelial Nectin-2 may not be related to a peculiar expression of Nectin-2 isoform in these cells. These data suggest that the DNAM-1 binding domain on Nectin-2 might not be functional in resting HUVECs by being in a low affinity state related to an absence of cis-dimerization or to a nonactive conformational state. DNAM-1 interaction with PVR is blocked by all anti-PVR mAbs tested. Some of these mAbs were also shown to inhibit DNAM-1–mediated NK cell lysis (2). Interestingly, these mAbs recognize either the V or the C domains of PVR, which suggests that they both contribute to the interaction with DNAM-1. These data contrast with those already described for Nectin transinteractions; indeed, we showed previously that V-to-V domain interactions play a prominent role during Nectin-1–Nectin-3 and Nectin-1–Nectin-4 transinteractions (7, 23).
Thus, today, PECAM-1 (26, 28–30), CD99 (24), JAMs (31–33), and now Nectins constitute four molecular families of endothelial cell adhesion molecules that have been described to participate to leukocyte diapedesis. Even though they may act together in some circumstances, these molecules may assume peculiar functions respective to their expression pattern, their spatio-temporal behavior at the intercellular junctions, or their role to deliver specific signals that regulate diapedesis.
It has been shown that NK cells migrate to injured tissues where they kill tumor or virus-infected target cells and interact with dendritic cells (34). It is conceivable that DNAM-1 molecules may regulate both migration of NK cells and NK cell–mediated cytolytic activity, depending on the level of HLA class I molecules expressed by encountered target cells. As described for JAMs (35), Nectins appear to be differentially expressed in endothelia. We observed that Nectin-2, but not PVR, is expressed on high endothelial venules (unpublished data). This suggests that Nectin-2 may take part in the process of lymphocyte homing to secondary lymphoid organs. Molecular basis of leukocyte diapedesis has only begun to be understood; recent studies highlighted that CD99 could act downstream of PECAM-1 during the diapedesis process (24), and that PECAM-1 could guide leukocyte diapedesis by recycling from vesicles to the zone of transmigration (30). Our immunofluorescence analysis shows that mAb-mediated disruption of PVR–DNAM-1 interactions induces a blockade of monocytes similar to that observed upon anti–PECAM-1 treatment (i.e., early during the diapedesis process; Fig. 7). This strongly suggests that PVR and DNAM-1 may take part in the first events of diapedesis together with PECAM-1. Different papers showed that Nectins, JAM-A, and PECAM-1 share common cytoplasmic partners that connect them to the cadherin system; Nectins and JAM-A interact with α-catenin via the F-actin binding protein AF-6 (36), and PECAM-1 directly interacts with β-catenin (37). Thus, it is possible that engagement of leukocytes through intercellular endothelial space triggers these molecules, which subsequently regulate VE-cadherin homophilic adhesion strength and induce its removal from cell-to-cell contact sites. Deciphering the sequence by which PECAM-1, CD99, JAMs, and Nectins play their respective role to the VE-cadherin gap formation will lead to a better understanding of the mechanisms that regulate leukocyte migration through endothelial cells.
Interestingly, cell adhesion molecules involved in the step of diapedesis have different behavior during the inflammation process. Among them, JAM-A is redistributed away from cell junctions to the apical pole upon endothelial activation (38). Recently, Ostermann et al. showed that endothelial luminal expression of JAM-A contributes to the LFA-1–dependent leukocyte recruitment (33). Nectins recruit JAM-A and enable its correct targeting to tight junctions in epithelial cells (36). Thus, we can postulate that Nectin-2, PVR, and JAM-A could form a molecular complex of adhesion molecules on endothelial cells that interact with DNAM-1 and LFA-1 during monocyte recruitment. It will be interesting to analyze the behavior of PVR and Nectin-2 upon endothelial activation.
Beyond this role, Nectins are expressed during development and in tumor cells; their potential role in tumor progression as well as in neovascularization and angiogenesis also deserve further investigation.
Acknowledgments
We thank V. Marin, E. Lecocq, F. Bardin, P. Gibier, A. Le Bivic Jean-Christophe Orsoni, D. Isnardon, and J.-R. Galindo for technical contributions. We are grateful to Drs. C. Stewart and C. Mawas for their help in preparing the manuscript.
This work was funded by Institut National de la Santé et de la Recherche Médicale, the Ligue Nationale Française Contre le Cancer, the Association pour la Recherche contre le Cancer, and the Associazione Italiana per la Ricerca sul Cancro.
Abbreviations used in this paper: AJ, adherens junction; DNAM-1, DNAX accessory molecule-1; HUVEC, human umbilical vein endothelial cell; JAM, junctional adhesion molecule; PECAM-1, platelet–endothelial cell adhesion molecule 1; PVR, poliovirus receptor; TEM, transendothelial migration.
References
- 1.Shibuya, A., D. Campbell, C. Hannum, H. Yssel, K. Franz-Bacon, T. McClanahan, T. Kitamura, J. Nicholl, G.R. Sutherland, L.L. Lanier, and J.H. Phillips. 1996. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity. 4:573–581. [DOI] [PubMed] [Google Scholar]
- 2.Bottino, C., R. Castriconi, D. Pende, P. Rivera, M. Nanni, B. Carnemolla, C. Cantoni, J. Grassi, S. Marcenaro, N. Reymond, et al. 2003. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 198:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendelsohn, C.L., E. Wimmer, and V.R. Racaniello. 1989. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 56:855–865. [DOI] [PubMed] [Google Scholar]
- 4.Lopez, M., F. Eberle, M.G. Mattei, J. Gabert, F. Birg, F. Bardin, C. Maroc, and P. Dubreuil. 1995. Complementary DNA characterization and chromosomal localization of a human gene related to the poliovirus receptor-encoding gene. Gene. 155:261–265. [DOI] [PubMed] [Google Scholar]
- 5.Eberle, F., P. Dubreuil, M.G. Mattei, E. Devilard, and M. Lopez. 1995. The human PRR2 gene, related to the human poliovirus receptor gene (PVR), is the true homolog of the murine MPH gene. Gene. 159:267–272. [DOI] [PubMed] [Google Scholar]
- 6.Reymond, N., J.P. Borg, E. Lecocq, J. Adelaide, G. Campadelli-Fiume, P. Dubreuil, and M. Lopez. 2000. Human nectin3/PRR3: a novel member of the PVR/PRR/nectin family that interacts with afadin. Gene. 255:347–355. [DOI] [PubMed] [Google Scholar]
- 7.Reymond, N., S. Fabre, E. Lecocq, J. Adelaide, P. Dubreuil, and M. Lopez. 2001. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J. Biol. Chem. 276:43205–43215. [DOI] [PubMed] [Google Scholar]
- 8.Geraghty, R.J., C. Krummenacher, G.H. Cohen, R.J. Eisenberg, and P.G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 280:1618–1620. [DOI] [PubMed] [Google Scholar]
- 9.Lopez, M., F. Cocchi, L. Menotti, E. Avitabile, P. Dubreuil, and G. Campadelli-Fiume. 2000. Nectin2alpha (PRR2alpha or HveB) and nectin2delta are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D. J. Virol. 74:1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takai, Y., and H. Nakanishi. 2003. Nectin and afadin: novel organizers of intercellular junctions. J. Cell Sci. 116:17–27. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi, K., H. Nakanishi, M. Miyahara, K. Mandai, K. Satoh, A. Satoh, H. Nishioka, J. Aoki, A. Nomoto, A. Mizoguchi, and Y. Takai. 1999. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 145:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez, M., M. Aoubala, F. Jordier, D. Isnardon, S. Gomez, and P. Dubreuil. 1998. The human poliovirus receptor related 2 protein is a new hematopoietic/endothelial homophilic adhesion molecule. Blood. 92:4602–4611. [PubMed] [Google Scholar]
- 13.Campadelli-Fiume, G., F. Cocchi, L. Menotti, and M. Lopez. 2000. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 10:305–319. [DOI] [PubMed] [Google Scholar]
- 14.Moretta, A., C. Bottino, M. Vitale, D. Pende, R. Biassoni, M.C. Mingari, and L. Moretta. 1996. Receptors for HLA class-I molecules in human natural killer cells. Annu. Rev. Immunol. 14:619–648. [DOI] [PubMed] [Google Scholar]
- 15.Kojima, H., H. Kanada, S. Shimizu, E. Kasama, K. Shibuya, H. Nakauchi, T. Nagasawa, and A. Shibuya. 2003. CD226 mediates platelet and megakaryocytic cell adhesion to vascular endothelial cells. J. Biol. Chem. 278:36748–36753. [DOI] [PubMed] [Google Scholar]
- 16.Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- 17.Johnson-Leger, C., M. Aurrand-Lions, and B.A. Imhof. 2000. The parting of the endothelium: miracle, or simply a junctional affair? J. Cell Sci. 113:921–933. [DOI] [PubMed] [Google Scholar]
- 18.Worthylake, R.A., and K. Burridge. 2001. Leukocyte transendothelial migration: orchestrating the underlying molecular machinery. Curr. Opin. Cell Biol. 13:569–577. [DOI] [PubMed] [Google Scholar]
- 19.Luscinskas, F.W., S. Ma, A. Nusrat, C.A. Parkos, and S.K. Shaw. 2002. Leukocyte transendothelial migration: a junctional affair. Semin. Immunol. 14:105–113. [DOI] [PubMed] [Google Scholar]
- 20.Vestweber, D. 2002. Regulation of endothelial cell contacts during leukocyte extravasation. Curr. Opin. Cell Biol. 14:587–593. [DOI] [PubMed] [Google Scholar]
- 21.Weber, C. 2003. Novel mechanistic concepts for the control of leukocyte transmigration: specialization of integrins, chemokines, and junctional molecules. J. Mol. Med. 81:4–19. [DOI] [PubMed] [Google Scholar]
- 22.Cocchi, F., L. Menotti, P. Mirandola, M. Lopez, and G. Campadelli-Fiume. 1998. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 72:9992–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fabre, S., N. Reymond, F. Cocchi, L. Menotti, P. Dubreuil, G. Campadelli-Fiume, and M. Lopez. 2002. Prominent role of the Ig-like V domain in trans-interactions of nectins. Nectin3 and nectin 4 bind to the predicted C-C'-C”-D beta-strands of the nectin1 V domain. J. Biol. Chem. 277:27006–27013. [DOI] [PubMed] [Google Scholar]
- 24.Schenkel, A.R., Z. Mamdouh, X. Chen, R.M. Liebman, and W.A. Muller. 2002. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat. Immunol. 3:143–150. [DOI] [PubMed] [Google Scholar]
- 25.Lu, C., M. Shimaoka, Q. Zang, J. Takagi, and T.A. Springer. 2001. Locking in alternate conformations of the integrin alphaLbeta2 I domain with disulfide bonds reveals functional relationships among integrin domains. Proc. Natl. Acad. Sci. USA. 98:2393–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller, W.A., S.A. Weigl, X. Deng, and D.M. Phillips. 1993. PECAM-1 is required for transendothelial migration of leukocytes. J. Exp. Med. 178:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shibuya, K., L.L. Lanier, J.H. Phillips, H.D. Ochs, K. Shimizu, E. Nakayama, H. Nakauchi, and A. Shibuya. 1999. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity. 11:615–623. [DOI] [PubMed] [Google Scholar]
- 28.Liao, F., J. Ali, T. Greene, and W.A. Muller. 1997. Soluble domain 1 of platelet–endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J. Exp. Med. 185:1349–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogen, S., J. Pak, M. Garifallou, X. Deng, and W.A. Muller. 1994. Monoclonal antibody to murine PECAM-1 (CD31) blocks acute inflammation in vivo. J. Exp. Med. 179:1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mamdouh, Z., X. Chen, L.M. Pierini, F.R. Maxfield, and W.A. Muller. 2003. Targeted recycling of PECAM from endothelial surface-connected compartments during diapedesis. Nature. 421:748–753. [DOI] [PubMed] [Google Scholar]
- 31.Martin-Padura, I., S. Lostaglio, M. Schneemann, L. Williams, M. Romano, P. Fruscella, C. Panzeri, A. Stoppacciaro, L. Ruco, A. Villa, et al. 1998. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 142:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Del Maschio, A., A. De Luigi, I. Martin-Padura, M. Brockhaus, T. Bartfai, P. Fruscella, L. Adorini, G. Martino, R. Furlan, M.G. De Simoni, and E. Dejana. 1999. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM). J. Exp. Med. 190:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostermann, G., K.S. Weber, A. Zernecke, A. Schroder, and C. Weber. 2002. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 3:151–158. [DOI] [PubMed] [Google Scholar]
- 34.Moretta, A. 2002. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat. Rev. Immunol. 2:957–964. [DOI] [PubMed] [Google Scholar]
- 35.Aurrand-Lions, M., C. Johnson-Leger, C. Wong, L. Du Pasquier, and B.A. Imhof. 2001. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood. 98:3699–3707. [DOI] [PubMed] [Google Scholar]
- 36.Fukuhara, A., K. Irie, H. Nakanishi, K. Takekuni, T. Kawakatsu, W. Ikeda, A. Yamada, T. Katata, T. Honda, T. Sato, et al. 2002. Involvement of nectin in the localization of junctional adhesion molecule at tight junctions. Oncogene. 21:7642–7655. [DOI] [PubMed] [Google Scholar]
- 37.Matsumura, T., K. Wolff, and P. Petzelbauer. 1997. Endothelial cell tube formation depends on cadherin 5 and CD31 interactions with filamentous actin. J. Immunol. 158:3408–3416. [PubMed] [Google Scholar]
- 38.Ozaki, H., K. Ishii, H. Horiuchi, H. Arai, T. Kawamoto, K. Okawa, A. Iwamatsu, and T. Kita. 1999. Cutting edge: combined treatment of TNF-alpha and IFN-gamma causes redistribution of junctional adhesion molecule in human endothelial cells. J. Immunol. 163:553–557. [PubMed] [Google Scholar]