

Abstract
Gram-positive organisms like Staphylococcus aureus are a major cause of morbidity and mortality worldwide. Humoral response molecules together with phagocytes play a role in host responses to S. aureus. The mannose-binding lectin (MBL, also known as mannose-binding protein) is an oligomeric serum molecule that recognizes carbohydrates decorating a broad range of infectious agents including S. aureus. Circumstantial evidence in vitro and in vivo suggests that MBL plays a key role in first line host defense. We tested this contention directly in vivo by generating mice that were devoid of all MBL activity. We found that 100% of MBL-null mice died 48 h after exposure to an intravenous inoculation of S. aureus compared with 45% mortality in wild-type mice. Furthermore, we demonstrated that neutrophils and MBL are required to limit intraperitoneal infection with S. aureus. Our study provides direct evidence that MBL plays a key role in restricting the complications associated with S. aureus infection in mice and raises the idea that the MBL gene may act as a disease susceptibility gene against staphylococci infections in humans.
Keywords: mannose-binding lectin, MBL, infection, neutropenia, innate immunity
Introduction
A basic function of innate immunity is restriction of the early proliferation of infectious agents (1, 2). Numerous molecules and effector cells conspire to restrict this initial spread of an infectious focus. Some examples of first line host defense molecules include antimicrobial peptides, natural antibodies, complement proteins, lipopolysaccharide-binding protein (LBP), soluble receptors, and collectins (3–5). The collectins are multimeric carbohydrate recognition domain-containing molecules with collagen stalks that include the pulmonary surfactant proteins A and D, conglutinin, CL-43, CL-46, and the mannose-binding lectin (MBL; 6–9)
MBL appears to be a prototypic pattern recognition molecule that is able to recognize the molecular patterns that decorate a wide range of microorganisms. Infectious agents that are recognized by MBL include certain Gram-positive and Gram-negative bacteria, yeast, parasites, mycobacteria, and viruses (7, 10). MBL was first surmised to play a role in host defense based on its overall structural similarity with the first complement component C1q (11, 12). Next, in vitro observations demonstrated that MBL could bind and opsonize bacteria as well as the yeast cell wall product mannan (13). The idea that a relative lack of MBL might predispose the host to infection was based on the description of an MBL-dependent opsonic defect in human serum that correlated with a phenotype of recurrent infection (14). These patients were found to have one of three amino acid substitution single nucleotide polymorphisms in exon 1 of the MBL gene that disrupts the collagen helix (15). It appears that the disordered collagen chain acts like a dominant negative that results in a decrease in circulating levels of MBL that can activate complement. More detailed analysis of the MBL gene has revealed at least seven distinct MBL haplotypes in humans, four of which (LYPB, LYQC, HYPD, and LXPA) dictate low serum levels (16). Interestingly, there is a high rate of haplotype variation in various human populations with a range of heterozygosity from 15% in Caucasians to 30% in certain African populations (17, 18).
Importantly, MBL seems to be able to distinguish species self as well as altered self, e.g., in the form of apoptotic cells from nonself (19). The specificity that allows the distinction of surfaces of virally infected cells and transformed cells from normal host cells depends on both fine recognition of molecular patterns and a macropattern (3). The macropattern appears to be dictated by the spatial orientation of the carbohydrate-binding domains and the differences in geometry of the sugars that adorn microorganisms versus host glycoproteins (3, 20, 21). MBL is able to activate complement via a novel mechanism that coopts the MBL-associated serine protease (MASP; 22, 23), MASP-2, which then mimics the classical pathway convertase to cleave the third complement component (C3; 23). In this way the MBL complement pathway is activated in an antibody-independent manner. Therefore, MBL has many functional properties that are reminiscent of an antibody and in fact MBL is considered an opsonin (13,14, 24–26).
The initial response to infection is a complex interaction between a variety of pattern recognition molecules that trigger the downstream physiological cascades of clotting, cytokine, and chemokine release and interface with effector cells such as neutrophils (27, 28). Neutrophils express complement receptors, MBL receptors (collectin receptors; 29, 30), and the receptor for LBP (31). Wright and Douglas (32) and Silverstein (33) linked humoral and cellular interactions and drew attention to the importance of cooperative interactions between neutrophils and opsonins in combating infection. More recent examples that have exploited the use of null animals to explore such interactions and are germane to this study include the interaction of LBP and neutrophils in resistance to i.p. Salmonella infection (34, 35). A similar synergistic interaction between neutrophils and MBL is suggested by clinical observations that chemotherapy-induced neutropenic patients with haplotypes that specify low serum MBL levels (9, 36, 37) appear more susceptible to infection (38). These clinical observations together with in vitro studies suggest that MBL plays a key role as an ante-antibody in first line host defense (39, 40) and support a role for MBL in combating infection in vivo.
To provide formal proof that MBL is indeed important in host defense in vivo, we set out to create a mouse model of MBL deficiency. Although humans and new world monkeys have a single MBL gene, two homologous forms of MBL, designated MBL-A and MBL-C, have been identified in rodents (41, 42). MBL-A and MBL-C, the respective gene products of the mbl1 and mbl2 genes, are 50% homologous (43), have distinct and overlapping binding specificities (43–45), are found predominantly in serum, and are able to bind MASPs to activate complement (43). The relative physiological role of these two proteins in vivo has not been clearly defined (43, 46–49). To address some of these questions we created MBL-A and MBL-C double KO (MBL-null) mice. These MBL-null mice lack MBL in serum and lack the MBL complement pathway. We chose to infect these mice with Staphylococcus aureus as this organism is a significant cause of bacteremia in humans worldwide (50, 51). Treatment of S. aureus infections is increasingly problematic with the emergence of widespread antibiotic resistance (52, 53). Although there are clinical identifiers that indicate likelihood of complication as a result of S. aureus infection, there is a paucity of data that pertains to genetic variation in host factors that confer resistance to S. aureus. We found that MBL-null mice are highly susceptible to an i.v. inoculation of S. aureus as at 48 h after inoculation all MBL-null mice had died compared with 55% survival of WT mice. In contrast, i.p. inoculation of S. aureus did not result in enhanced infectious complications in MBL-null mice compared with WT mice unless mice were rendered neutropenic but not neutropenic WT mice. The neutropenic MBL-null mice displayed enhanced bacterial accumulation in organs and had persistent bacteremia 10 d after inoculation. Our results are consistent with a proposed role for MBL in first line host defense.
Materials and Methods
Generation of MBL-null Mice.
A genomic DNA clone encoding MBL-C was isolated from a 129SvJ library in Lambda Fix II vector (Stratagene) and mapped (54). The MBL-C gene was disrupted by introducing the neomycin resistance gene (neor) into exon 6. The MBL-C gene-targeting construct consists of a 4.5-kb PmeI-NotI fragment toward the 5′ end followed by neor, and then a 1.5-kb PacI-SrfI fragment toward the 3′ end in a KO3 vector (see Fig. 1 a; provided by K. Moore, Massachusetts General Hospital, Boston, MA). 17 out of 111 embryonic stem cell clones underwent homologous recombination. This was confirmed by Southern blot analysis (not depicted). The positive embryonic stem cell clones were injected into C57Bl/6J blastocysts at the Transgenic and Knockout Mouse Core Facility at Massachusetts General Hospital, directed by Dr. E. Li. Genotyping was performed by PCR. A colony of MBL-C KO mice was expanded and some were crossed with MBL-A KO (48) mice to create MBL-null mice. All animal experiments were performed under a protocol approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital.
Figure 1.
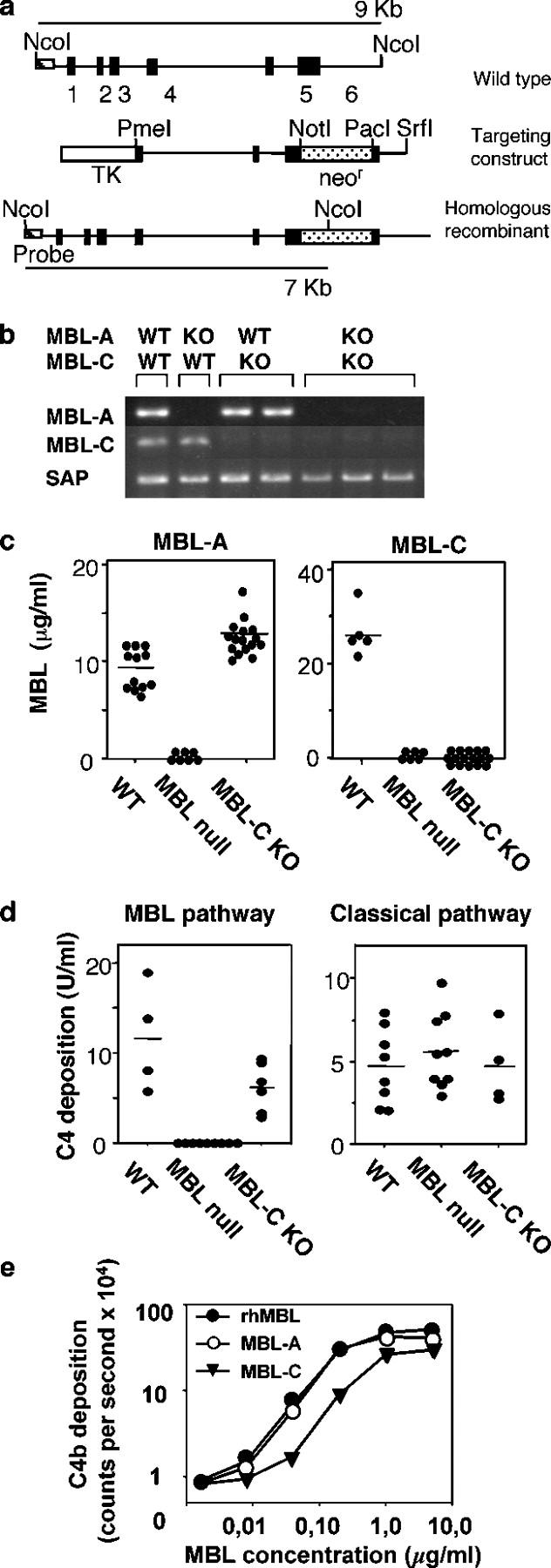
Generation and characterization of MBL-null mice. (a) MBL-C targeting construct. Genomic organization of MBL-C is shown and compared with the targeting vector and homologous recombinant. (b) RT-PCR analysis of transcript for MBL-A, MBL-C, and serum amyloid protein (SAP) in liver. (c) Serum levels of MBL-A and MBL-C in WT, MBL-C KO, and MBL-null mice. •, individual mice; bars, the mean value for each group. (d) C4 cleaving activity of serum. The capacity of serum to activate C4 via the MBL complement pathway (left) or classical pathway (right) was assayed as described above. •, individual mice; bars, the mean value for each group. (e) C4 cleaving activity. Comparison of rhMBL with purified MBL-A and MBL-C. •, rhMBL; ○, MBL-A; ▾, MBL-C.
RT-PCR.
RT-PCR has been described (48).
Detection and Assay of MBLs.
The assay for MBL has been described (46). Complement component 4 (C4) cleaving activity of purified MBLs and recombinant human MBL (rhMBL) was measured by previously reported methods (55) with modification. In brief, microtiter wells were coated with mannan and a diluted mixture of different amounts of MBLs in 2.5% MBL-null mouse serum was added to the wells. After incubation at 37°C and rinsing, deposited C4 fragments were detected with biotinylated monoclonal anti–mouse C4 followed by europium-labeled streptavidin and measurement by time-resolved fluorometry. C4 converting activity in mouse serum was measured for the MBL complement pathway and the classical pathway by a modification of the above methods using microtiter wells that were coated with mannan or human IgG, respectively. Diluted serum samples were added to the wells at 4°C to avoid complement activation. After incubation at 4°C and rinsing, human C4 was added and incubated at 37°C. The wells were rinsed and deposited C4 fragments were detected with biotinylated rabbit anti–human C4c antibody followed by alkaline phosphatase–conjugated biotin–avidin complex and p-nitrophenyl phosphate substrate. OD 415 nm was measured.
S. aureus Infection.
All mice were between 6 and 12 wk old, from generations F4–F11, and were maintained on a mixed background of 129Sv × C57B/6J. Age- and gender-matched mice were used in each experiment. The strains of S. aureus used were Reynolds capsular serotype 5 (S. aureus CP5; provided by J.C. Lee, Brigham and Women's Hospital, Boston, MA; 56, 57) and bioluminescent S. aureus Xen 8.1 (biolumi-S. aureus; provided by K.P. Francis, Xenogen Corp., Alameda, CA), which is a modification of S. aureus 8325–4 (58). The biolumi-S. aureus was used for studies of in vivo imaging, whereas the rest of the studies were performed with the S. aureus CP5. S. aureus was grown overnight in Columbia media with 2% NaCl, washed once, and resuspended in saline. Mice were inoculated i.v. in the tail vein with 5 × 106, 5 × 107, or 5 × 108 CFU/0.2 ml saline/mouse, evaluated for complications of infection, and 5 × 107 CFU/mouse was chosen as the optimal dose. Dose response for i.p. inoculation was performed with 4 × 105, 4 × 106, and 4 × 107 CFU/0.5 ml saline/mouse, and 2 × 106 CFU/mouse was the optimal dose. Neutropenia was induced by i.p. injection of cyclophosphamide (CY) at 150 and 100 mg/kg at 4 and 1 d before S. aureus inoculation, respectively.
For reconstitution experiments, 75 μg rhMBL (provided by NatImmune A/S) in 0.2 ml saline/mouse was injected i.p. 1 h before the inoculation and then daily for 3 d after inoculation because a half-life of rhMBL via i.p. injection was 14–20 h (unpublished data). Serum levels of rhMBL measured at 1 d after injection ranged between 5 and 11 μg/ml, which is in the physiological range in mouse.
In Vivo Bioluminescence Imaging.
The low light imaging system (Hamamatsu Photonics KK) has been previously described in detail (59).
Bacterial Load in Blood and Organs.
Blood was collected from the tail vein and immediately mixed with heparin. Organs were harvested from killed mice, weighed, and homogenized in saline (2 ml for liver and 1 ml for the other organs). Serial dilutions of the blood and the organ homogenates were cultured on tryptic soy agar plates supplemented with 5% sheep blood plates (TSA-II) overnight at 37°C. CFUs were calculated as CFU/ml for blood and CFU/g of wet weight for organs.
Bacterial Growth Assay in Plasma, Serum, and Whole Blood.
Plasma and serum were collected from hirudin-treated blood and coagulated blood at room temperature for 2 h, respectively. 60 μl serum, plasma, or hirudin-treated blood was mixed with S. aureus CP5 (105 CFU/ml) in a 100-μl reaction volume. After the mixtures were cultured at 37°C for 2 h, 10-μl samples were removed, diluted, and plated on TSA-II plates. CFUs were determined after overnight culture.
Cytokine Assay.
TNF-α and IL-6 were measured by ELISA kits (R&D Systems) according to the manufacturer's instructions as previously described (48).
Phagocytosis Assays.
Resident peritoneal macrophages were obtained by peritoneal lavage. FITC-labeled S. aureus CP5 (FITC-S. aureus) was opsonized in 40% serum (vol/vol in HBSS) at 37°C for 30 min, washed, and suspended in 100 μl HBSS to a concentration of 2.5 × 108 cells/ml. 1.25 × 105 macrophages were mixed with 1.25 × 107 opsonized FITC-S. aureus in 100 μl HBSS and incubated at 37°C for 30 min. The extracellular fluorescence was quenched by the addition of 200 μl PBS containing 0.04% trypan blue and 1% formaldehyde, pH 5.5, and ingested bacteria were scored by flow cytometry (60). Triplicate experiments were repeated twice. For in vivo phagocytosis assay, 2 × 107 FITC-S. aureus were inoculated i.p. and 10 min thereafter peritoneal cells were collected individually by peritoneal lavage and washed once before quenching. Flow cytometry assays were performed on a FACSCalibur™ (BD Biosciences). Results were analyzed using CELLQuest™ software.
Statistical Analysis.
Data of abscess formation was assessed by χ2-test (JMP5 software; SAS Institute). Data of bacterial loads was analyzed by ANOVA using Statview (SAS Institute).
Results
Generation and Characterization of MBL-null Mice.
MBL-null mice were generated by crossing MBL-A KO and MBL-C KO mice. MBL-C KO mice were created by introducing neor gene into exon 5 (Fig. 1 a). MBL-A KO mice were generated previously (48). Disruption of the MBL genes was confirmed by the lack of mRNA for MBLs in the liver, the principle site of MBL synthesis (49), and undetectable MBLs in serum (Fig. 1, b and c). MBL-A KO, MBL-C KO, and MBL-null mice were healthy, viable, fertile, and appeared normal with no obvious developmental defects (not depicted). Histological examination of lung, liver, spleen, lymph node, kidney, brain, and intestine derived from mice 6–10 wk old did not reveal any obvious abnormality (unpublished data).
Next, we examined the MBL complement pathway in both MBL-C KO and MBL-null mice. This third novel pathway of complement activation requires that MBL engages a ligand to trigger the activation of MASPs that generate the C3 convertase, C4bC2b (22, 23). The MBL-dependent deposition of C4b is therefore an accurate measurement of this pathway. Previously we reported that serum levels of MBL-C in MBL-A KO mice were similar to levels in WT mice and that the MBL complement pathway in these MBL-A KO mice was half that of WT mice (48). These results suggested that both MBL-A and MBL-C contributed to this pathway in mice. MBL-C KO mice had slightly elevated serum levels of MBL-A compared with WT mice (Fig. 1 c). This result is likely explained by the fact that MBL-A is an acute phase reactant. However, as was observed in MBL-A KO mice, the MBL complement pathway was reduced by 50% in MBL-C mice (Fig. 1 d). To investigate the relative complement-activating ability of MBL-A and MBL-C, we performed a dose response using purified MBL-C and MBL-A (43) that were added back to MBL-null mouse serum, and then measured MBL-dependent C4 deposition. The specific activity of MBL-A was approximately four times that of MBL-C (Fig. 1 e). However, the serum levels of MBL-C were threefold those of MBL-A in WT mice (Fig. 1 c). Taken together, these results indicate that MBL-A and MBL-C have equivalent total activity in serum. Importantly and relevant to later reconstitution experiments, rhMBL was active in mouse serum in vitro and its specific activity was equal to that of mouse MBL-A and four times that of MBL-C (Fig. 1 e). This suggests that in so far as complement activation is concerned, MBL-A and MBL-C are redundant. As expected, the MBL complement pathway was not functional in MBL-null mice as neither MBL-A nor MBL-C is found in the serum (Fig. 1 d, left). As shown in Fig. 1 d, the classical pathway was unaffected in MBL-null mice.
MBL-null Mice Are Highly Susceptible to S. aureus Bacteremia.
Resistance against S. aureus is multifactorial. The products of neutrophils and platelets, complement and peptidoglycan recognition proteins like Toll-like receptor 2, are key components in the initial armamentarium against this pathogen (61, 62). Furthermore, it has recently been shown in vitro that complement-dependent killing of S. aureus is mediated via the MBL complement pathway rather than the classical or the alternative complement pathways (25). To evaluate the in vivo role of MBL against infection with S. aureus, WT and MBL-null mice were inoculated i.v. with 5 × 107 CFU S. aureus/mouse and survival was monitored.
At 48 h the mortality was 100% for MBL-null mice compared with 55% survival for WT mice (Fig. 2). Furthermore, pretreatment of MBL-null mice with rhMBL partially rescued the phenotype in that the survival of MBL-null mice was 45% by 48 h (Fig. 2). The phenotype of MBL-A KO and MBL-C KO mice was similar to that of WT mice (unpublished data). These results suggest that these two forms of MBL play a redundant role in resistance to S. aureus infection and that only when both proteins are absent the susceptibility to S. aureus is revealed.
Figure 2.

Increased mortality in MBL-null mice from S. aureus infection. S. aureus was inoculated i.v. and survival was followed as described in Materials and Methods. Numbers of mice used were 15 WT, 14 MBL-null, and 9 MBL-null plus rhMBL. *, P < 0.0001.
Next, we investigated whether the enhanced susceptibility of MBL-null mice to S. aureus infection was a result of altered distribution and growth of the bacteria in the blood, kidney, spleen, liver, and lung. One and a half logs more CFU/ml was found in the blood of MBL-null mice compared with WT mice at 24 h after inoculation (Fig. 3). It was not possible to examine the later kinetics of bacteremia in MBL-null mice as all had died by 48 h, but longer studies in WT mice that survived the infection indicated that these mice sterilize the blood several days after inoculation (not depicted). There were statistically significant higher bacterial loads in the kidney, spleen, and liver in MBL-null mice compared with WT mice at 24 h after inoculation (Fig. 3).
Figure 3.

Increased bacterial loads in blood and organs of MBL-null mice. Bacterial titers were assayed at 24 h after i.v. inoculation of S. aureus as described in Materials and Methods. Six mice were in each group. Open bars, WT; closed bars, MBL-null. Bars indicate mean ± SD. *, P ≤ 0.05; **, P < 0.01.
To evaluate the relative contribution of direct complement-mediated lysis and MBL-dependent opsonophagocytosis, we incubated S. aureus in plasma and serum of WT, MBL-null, and C3-null mice. We found that none of the plasma (Fig. 4 a) and sera (not depicted) restricted the growth of bacteria at 10 min or 2 h after inoculation. In addition, there was no difference in the growth rate at all three conditions (Fig. 4 a). By contrast, ex vivo whole blood killing assay revealed that after 2 h of incubation the growth of S. aureus was restricted in whole blood from WT mice compared with 10 min of incubation, whereas S. aureus continued to grow in whole blood from MBL-null mice (Fig. 4 b). These results indicate that phagocytes, MBL, and complement are required for S. aureus killing.
Figure 4.
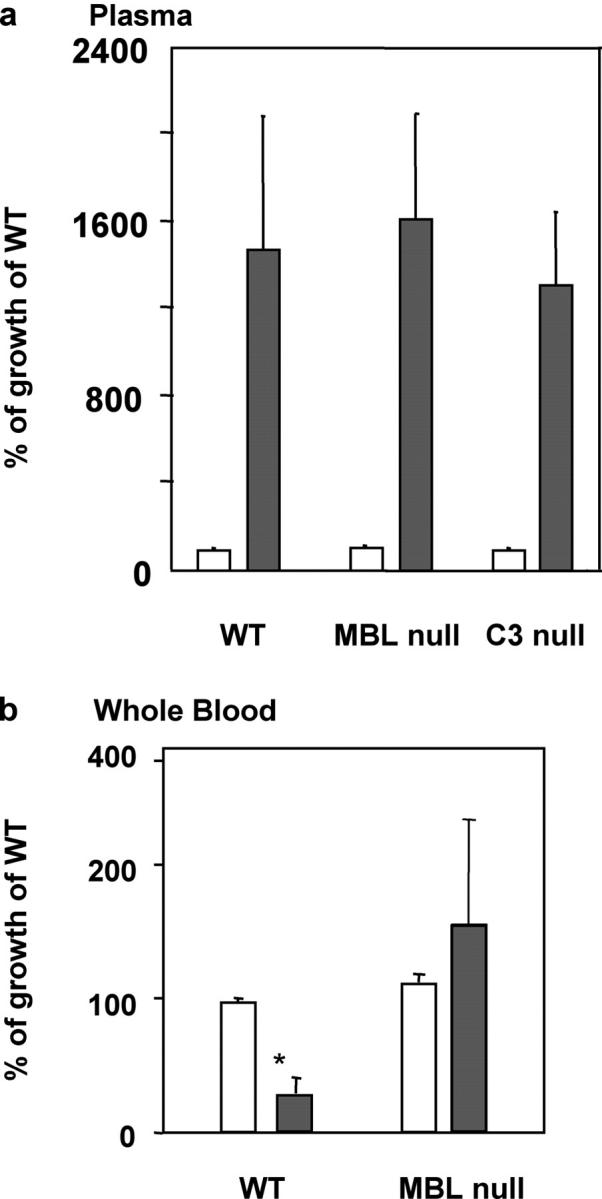
Restricted bacterial growth in blood of WT mice and enhanced growth in blood but not in plasma of MBL-null mice. Open bars, at 10 min; closed bars, at 2 h. (a) Bacterial growth in plasma. Results are shown as a percentage of bacterial growth in WT plasma at 10 min. Pooled plasma was used and the assay was performed in triplicates as described in Materials and Methods. Bars indicate mean ± SD. (b) Results are shown as a percentage of bacterial growth in WT blood at 10 min. Blood was collected from four mice individually and the assay was performed in triplicates as described in Materials and Methods. Bars indicate mean ± SD. *, P < 0.05
Role of MBL in Cytokine Response to S. aureus Infection.
The levels of TNF-α and IL-6 in the blood of MBL-null and WT mice were determined at 2 and 24 h after the i.v. inoculation. Both TNF-α and IL-6 (P < 0.0001) were reduced in the serum of MBL-null mice compared with WT mice at 2 h (Fig. 5). In contrast, at 24 h there was a 15-fold increase in TNF-α (P < 0.05) and an eightfold increase in IL-6 (P < 0.0005) in the serum of MBL-null mice compared with WT mice (Fig. 5). Preliminary in vitro studies with bone marrow–derived macrophages from MBL-null mice that were cultured with heat-killed S. aureus showed enhancement of IL-6 secretion at 24 h. A similar trend was also observed for TNF-α secretion (unpublished data).
Figure 5.

Cytokine production after S. aureus infection. Levels of cytokine induction was less at 2 h but more at 24 h in MBL-null mice compared with WT mice after S. aureus i.v. inoculation. Six mice were used in each group. Bars indicate mean ± SE. *, P < 0.05; **, P < 0.001.
MBL and Neutrophils Are Required to Combat i.p. Challenge of S. aureus.
The role of MBL in restricting tissue infection as it developed in the peritoneal cavity was evaluated. We adapted a modified rat infection model of S. aureus (57) to mice by administering the bacteria i.p. to achieve a slower seeding into the blood and tissues. In this way we could assess the role of MBL in combating infection in inflamed body cavities. Bacteremia and abscess formation were evaluated at various time points up to 10 d after i.p. inoculation of S. aureus ranging between 4 × 105 and 4 × 107 CFU/mouse. Even the highest dose of bacterial inoculation did not show difference in survival between WT, MBL-A KO, and MBL-null mice. We chose a dose of 2 × 106 CFU/mouse and compared abscess formation in WT, MBL-A KO, and MBL-null mice. There was no abscess formation in organs that we examined in any of the mice tested (Fig. 6). We did not test MBL-C KO mice as we assumed that they would be similar to MBL-A KO mice based on equivalence of MBL-A– and MBL-C–dependent complement pathway activity in the serum (Fig. 1 e). Of note, MBL is detectable in the peritoneal cavity of WT mice within hours of an inflammatory challenge (unpublished data).
Figure 6.
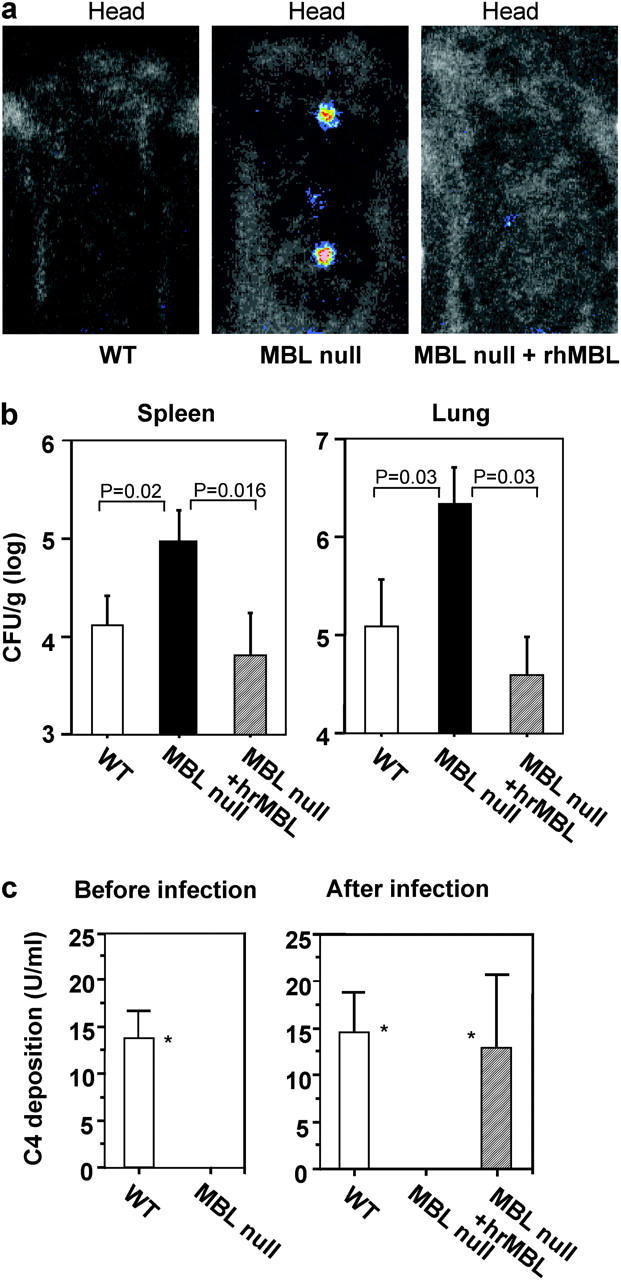
Increased S. aureus infection in MBL-null mice and rescued MBL complement pathway by rhMBL in MBL-null mice. (a) In vivo imaging of mice at 48 h after inoculation of the biolumi-S. aureus was performed as described in Materials and Methods. Representative pictures from WT, MBL-null, and MBL-null mice that were reconstituted with rhMBL (MBL null plus rhMBL) are shown. (b) Increased level of bacteria in organs from MBL-null mice. Organs were harvested at 96 h after the infection with biolumi-S. Aureus and bacterial load was measured as described in Materials and Methods. Bars indicate mean ± SD. Numbers of mice used: WT, 8; MBL-null, 7; MBL-null plus rhMBL, 7. (c) MBL complement pathway activity before and after S. aureus CP5 infection. Plasma was collected at 4 d before as a base line and 4 d after S. aureus inoculation and analyzed for C4 deposition activity on mannan as described in Materials and Methods. Numbers of mice used before infection: WT, 12; MBL-null, 19. Numbers of mice used after infection: WT, 12; MBL-null, 10; MBL-null plus rhMBL, 9. Two experiments were combined. Bars indicate mean ± SD. *, P = 0.002.
Clinical observations indicate that cancer patients with chemotherapy-induced neutropenia who have MBL haplotypes that specify low serum levels of MBL have an increased incidence of infections compared with similar populations of MBL-sufficient patients (38, 63, 64). We decided to simulate the clinical situation of febrile neutropenia to test this observation under controlled experimental conditions. MBL-null and WT mice were rendered neutropenic by i.p. injection of CY. 4 d after injection of CY all mice were neutropenic, at which time these mice were inoculated with S. aureus. By 10 d after inoculation, 21 out of 29 neutropenic MBL-null mice developed significant numbers of visible abscesses in organs compared with 3 out of 15 neutropenic WT mice (Table I, P = 0.0003).
Table I.
Abscess Formation in Organs
| Numbers of mice with abscess formation per total mice in each group
|
|||||
|---|---|---|---|---|---|
| Kidney | Liver | Lung | Spleen | Any combination | |
| WT + CY | 1/15 | 3/15 | 1/15 | 0/15 | 3/15 |
| MBL-A KO + CY | 1/7 | 2/7 | 0/7 | 0/7 | 2/7 |
| MBL-C KO + CY | 0/9 | 3/9 | 0/9 | 0/9 | 3/9 |
| MBL-null + CY | 12/29 (0.010)a | 17/29 (0.012) | 9/29 (0.049) | 2/29 | 21/29 (0.0003) |
| WT | 0/8 | 0/8 | 0/8 | 0/8 | 0/8 |
| MBL-A KO | 0/7 | 0/7 | 0/7 | 0/7 | 0/7 |
| MBL-null | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 |
Numbers in parentheses indicate p-values against WT + CY mice group.
The most frequent target organ in MBL-null mice was the liver (17 out of 29) followed by the kidney (12 out of 29) and the lung (9 out of 29), whereas the spleen seemed relatively resistant to later infection (2 out of 29; Table I). There was no statistically significant difference between the neutropenic MBL-A KO, neutropenic MBL-C KO, and neutropenic WT mice, indicating that MBL-A and MBL-C are redundant under these circumstances.
Neutropenic MBL-null Mice Accumulate More Bacteria in Blood and Organs Compared with Neutropenic WT Mice by Day 4 of Infection.
The kinetics of bacterial proliferation after infection in the peritoneal cavity was assayed. In particular, we aimed to test the role of MBL in the complicated interplay between the bacteria and host. For S. aureus infection, this revolves around the ability of the host to adapt and resist the broad array of bacterially derived pathogenicity factors. To this end, neutropenic WT and neutropenic MBL-null mice were inoculated i.p. with biolumi-S. aureus and the resulting infection was followed for 1 h and then daily after inoculation in real time by in vivo bioluminescence imaging. The accumulation of bacteria in the kidney and in the paratracheal lymph nodes was evident by day 2 in neutropenic MBL-null mice compared with no accumulation in neutropenic WT mice (Fig. 6 a). For more definitive proof, bacterial titers in blood and organ were determined to quantitate biolumi-S. aureus. There was no significant difference in bacterial titers in the blood until day 3 after inoculation. However, by day 4 there were two log differences between neutropenic WT mice with 105 CFU/ml and neutropenic MBL-null mice with 2 × 107 CFU/ml (P = 0.037). By day 8 the neutropenic WT mice had sterilized their blood, whereas there was persistent bacteremia in the neutropenic MBL-null mice despite a recovery of circulating neutrophils (unpublished data).
There was a 10–100-fold higher number of S. aureus in the spleens and lungs of neutropenic MBL-null mice compared with neutropenic WT mice, respectively (Fig. 6 b). These bacterial titers in organs were consistent with the abscess formation data presented above.
The Phenotype Is Reversed by Treatment of Neutropenic MBL-null Mice with rhMBL.
We investigated the effect of treating neutropenic MBL-null mice with rhMBL. Neutropenic MBL-null mice that received rhMBL had no detectable collections of biolumi-S. aureus in their organs (Fig. 6 a). Bacterial culture of organs confirmed that rhMBL-treated neutropenic MBL-null mice had 20–100 times less accumulation of bacteria in the spleens and lungs, respectively, compared with untreated neutropenic MBL-null mice (Fig. 6 b).
As expected, the MBL complement pathway was intact in neutropenic WT mice but not in neutropenic MBL-null mice before infection (Fig. 6 c, left). The MBL complement pathway was restored in vivo in neutropenic MBL-null mice that received rhMBL (Fig. 6 c, right). These findings indicate that the reconstitution of the MBL complement pathway directly correlated with a decrease in bacterial accumulation in the tissues.
Decreased Bacterial Phagocytosis by Resident Peritoneal Macrophages in MBL-null Mice.
In this study we observed that neutropenic MBL-null mice are more susceptible to i.p. infection than neutropenic WT mice. CY-treated WT and MBL-null mice, in addition to being neutropenic, had an 80% decrease in circulating monocytes and resident peritoneal macrophages, indicating that the effects of chemotherapy did not affect neutrophils alone. We reasoned that despite the reduction in the number, resident peritoneal macrophages, together with MBL, play a key role in restricting the early infection in the absence of neutrophils. We detected MBL in the peritoneal cavity of WT mice within hours of infection (unpublished data). Resident peritoneal macrophages were harvested 10 min after inoculation of FITC-S. aureus into the peritoneal cavity of WT and MBL-null mice, and phagocytosis as bacterial uptake was analyzed by FACS®. There was a 40% reduction in bacterial phagocytosis by resident peritoneal macrophages from MBL-null mice compared with those from WT mice (Fig. 7). In addition, the bacterial phagocytosis in vitro by peritoneal macrophages from WT mice was 35% less when FITC-S. aureus was opsonized with serum from MBL-null mice compared with that from WT mice.
Figure 7.

Decreased macrophage phagocytosis in MBL-null mice. Phagocytosis was assayed in vivo (a) and in vitro (b) as described in Materials and Methods. Phagocytosis is shown by mean fluorescence intensity for the ingested FITC-S. aureus. Bars indicate mean ± SE. **, P = 0.003; *, P = 0.009.
Discussion
In this study we demonstrate that mice that lack MBL-A and MBL-C (Fig. 1 c) and thereby do not have a functional MBL complement pathway (Fig. 1 d) are highly susceptible to infection with S. aureus. All MBL-null mice died by 48 h after i.v. inoculation of S. aureus (Fig. 2) and there was a corresponding 10–100-fold increase in S. aureus accumulation in blood, liver, spleen, kidney, and lung of MBL-null mice compared with those of WT mice 24 h after inoculation (Fig. 3). The persistent bacteremia led to overwhelming sepsis and the demise of the MBL-null mice. S. aureus was killed in whole blood of WT mice, whereas the bacteria grew in that of MBL-null mice (Fig. 4). In addition, pretreatment of MBL-null mice with rhMBL reversed the susceptibility phenotype. These results suggest that MBL under the conditions of this mouse model has a nonredundant role in restricting the spread of S. aureus from the blood to the tissues.
MBL and MASPs circulate in the serum as complexes (65). Once MBL recognizes a microbial surface, MASP-2 coops the classical complement pathway convertase, which in turn leads to the deposition of the cleaved third complement component C3b (22, 23). The C3b serves as a ligand for complement receptors that are expressed on phagocytes. In addition to the enhancement of opsonophagocytosis, activation of the MBL complement pathway may result in the assembly of the membrane attack complex with the resultant fluid phase lysis of the target microorganism. MBL may also directly opsonize targets for clearance by phagocytic cells that express collectin receptors. A recent study evaluated the relative importance of MBL-dependent clearance mechanisms in vitro as it pertains to S. aureus (25). Importantly, that study revealed that the MBL complement pathway, but not the alternative complement pathway, was required for antibody-independent C3 deposition on S. aureus. In addition, it was observed that C3 opsonization of S. aureus resulted in enhanced uptake of organisms by human neutrophils. Furthermore, MBL alone in the absence of complement was also able to facilitate enhanced uptake of bacteria by phagocytic cells. We attempted to address the relative importance of these three mechanisms in defense against S. aureus. Plasma and serum from WT, MBL-null, and C3-null mice were not effective in restricting the growth of S. aureus (Fig. 4), indicating that S. aureus is resistant to direct attack via MBL, complement, or the combination of MBL and complement. However, phagocytosis of S. aureus in whole blood was highly dependent of MBL (Fig. 4). Taken together, our results indicate that MBL-initiated opsonophagocytosis by both neutrophils and macrophages is an important first line host defense against S. aureus. Our studies did not address the relative role of MBL alone versus MBL-mediated complement activation in mediating opsonophagocytosis. We hope to address this point in part by creating C3 × MBL-null animals. Uptake of S. aureus by phagocytes can also occur via macrophage scavenger receptors in an opsonin-independent manner (66).
The net effect uptake by PMNs results in the triggering of the oxidative burst and the release of peptides and proteins like phospholipase A2 (67), cathelicidins (68), defensins (69), and cathepsin G (70), all of which can directly kill staphylococci. It would be interesting to compare MBL-dependent release of these mediators with MBL-independent release of these effectors as the next step in better understanding of the MBL-dependent killing of S. aureus.
This study demonstrated that MBL not only acts as an opsonin, but also stimulates a proinflammatory response. We noted that there was a muted cytokine response as defined by IL-6 and TNF-α levels in blood of MBL-null animals at 2 h after inoculation (Fig. 5). The failure to contain the infection led to sepsis and death of all the MBL-null mice by 48 h with high levels of IL-6 and TNF-α (Fig. 5) commensurate with an exaggerated host response to bacterial sepsis. These in vivo findings correlate with our in vitro findings in that S. aureus preincubated with serum from MBL-null mice was relatively ineffective in triggering cytokine release via macrophages (unpublished data). Based on past studies (71) we postulate that the release of appropriate amounts of TNF-α stimulates neutrophils and monocytes to possess enhanced killing activity against S. aureus, as this was described in an in vitro study (72). We hypothesized that the relative lack of TNF-α resulted in less than optimal stimulation of neutrophils and together with the failure to activate MBL complement pathway contributed to the enhanced susceptibility of MBL-null mice to infection.
We wished to evaluate the intertwining role of MBL and neutrophils further, and reasoned that local anti-staphylococci defenses would combat a low multiplicity of infection in the first instance in this body compartment. Such defense mechanisms would be supplemented during the acute inflammatory response that would include both neutrophils and MBL. Our findings that MBL-null and WT mice were equally resistant to sublethal challenge of S. aureus (Table I) demonstrate that MBL complement pathway alone is redundant in first line host defense against staphylococci in the peritoneal cavity. This is in contrast to infection in blood where MBL appears to have a nonredundant role. The well-documented anti-staphylococcal action of platelets, phagocytic cells, and their products appear sufficient to compensate for the lack of MBL. However, the increased abscess formation in neutropenic MBL-null mice compared with neutropenic WT mice indicates that MBL, neutrophils, and resident peritoneal macrophages together provide an effective barrier to i.p. infection with S. aureus. It appears that in the absence of neutrophils and MBL, the pathogen obtains a growth advantage and spreads beyond the initial infectious nidus in the peritoneal cavity to the paratracheal lymph nodes (Fig. 6 a). In the neutropenic MBL-null mice systemic bacterial infection ensues within days after the infectious inoculum. Organs are seeded with pathogenic bacteria and abscess formation is observed in 21 out of 29 neutropenic MBL mice versus 3 out of 15 neutropenic WT mice (Table I). The restoration of MBL with exogenous MBL was required to contain the infection both locally and systemically and correlated with the restoration of the MBL complement pathway in vivo (Fig. 6 c). Therefore, MBL fulfills the requirements that might be expected of a molecule involved in innate immunity as it is required to limit an early response to infection in the blood. Additionally, MBL is important in containing the spread of an i.p. infection, at least under the experimental conditions described in this study. Other serum opsonins, like LBP, act in concert with neutrophils and have a selective action against certain Gram-negative pathogens that might be dependent on the route of infection (35). In this regard, LBP-null mice are uniquely susceptible to i.p. but not per oral or i.v. infection with Salmonella typhimurium and S. aureus (35). On the other hand, MBL binds a broader range of bacteria, fungi, certain parasites, and viruses (7). The availability of MBL-null mice will be useful in adjudicating the relative role of MBL in maintaining the balance between the host and other bacterial, viral, and protozoan pathogens.
The establishment of an infection reflects a balance between the virulence of the organism and susceptibility of the individual. We did not examine a wide range of S. aureus clinical isolates in this study but it is clear that S. aureus uses cell-associated products, secreted exotoxins, and regulatory loci to enhance and modify its pathogenicity. Treatment of S. aureus infections has become difficult given the emergence of widespread antibiotic resistance (52). Clinical isolates resistant to methicillin now appear to be resistant to multiple antibiotics with the recent appearance of full resistance to vancomycin (50). This raises the possibility that rhMBL may have an adjuvant role clinically together with antibiotics against antibiotic-resistant staphylococci. The idea that MBL might be an infection susceptibility gene is supported by the earliest studies by Super et al. (14) and Sumiya et al. (15). Since that time, numerous studies have indicated an association of low MBL levels with recurrent infections in both adults and children. In one study a higher proportion of patients with invasive pneumococcal disease were homozygous for MBL variant genes compared with age- and race-matched controls (73). The interdependence of MBL and neutrophils in combating infection also has a clinical corollary. Neth et al. (63) found that children with febrile neutropenia after chemotherapy who had low secretor MBL haplotypes stayed in the hospital an average of 2 d longer than those who were MBL sufficient. Two other studies in adult cancer patients demonstrated an association between low MBL levels and clinically significant events in the setting of febrile neutropenia, whereas one study failed to demonstrate this association (38). The increased susceptibility to infection of patients with cystic fibrosis, chronic granulomatous disease, complement deficiency, common variable immunodeficiency, and antibody subclass deficiency who have low MBL secretor haplotypes provides further credence to the idea that MBL synergies with other modalities of host defense (6). Taken together, these examples illustrate that MBL acts in concert with other modalities of the innate immune system to alter the balance between the host and the pathogen. Our study adds credence to this paradigm but also indicates that if the infection is blood born, MBL has a nonredundant role, at least in the context of S. aureus bacteremia. However, for tissue, infectious susceptibility required not only MBL deficiency, but also a second hit, which in this instance was neutropenia.
It would be interesting to assess the association of MBL variants and complications of S. aureus bacteremia in large cohort human studies. Given the results of this study one might predict that MBL levels might be an additional clinical identifier that is a biological marker of host resistance. Finally, our study provides direct in vivo evidence that MBL does indeed function as an “ante-antibody” in first line host defense in that the MBL together with MBL complement pathway is an important host factor that protects against the bacterial infection.
Acknowledgments
The authors thank members of the Laboratory of Developmental Immunology at the Massachusetts General Hospital for helpful discussion and critical reading of this manuscript. The authors also thank Dr. Jean C. Lee, Channing Laboratory, the Brigham and Women's Hospital, Harvard Medical School and Dr. Kevin P. Francis, Xenogen Corp., for providing S. aureus CP5 and Bioluminescent S. aureus Xen 8.1, respectively. The authors also thank NatImmune A/S and Dr. K Moore, Lipid Metabolism Unit at the Massachusetts General Hospital for providing rhMBL and KO3 vector, respectively. The authors acknowledge the Transgenic Knockout Mouse Core facility for generation of the MBL-C–null mice at the Massachusetts General Hospital, in particular, Dr. En Li, Mrs. Lian Wu, and Mong Lei. We also acknowledge the Department of Molecular Biology for use of the FACSCalibur™ System.
This research is supported by grant NIH RO1AI42788. Conflict of interest: R.A.B. Ezekowitz, S. Thiel, and J.C. Jensenius have a financial interest in NatImmune, which is the company that supplied the rhMBL.
L. Shi and K. Takahashi contributed equally to this work.
Abbreviations used in this paper: CY, cyclophosphamide; LBP, lipopolysaccharide-binding protein; MASP, mannose-binding lectin–associated serine protease; MBL, mannose-binding lectin; rhMBL, recombinant human MBL.
References
- 1.Janeway, C.A. 1989. Approaching the asymptote: evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54:1–13. [DOI] [PubMed] [Google Scholar]
- 2.Ezekowitz, R.A.B., and J. Hoffmann. 1998. Innate immunity: the blossoming of innate immunity (editorial overview). Curr. Opin. Immunol. 10:9–11. [Google Scholar]
- 3.Hoffmann, J.A., F.C. Kafatos, C.A. Janeway, and R.A. Ezekowitz. 1999. Phylogenetic perspectives in innate immunity. Science. 284:1313–1318. [DOI] [PubMed] [Google Scholar]
- 4.Tobias, P.S., and R.J. Ulevitch. 1994. Lipopolysaccharide-binding protein and CD14 in the lipopolysaccharide-dependent activation of cells. Chest. 105:48s–50s. [DOI] [PubMed] [Google Scholar]
- 5.Lu, J., C. Teh, U. Kishore, and K.B. Reid. 2002. Collectins and ficolins: sugar pattern recognition molecules of the mammalian innate immune system. Biochim. Biophys. Acta. 1572:387–400. [DOI] [PubMed] [Google Scholar]
- 6.Holmskov, U., S. Thiel, and J.C. Jensenius. 2003. Collections and ficolins: humoral lectins of the innate immune defense. Annu. Rev. Immunol. 21:547–578. [DOI] [PubMed] [Google Scholar]
- 7.Fraser, I.P., H. Koziel, and R.A. Ezekowitz. 1998. The serum mannose-binding protein and the macrophage mannose receptor are pattern recognition molecules that link innate and adaptive immunity. Semin. Immunol. 10:363–372. [DOI] [PubMed] [Google Scholar]
- 8.Holmskov, U., R. Malhotra, R.B. Sim, and J.C. Jensenius. 1994. Collectins: collagenous C-type lectins of the innate immune defense system. Immunol. Today. 15:67–74. [DOI] [PubMed] [Google Scholar]
- 9.Turner, M.W., and R.M. Hamvas. 2000. Mannose-binding lectin: structure, function, genetics and disease associations. Rev. Immunogenet. 2:305–322. [PubMed] [Google Scholar]
- 10.Epstein, J., Q. Eichbaum, S. Sheriff, and R.A.B. Ezekowitz. 1996. The collectins in innate immunity. Curr. Opin. Immunol. 8:29–35. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda, K., T. Sannoh, N. Kawasaki, T. Kawasaki, and I. Yamashina. 1987. Serum lectin with known structure activates complement through the classical pathway. J. Biol. Chem. 262:7451–7454. [PubMed] [Google Scholar]
- 12.Drickamer, K., M.S. Dordal, and L. Reynolds. 1986. Mannose-binding proteins isolated from rat liver contain carbohydrate-recognition domains linked to collagenous tails. Complete primary structures and homology with pulmonary surfactant apoprotein. J. Biol. Chem. 261:6878–6887. [PubMed] [Google Scholar]
- 13.Kuhlman, M., K. Joiner, and R.A. Ezekowitz. 1989. The human mannose-binding protein functions as an opsonin. J. Exp. Med. 169:1733–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Super, M., S. Thiel, J. Lu, R.J. Levinsky, and M.W. Turner. 1989. Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet. 2:1236–1239. [DOI] [PubMed] [Google Scholar]
- 15.Sumiya, M., M. Super, P. Tabona, R.J. Levinsky, T. Arai, M.W. Turner, J.A. Summerfield, S. Thiel, and J. Lu. 1991. Molecular basis of opsonic defect in immunodeficient children. Lancet. 337:1569–1570. [DOI] [PubMed] [Google Scholar]
- 16.Madsen, H.O., M.L. Satz, B. Hogh, A. Svejgaard, and P. Garred. 1998. Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J. Immunol. 161:3169–3175. [PubMed] [Google Scholar]
- 17.Super, M., S.D. Gillies, S. Foley, K. Sastry, J.E. Schweinle, V.J. Silverman, and R.A. Ezekowitz. 1992. Distinct and overlapping functions of allelic forms of human mannose binding protein. Nat. Genet. 2:50–55. [DOI] [PubMed] [Google Scholar]
- 18.Madsen, H.O., P. Garred, S. Thiel, J.A. Kurtzhals, L.U. Lamm, L.P. Ryder, and A. Svejgaard. 1995. Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J. Immunol. 155:3013–3020. [PubMed] [Google Scholar]
- 19.Ogden, C.A., A. deCathelineau, P.R. Hoffmann, D. Bratton, B. Ghebrehiwet, V.A. Fadok, and P.M. Henson. 2001. C1q and mannose binding lectin engagement of cell surface calreticulin and cd91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 194:781–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheriff, S., C.Y. Chang, and R.A. Ezekowitz. 1994. Human mannose-binding protein carbohydrate recognition domain trimerizes through a triple alpha-helical coiled-coil. Nat. Struct. Biol. 1:789–794. (erratum published 3:103). [DOI] [PubMed] [Google Scholar]
- 21.Weis, W.I., M.E. Taylor, and K. Drickamer. 1998. The C-type lectin superfamily in the immune system. Immunol. Rev. 163:19–34. [DOI] [PubMed] [Google Scholar]
- 22.Matsushita, M., and T. Fujita. 1992. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J. Exp. Med. 176:1497–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiel, S., T. Vorup-Jensen, C.M. Stover, W. Schwaeble, S.B. Laursen, K. Poulsen, A.C. Willis, P. Eggleton, S. Hansen, U. Holmskov, et al. 1997. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 386:506–510. [DOI] [PubMed] [Google Scholar]
- 24.Valdimarsson, H., M. Steffansson, T. Vikingsdottir, G.J. Arason, C. Koch, S. Thiel, and J.C. Jensenius. 1998. Reconstitution of opsonizing activity by infusion of mannan-binding lectin (MBL) to MBL-deficient humans. Scand. J. Immunol. 48:116–123. [DOI] [PubMed] [Google Scholar]
- 25.Neth, O., D.L. Jack, M. Johnson, N.J. Klein, and M.W. Turner. 2002. Enhancement of complement activation and opsonophagocytosis by complexes of mannose-binding lectin with mannose-binding lectin-associated serine protease after binding to Staphylococcus aureus. J. Immunol. 169:4430–4436. [DOI] [PubMed] [Google Scholar]
- 26.Super, M., R.J. Levinsky, M.W. Turner, S. Thiel, and J. Lu. 1990. Association of low levels of mannan-binding protein with a common defect of opsonisation. Clin. Exp. Immunol. 79:144–150. 2311294 [Google Scholar]
- 27.Matsukawa, A., C.M. Hogaboam, N.W. Lukacs, P.M. Lincoln, H.L. Evanoff, and S.L. Kunkel. 2000. Pivotal role of the CC chemokine, macrophage-derived chemokine, in the innate immune response. J. Immunol. 164:5362–5368. [DOI] [PubMed] [Google Scholar]
- 28.Chinnaiyan, A.M., M. Huber-Lang, C. Kumar-Sinha, T.R. Barrette, S. Shankar-Sinha, V.J. Sarma, V.A. Padgaonkar, and P.A. Ward. 2001. Molecular signatures of sepsis: multiorgan gene expression profiles of systemic inflammation. Am. J. Pathol. 159:1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmskov, U.L. 2000. Collectins and collectin receptors in innate immunity. APMIS Suppl. 100:1–59. [PubMed] [Google Scholar]
- 30.Wright, J.R. 1997. Immunomodulatory functions of surfactant. Physiol. Rev. 77:931–962. [DOI] [PubMed] [Google Scholar]
- 31.Wright, S.D., R.A. Ramons, P.S. Tobias, R.J. Ulevitch, and J.C. Mathison. 1990. CD14 serves as the cellular receptor for complexes of lipopolysaccharide with lipopolysaccharide binding protein. Science. 249:1431–1433. [DOI] [PubMed] [Google Scholar]
- 32.Wright, A.E., and S.R. Douglas. 1904. Opsonins. Proc R. Soc. Ser. 73:128–142. [Google Scholar]
- 33.Silverstein, A.M. 2003. Cellular versus humoral immunology: a century-long dispute. Nat. Immunol. 4:425–428. [DOI] [PubMed] [Google Scholar]
- 34.Yang, K.K., B.G. Dorner, U. Merkel, B. Ryffel, C. Schutt, D. Golenbock, M.W. Freeman, R.S. Jack, J. Fierer, M.A. Swancutt, et al. 2002. Neutrophil influx in response to a peritoneal infection with Salmonella is delayed in lipopolysaccharide-binding protein or CD14-deficient mice. J. Immunol. 169:4475–4480. [DOI] [PubMed] [Google Scholar]
- 35.Fierer, J., M.A. Swancutt, D. Heumann, and D. Golenbock. 2002. The role of lipopolysaccharide binding protein in resistance to Salmonella infections in mice. J. Immunol. 168:6396–6403. [DOI] [PubMed] [Google Scholar]
- 36.Garred, P., H.O. Madsen, J.A. Kurtzhals, L.U. Lamm, S. Thiel, A.S. Hey, and A. Svejgaard. 1992. Diallelic polymorphism may explain variations of the blood concentration of mannan-binding protein in Eskimos, but not in black Africans. Eur. J. Immunogenet. 19:403–412. [DOI] [PubMed] [Google Scholar]
- 37.Summerfield, J.A., M. Sumiya, M. Levin, and M.W. Turner. 1997. Mannose-binding protein gene mutations are associated with childhood infection in a consecutive hospital series. BMJ. 314:1229–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterslund, N.A., C. Koch, J.C. Jensenius, and S. Thiel. 2001. Association between deficiency of mannose-binding lectin and severe infections after chemotherapy. Lancet. 358:637–638. [DOI] [PubMed] [Google Scholar]
- 39.Turner, M.W. 1998. Mannose-binding lectin (MBL) in health and disease. Immunobiology. 199:327–339. [DOI] [PubMed] [Google Scholar]
- 40.Ezekowitz, R.A.B. 1991. Ante-antibody immunity. Curr. Biol. 1:60–62. [DOI] [PubMed] [Google Scholar]
- 41.Mogues, T., T. Ota, A.I. Tauber, and K.N. Sastry. 1996. Characterization of two mannose-binding protein cDNAs from rhesus monkey (Macaca mulatta): structure and evolutionary implications. Glycobiology. 6:543–550. [DOI] [PubMed] [Google Scholar]
- 42.Laursen, S.B., T.S. Dalgaard, S. Thiel, B.L. Lim, T.V. Jensen, H.R. Juul-Madsen, A. Takahashi, T. Hamana, M. Kawakami, and J.C. Jensenius. 1998. Cloning and sequencing of a cDNA encoding chicken mannan-binding lectin (MBL) and comparison with mammalian analogues. Immunology. 93:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hansen, S., S. Thiel, A. Willis, U. Holmskov, and J.C. Jensenius. 2000. Purification and characterization of two mannan-binding lectins from mouse serum. J. Immunol. 164:2610–2618. [DOI] [PubMed] [Google Scholar]
- 44.Lee, R.T., Y. Ichikawa, M. Fay, K. Drickamer, M.C. Shao, and Y.C. Lee. 1991. Ligand-binding characteristics of rat serum-type mannose-binding protein (MBP-A). Homology of binding site architecture with mammalian and chicken hepatic lectins. J. Biol. Chem. 266:4810–4815. [PubMed] [Google Scholar]
- 45.Ng, K.K., K. Drickamer, and W.I. Weis. 1996. Structural analysis of monosaccharide recognition by rat liver mannose-binding protein. J. Biol. Chem. 271:663–674. [DOI] [PubMed] [Google Scholar]
- 46.Liu, H., L. Jensen, S. Hansen, S.V. Petersen, K. Takahashi, A.B. Ezekowitz, F.D. Hansen, J.C. Jensenius, and S. Thiel. 2001. Characterization and quantification of mouse mannan-binding lectins (MBL-A and MBL-C) and study of acute phase responses. Scand. J. Immunol. 53:489–497. [DOI] [PubMed] [Google Scholar]
- 47.Lee, S.J., G. Gonzalez-Aseguinolaza, and M.C. Nussenzweig. 2002. Disseminated candidiasis and hepatic malarial infection in mannose-binding-lectin-A-deficient mice. Mol. Cell. Biol. 22:8199–8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi, K., J. Gordon, H. Liu, K. Sastry, J. Epstein, M. Motwani, I. Laursen, S. Thiel, J. Jensenius, M. Carroll, et al. 2002. Lack of mannose-binding lectin-A enhances survival in a mouse model of acute septic peritonitis. Microbes Infect. 4:773–784. [DOI] [PubMed] [Google Scholar]
- 49.Uemura, K., M. Saka, T. Nakagawa, N. Kawasaki, S. Thiel, J.C. Jensenius, and T. Kawasaki. 2002. L-MBP is expressed in epithelial cells of mouse small intestine. J. Immunol. 169:6945–6950. [DOI] [PubMed] [Google Scholar]
- 50.Diekema, D.J., M.A. Pfaller, R.N. Jones, F.J. Schmitz, J. Smayevsky, J. Bell, and M. Beach. 2002. Age-related trends in pathogen frequency and antimicrobial susceptibility of bloodstream isolates in North America: SENTRY Antimicrobial Surveillance Program, 1997-2000. Int. J. Antimicrob. Agents. 20:412–418. [DOI] [PubMed] [Google Scholar]
- 51.Chang, F.Y., J.E. Peacock, Jr., D.M. Musher, P. Triplett, B.B. MacDonald, J.M. Mylotte, A. O'Donnell, M.M. Wagener, and V.L. Yu. 2003. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore). 82:333–339. [DOI] [PubMed] [Google Scholar]
- 52.Melzer, M., S.J. Eykyn, W.R. Gransden, and S. Chinn. 2003. Is methicillin-resistant Staphylococcus aureus more virulent than methicillin-susceptible S. aureus? A comparative cohort study of British patients with nosocomial infection and bacteremia. Clin. Infect. Dis. 37:1453–1460. [DOI] [PubMed] [Google Scholar]
- 53.Fowler, V.G., Jr., M.K. Olsen, G.R. Corey, C.W. Woods, C.H. Cabell, L.B. Reller, A.C. Cheng, T. Dudley, and E.Z. Oddone. 2003. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch. Intern. Med. 163:2066–2072. [DOI] [PubMed] [Google Scholar]
- 54.Sastry, R., J.S. Wang, D.C. Brown, R.A. Ezekowitz, A.I. Tauber, and K.N. Sastry. 1995. Characterization of murine mannose-binding protein genes Mbl1 and Mbl2 reveals features common to other collectin genes. Mamm. Genome. 6:103–110. [DOI] [PubMed] [Google Scholar]
- 55.Petersen, S.V., S. Thiel, L. Jensen, R. Steffensen, and J.C. Jensenius. 2001. An assay for the mannan-binding lectin pathway of complement activation. J. Immunol. Methods. 257:107–116. [DOI] [PubMed] [Google Scholar]
- 56.Lee, J.C., M.J. Betley, C.A. Hopkins, N.E. Perez, and G.B. Pier. 1987. Virulence studies, in mice, of transposon-induced mutants of Staphylococcus aureus differing in capsule size. J. Infect. Dis. 156:741–750. [DOI] [PubMed] [Google Scholar]
- 57.Lee, J.C., J.S. Park, S.E. Shepherd, V. Carey, and A. Fattom. 1997. Protective efficacy of antibodies to the Staphylococcus aureus type 5 capsular polysaccharide in a modified model of endocarditis in rats. Infect. Immun. 65:4146–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Francis, K.P., D. Joh, C. Bellinger-Kawahara, M.J. Hawkinson, T.F. Purchio, and P.R. Contag. 2000. Monitoring bioluminescent Staphylococcus aureus infections in living mice using a novel luxABCDE construct. Infect. Immun. 68:3594–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamblin, M.R., D.A. O'Donnell, N. Murthy, C.H. Contag, and T. Hasan. 2002. Rapid control of wound infections by targeted photodynamic therapy monitored by in vivo bioluminescence imaging. Photochem. Photobiol. 75:51–57. [DOI] [PubMed] [Google Scholar]
- 60.Ramet, M., P. Manfruelli, A. Pearson, B. Mathey-Prevot, and R.A. Ezekowitz. 2002. Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E. coli. Nature. 416:644–648. [DOI] [PubMed] [Google Scholar]
- 61.Cunnion, K.M., H.M. Zhang, and M.M. Frank. 2003. Availability of complement bound to Staphylococcus aureus to interact with membrane complement receptors influences efficiency of phagocytosis. Infect. Immun. 71:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Molne, L., M. Verdrengh, and A. Tarkowski. 2000. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect. Immun. 68:6162–6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neth, O., I. Hann, M.W. Turner, and N.J. Klein. 2001. Deficiency of mannose-binding lectin and burden of infection in children with malignancy: a prospective study. Lancet. 358:614–618. [DOI] [PubMed] [Google Scholar]
- 64.Mullighan, C.G., S. Heatley, K. Doherty, F. Szabo, A. Grigg, T.P. Hughes, A.P. Schwarer, J. Szer, B.D. Tait, L. Bik To, et al. 2002. Mannose-binding lectin gene polymorphisms are associated with major infection following allogeneic hemopoietic stem cell transplantation. Blood. 99:3524–3529. [DOI] [PubMed] [Google Scholar]
- 65.Dahl, M.R., S. Thiel, M. Matsushita, T. Fujita, A.C. Willis, T. Christensen, T. Vorup-Jensen, and J.C. Jensenius. 2001. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 15:127–135. [DOI] [PubMed] [Google Scholar]
- 66.Thomas, C.A., Y. Li, T. Kodama, H. Suzuki, S.C. Silverstein, and J. El Khoury. 2000. Protection from lethal Gram-positive infection by macrophage scavenger receptor–dependent phagocytosis. J. Exp. Med. 191:147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laine, V.J., D.S. Grass, and T.J. Nevalainen. 1999. Protection by group II phospholipase A2 against Staphylococcus aureus. J. Immunol. 162:7402–7408. [PubMed] [Google Scholar]
- 68.Midorikawa, K., K. Ouhara, H. Komatsuzawa, T. Kawai, S. Yamada, T. Fujiwara, K. Yamazaki, K. Sayama, M.A. Taubman, H. Kurihara, et al. 2003. Staphylococcus aureus susceptibility to innate antimicrobial peptides, beta-defensins and CAP18, expressed by human keratinocytes. Infect. Immun. 71:3730–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakajima, Y., J. Ishibashi, F. Yukuhiro, A. Asaoka, D. Taylor, and M. Yamakawa. 2003. Antibacterial activity and mechanism of action of tick defensin against Gram-positive bacteria. Biochim. Biophys. Acta. 1624:125–130. [DOI] [PubMed] [Google Scholar]
- 70.Katzif, S., D. Danavall, S. Bowers, J.T. Balthazar, and W.M. Shafer. 2003. The major cold shock gene, cspA, is involved in the susceptibility of Staphylococcus aureus to an antimicrobial peptide of human cathepsin G. Infect. Immun. 71:4304–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferrante, A., A.J. Martin, E.J. Bates, D.H. Goh, D.P. Harvey, D. Parsons, D.A. Rathjen, G. Russ, and J.M. Dayer. 1993. Killing of Staphylococcus aureus by tumor necrosis factor-alpha-activated neutrophils. The role of serum opsonins, integrin receptors, respiratory burst, and degranulation. J. Immunol. 151:4821–4828. [PubMed] [Google Scholar]
- 72.Bates, E.J., A. Ferrante, and L.J. Beard. 1991. Characterization of the major neutrophil-stimulating activity present in culture medium conditioned by Staphylococcus aureus-stimulated mononuclear leukocytes. Immunology. 72:448–450. [PMC free article] [PubMed] [Google Scholar]
- 73.Roy, S., K. Knox, S. Segal, D. Griffiths, C.E. Moore, K.I. Welsh, A. Smarason, N.P. Day, W.L. McPheat, D.W. Crook, et al. 2002. MBL genotype and risk of invasive pneumococcal disease: a case-control study. Lancet. 359:1569–1573. [DOI] [PubMed] [Google Scholar]