

Abstract
Survivin has received great attention due to its expression in many human tumors and its potential as a therapeutic target in cancer. Survivin expression has been described to be cell cycle–dependent and restricted to the G2-M checkpoint, where it inhibits apoptosis in proliferating cells. In agreement with this current view, we found that survivin expression was high in immature neutrophils, which proliferate during differentiation. In contrast with immature cells, mature neutrophils contained only little or no survivin protein. Strikingly, these cells reexpressed survivin upon granulocyte/macrophage colony-stimulating factor (CSF) or granulocyte CSF stimulation in vitro and under inflammatory conditions in vivo. Moreover, survivin-deficient mature neutrophils were unable to increase their lifespan after survival factor exposure. Together, our findings demonstrate the following: (a) overexpression of survivin occurs in primary, even terminally differentiated cells and is not restricted to proliferating cells; and (b) survivin acts as an inhibitor of apoptosis protein in a cell cycle–independent manner. Therefore, survivin plays distinct and independent roles in the maintenance of the G2-M checkpoint and in apoptosis control, and its overexpression is not restricted to proliferating cells. These data provide new insights into the regulation and function of survivin and have important implications for the pathogenesis, diagnosis, and treatment of inflammatory diseases and cancer.
Keywords: antisense, cancer, cytokines, differentiation, infection
Introduction
Survivin is a member of the inhibitor of apoptosis protein (IAP) family (1). Overexpression of survivin protects cells from death induced by different apoptotic stimuli and, conversely, targeting survivin expression was shown to increase apoptosis and chemosensitivity (2). However, the concept that survivin is solely an antiapoptotic protein has recently been challenged by findings in yeast and Caenorhabditis elegans, where it appears to act as a regulator of cell division (3, 4). Early studies reported that survivin expression occurs at the G2-M boundary of the cell cycle and interference with its function leads to cell division defects, resulting in multinucleated polyploid cells (5). More recent studies have also shown that survivin shuttles between the nucleus and the cytoplasm (6), where it effectively inhibits apoptosis, most likely by binding to second mitochondrial activator of caspase (Smac; reference 7), but not to caspases (8, 9). Altogether, these data provide evidence that survivin acts as a link between the apoptosis machinery and the checkpoints that control mitotic progression. Thus, deregulated expression of survivin has oncogenic potential by enabling cells to overcome the G2-M checkpoint and progress through mitosis, and by providing cells with a mechanism to evade checkpoint-controlled apoptosis.
Independent of its mode of action, survivin has garnered great interest due to its differential pattern of tissue expression. Although fetal tissues abundantly express survivin mRNA and protein, it has been reported to be absent from most normal adult tissues. Interestingly, the majority of human tumors express survivin protein at high levels, suggesting that reactivation of the survivin gene occurs frequently during carcinogenesis and malignant progression (10, 11). If indeed survivin inactivates Smac (7), its overexpression in tumor cells might result in resistance to a broad range of apoptotic stimuli, including chemo- and radiation therapy. Based on these considerations, survivin has been suggested as an attractive target for cancer therapy (12).
In this paper, we demonstrate that survivin is highly expressed in immature neutrophils, whereas in mature neutrophils, survivin expression is absent or only marginal. Strikingly, neutrophil hematopoietic growth factors reactivated the survivin gene in these terminally differentiated cells, which are unable to resume cell cycle progression at G2-M, and high levels of survivin were expressed in mature neutrophils under inflammatory conditions in vivo. Using genetically modified mouse and human neutrophils, we demonstrate that survivin has an exclusive antiapoptotic function in these cells. Moreover, our data question the value of survivin as a specific target for cancer therapy. Instead, survivin may be a new therapeutic target for the development of antiinflammatory drugs.
Materials and Methods
Reagents.
Human GM-CSF was purchased from Novartis Pharma GmbH and human G-CSF was obtained from Aventis Pharma AG. FITC-conjugated anti-CD14 and anti-CD15, allophycocyanin-conjugated anti-CD34 and anti-CD11b, as well as PE-conjugated anti-CD7, anti-CD16, and anti-CD36 mAbs were obtained from Becton Dickinson and BD Biosciences. From the same companies, we received nonconjugated anti-CD15, anti–IAP-1, anti-XIAP, anti–Mcl-1, and anti–caspase-3 mAbs as well as mouse IL-3 and the annexin V apoptosis detection kit. Polyclonal rabbit antisurvivin, anti–IAP-2, and anti-NAIP Abs were ordered from R&D Systems. Anti–β-actin mAb was obtained from Sigma-Aldrich and anti–glyceraldehyde-3–phosphate dehydrogenase (GAPDH) mAb was obtained from Chemicon International, Inc. HRP-conjugated secondary Abs were obtained from Amersham Biosciences. Rhodamine–tetramethyl rhodamine isothiocyanate (TRITC) and FITC-conjugated goat anti–rabbit and donkey anti–mouse secondary Abs were purchased from Jackson ImmunoResearch Laboratories. Anti-PE secondary Ab microbeads were obtained from Miltenyi Biotec. Myeloperoxidase (MPO) staining solution was provided by the pharmacy of the University Hospital Bern. PHA was obtained from Roche Diagnostics and anti-CD3 mAb was obtained from DakoCytomation. All other chemicals were, unless stated otherwise, obtained from Sigma-Aldrich.
The 20-mer first generation antisense oligonucleotide 4003 targeting the survivin mRNA has been described previously (13). The second generation 2′-O-(2-methoxy)-ethyl-modified (2′-MOE) gapmer version of 4003 (as) and its three-base mismatch sequence control (ms) were synthesized using an automated DNA synthesizer as described previously (model 394B; Applied Biosystems; reference 14). Sequences were as follows: antisense survivin, 5′-cscscsasgsCsCsTsTsCsCsAsGsCsTscscststsg-3′; and mismatch survivin, 5′-cscstsasgsCsCsTsTsCsCsAsGsGsTscscstsasg-3′. Small letters refer to 2′-O-(2-methoxy)-ethyl-modified nucleotides, capital letters to DNA, and “s” refers to phosphorothioate linkages.
Cells.
Bone marrow aspirates were obtained from cancer patients at staging or restaging and analyzed by an experienced hematologist. Only samples with normal cellular morphology and distribution were considered in this work. The patients did not receive any chemotherapeutic or immunosuppressive treatment at the time of investigation. To induce sedimentation of the erythrocytes, an equal volume of 2% dextran T-500 (Amersham Biosciences) in 0.9% NaCl was added to the bone marrow aspirates. The bone marrow leukocytes were layered on top of a two-step discontinuous Percoll density gradient and centrifuged at 1,000 g at 4°C for 20 min through which a mixed population of immature neutrophils was obtained (15). Because this population of cells also contained erythroid and lymphoid precursors, we developed a new simple technique to improve the purity of the immature neutrophil population based on negative selection. The cells were incubated with a combination of anti-CD7 and anti-CD36 mAbs and subsequently with secondary Ab microbeads. The labeled cells were depleted by passing them through a magnetic cell separation system (Miltenyi Biotec) with LS+/VS+ column in the field of a permanent magnet. The resulting cell population contained >97% cells of the neutrophil lineage as determined by MPO staining (16), analysis of lineage-associated surface proteins (17), as well as by staining with Diff-Quik (Medion GmbH) and light microscopy.
Peripheral blood neutrophils were purified from healthy normal individuals or patients suffering from cystic fibrosis (CF) and associated Pseudomonas aeruginosa infection by Ficoll-Hypaque centrifugation (18, 19). The resulting cell populations contained >95% neutrophils. Written consent was obtained from all patients and control individuals who donated bone marrow aspirates and blood, respectively. The study was approved by the local ethics committee.
Peripheral blood neutrophils from heterozygous survivin+/− and normal survivin+/+ mice (20) were isolated by centrifugation over a Histopaque 1119 and Histopaque 1077 gradient (Sigma-Aldrich) using a standard protocol (21) 24 h after i.p. administration of 30 μg LPS. The purity of the resulting mouse neutrophil population was between 82 and 85%. Blood samples of two mice were pooled to obtain sufficient numbers of neutrophils to set up one ex vivo viability experiment.
Characterization of Purified Bone Marrow Neutrophils.
After purification, cells were stained with saturated concentrations of fluorescence-labeled anti-CD7, anti-CD11b, anti-CD14, anti-CD15, anti-CD16, anti-CD34, anti-CD36, and appropriate control mAbs according to standard protocols for flow cytometric analysis (FACS Calibur™; Becton Dickinson). Based on the expression of lineage-associated surface proteins and light scattering properties, bone marrow neutrophils were assigned to six distinct maturational stages (17). The distribution of the different maturation stages was quite similar among the 17 normal bone marrow populations used in this work (see Table I). The percentage of segmented mature neutrophils was usually <10%. The purified mixed neutrophilic population was scarcely contaminated (<3%) with CD34+ progenitor cells, CD7+ lymphoid cells, CD36+ erythroid cells, and CD14+ monocytes.
Table I.
Relative Cellular Distribution of Purified Bone Marrow Neutrophils
| No. | CD15low
CD16− CD11b− (myeloblasts)a |
CD15+
CD16− CD11b− (promyelocytes)a |
CD15+
CD16− CD11blow (early myelocytes)a |
CD15+
CD16− CD11b+ (myelocytes)a |
CD15+
CD16low CD11b+ (metamyelocytes and band cells)a |
CD15+
CD16+ CD11b+ (mature neutrophils)a |
|---|---|---|---|---|---|---|
| % | % | % | % | % | % | |
| 1 | 5 | 10 | 43 | 22 | 14 | 6 |
| 2 | 28 | 9 | 20 | 5 | 35 | 3 |
| 3 | 11 | 18 | 32 | 6 | 30 | 3 |
| 4 | 21 | 21 | 42 | 2 | 11 | 3 |
| 5 | 17 | 4 | 7 | 2 | 53 | 17 |
| 6 | 7 | 4 | 25 | 23 | 32 | 9 |
| 7 | 9 | 21 | 38 | 18 | 12 | 2 |
| 8 | 9 | 6 | 48 | 14 | 20 | 3 |
| 9 | 15 | 13 | 12 | 2 | 43 | 15 |
| 10 | 5 | 6 | 22 | 4 | 34 | 29 |
| 11 | 19 | 11 | 26 | 10 | 23 | 11 |
| 12 | 26 | 27 | 25 | 1 | 20 | 1 |
| 13 | 16 | 12 | 26 | 5 | 32 | 9 |
| 14 | 12 | 3 | 15 | 3 | 59 | 8 |
| 15 | 24 | 2 | 21 | 11 | 39 | 3 |
| 16 | 7 | 4 | 23 | 13 | 47 | 6 |
| 17 | 13 | 1 | 10 | 16 | 45 | 15 |
| mean (SEM) | 14.4 (1.8) | 10.1 (1.9) | 25.6 (2.9) | 9.2 (1.8) | 32.3 (3.5) | 8.4 (1.8) |
The majority of the cells represent the indicated cell type. However, some overlap between different maturation stages can occur. For instance, some early myelocytes are also present within the subpopulation indicated as “promyelocytes,” and metamyelocytes are usually seen in the subpopulation defined as “myelocytes.”
Cell Cultures.
Human immature and mature neutrophils were cultured at 106/ml in complete culture medium (RPMI 1640 containing 10% FCS) and, where indicated, treated with 50 ng/ml GM-CSF, 25 ng/ml G-CSF, or 500 ng/ml IL-3. Survivin antisense oligonucleotides (as) or survivin mismatch control oligonucleotides (ms) were used at the indicated concentrations. When applied in combination with cytokines, the oligonucleotides were given 2 h before addition of cytokines.
Determination of Cell Death and Apoptosis.
Cell death was assessed at the indicated times by uptake of 1 μM ethidium bromide and flow cytometric analysis (FACS Calibur™; references 21, 22). To determine whether cell death was due to apoptosis, redistribution of phosphatidylserine in the absence of propidium iodide uptake was measured by flow cytometry (22). Neutrophil apoptosis was also assessed by oligonucleosomal DNA fragmentation (23, 24).
Immunoblotting.
Gel electrophoresis and immunoblotting were performed as described previously (24). In brief, after electrotransfer of the separated proteins, the filters were incubated overnight with antisurvivin (1/1,000), anti-IAP-2 (1/666), anti-NAIP (1/1,000), anti-XIAP (1/250), anti–IAP-1 (1/333), anti–Mcl-1 (1/1,000), or anti–caspase-3 (1/1,000) Abs at 4°C in TBS/0.1% Tween 20/3% nonfat dry milk. The final concentrations of the primary Abs ranged between 1 and 3 μg/ml. 20 ng of purified full-length recombinant human survivin (R&D Systems) was used as positive control for antisurvivin immunoblotting. For loading controls, stripped filters were incubated with anti–β-actin (1/10,000) or anti-GAPDH (1/2,000) mAbs. Filters were washed in TBS/0.1% Tween 20 for 30 min and incubated with the appropriate HRP-conjugated secondary Ab (Amersham Biosciences) in TBS/0.1% Tween 20/5% nonfat dry milk for 1 h. Filters were developed by an ECL technique (ECL-Kit; Amersham Biosciences) according to the manufacturer's instructions.
Densitometry Analysis.
In some experiments, protein expression levels were analyzed by densitometry (24). OD of survivin, XIAP, and Mcl-1 bands divided by the OD of the corresponding β-actin band are expressed as percentage of OD (protein of interest)/OD (β-actin) of freshly isolated neutrophils that was defined as 100%.
Real-Time PCR.
107 neutrophils were washed with PBS, and total cellular RNA was isolated according to the RNeasy protocol (QIAGEN), which includes DNase digestion. RNA was reverse transcribed with the first strand cDNA synthesis kit (Amersham Biosciences) by using random hexanucleotides according to the manufacturer's instructions. Specific real-time monitoring of PCR amplification of survivin cDNA was performed with a 1/100 dilution of neutrophil cDNA using the S2 primers and probe as described previously (13). Quantification of survivin mRNA levels was performed using ribosomal RNA as internal standard for all samples. For comparison, survivin expression in A549 lung cancer cells was quantified and taken as a 100% reference value.
Enzymatic Caspase Assay.
2.5 × 106 neutrophils were cultured in the presence or absence of 50 ng/ml GM-CSF, 25 ng/ml G-CSF, antisense, or mismatch survivin oligonucleotides at the indicated concentrations for 10 h, washed with cold PBS, and subsequently lysed in 50 μl of cell lysis buffer (50 mM Hepes, pH 7.4/0.1% Chaps/5 mM DTT/0.1 mM EDTA) using a Teflon glass homogenizer (VWR International) on ice for 10 min. After a 10-min centrifugation step at 10,000 g at 4°C, an aliquot of the supernatant was saved for caspase-3 immunoblotting, and caspase-3–like activity was measured in 10 μl supernatants as enzymatic conversion of the colorimetric substrate Ac-DEVD-pNA at 405 nm according to the manufacturer's instructions (QuantiZyme caspase-3 cellular activity assay kit; BIOMOL Research Laboratories, Inc.).
Confocal Laser Scanning Microscopy.
106 cells/ml cytospins were made from freshly purified cells on noncoated slides. Cells were fixed in 4% paraformaldehyde at room temperature for 10 min and washed three times in PBS, pH 7.4. Permeabilization of cells was performed with 0.05% saponin in buffer A (3% BSA in PBS) at room temperature for 5 min and with acetone at −20°C for 15 min. To prevent nonspecific binding, slides were incubated in blocking buffer (25% human immunoglobulins, 25% normal goat serum, 25% normal donkey serum, and 25% BSA) at room temperature for 1 h. Indirect immunostainings of CD15, survivin, and caspase-3 were performed at room temperature for 1 h by using the following primary Abs: anti-CD15 mAb (1/20; diluted in buffer A), rabbit antisurvivin polyclonal Ab (1/50), and anti–caspase-3 mAb (1/50). Mouse and rabbit control antibodies, respectively, were used at the same concentrations in each experiment.
Immunofluorescent stainings were also performed on 5-μm-thick paraformaldehyde-fixed paraffin-embedded tissue sections from CF, appendicitis, and ulcerative colitis patients. Slides were dried at 52°C for 2 h and deparaffinized using NeoClear Solution (Merck), ethanol (100, 90, 80, 60, and 40%), and water at room temperature. After microwave treatment in TE-buffer (10 mM Tris, pH 8.0, and 1 mM EDTA), slides were washed in water, blocked, and stained with primary Ab as aforementioned.
After incubation with primary Ab, cells and tissues, respectively, were incubated with appropriate TRITC- and FITC-conjugated secondary Abs (1/100) in the dark at room temperature for 1 h. The antifading agent Mowiol (Calbiochem) was added. Slides were covered by coverslips and analyzed by confocal laser scanning microscopy (LSM models 410 and 510; Carl Zeiss MicroImaging, Inc.) equipped with Ar and HeNe lasers.
Immunohistochemistry.
Aside from immunofluorescence analysis, survivin expression on tissue neutrophils was also analyzed by regular immunohistochemistry on paraffin-embedded tissue sections from appendicitis patients according to previously established protocols with slight modifications (25, 26). In brief, sections were deparaffinized, and a subsequent antigen retrieval treatment was performed. Staining with antisurvivin (1/50) was performed using EnVision+System peroxidase kit (AEC; DakoCytomation). A rabbit control Ab was used at the same concentration as a negative control. Binding of the primary Ab was detected by a goat anti–rabbit Ab that was available in the kit. Sections were slightly counterstained with hematoxylin, mounted, and examined under a microscope (Axiovert model 35; Carl Zeiss MicroImaging, Inc.) at original magnifications (400 and 1,000).
Proliferation Assays.
Proliferation of mature and immature neutrophils (106/ml) was analyzed in the presence and absence of GM-CSF and G-CSF, respectively. PBMCs were used as controls. PBMCs were stimulated with 10 μg/ml PHA and 5 μg/ml anti-CD3 mAb, respectively. Total culture times were 48 h (mature neutrophils and PBMCs) and 72 h (immature neutrophils). Pulsing the cells with 1 μCi/ml (methyl-[3H]thymidine; Hartmann Analytic) was performed for 16 h and its incorporation was measured by using a liquid scintillation counter (Wallac ADL).
Statistical Analysis.
An analysis of variance test and Student's t test were used to compare mean levels. P < 0.05 was considered statistically significant. Mean levels are presented together with SEM. In the antisense oligonucleotide experiments, antisense and mismatch survivin-treated cells were compared at the indicated concentrations.
Results
Spontaneous Apoptosis Is Largely Delayed in Immature Compared with Mature Neutrophils.
Purification of immature neutrophils from morphologically normal human bone marrow aspirates yielded a pure population of CD34− CD36−CD7−, but also MPO positive cells (Fig. 1, >97%). However, the cell population contained a mix of neutrophils representing different maturation stages (Table I) . When culturing these mostly immature cells, we observed that they did not rapidly undergo cell death (Fig. 2 A) and apoptosis (Fig. 2 B) as seen in mature neutrophils. Because caspase-3 has been described as a key effector caspase in neutrophil apoptosis (27–29), we measured caspase-3 cleavage by immunoblotting. Both freshly purified immature and mature neutrophils contained significant amounts of procaspase-3 (Fig. 2 C). However, the active 17-kD fragment was not detectable in the immature population. Culturing the mature cells was associated with a partial cleavage of procaspase-3 and in the appearance of the p17 fragment usually after 4–8 h of culture. At later time points, cleavage of procaspase-3 continued until the molecule was no longer detectable. In contrast, culturing of immature cells up to 72 h did not result in cleavage of procaspase-3 (Fig. 2 C). Together, these data suggest that immature neutrophils have much lower caspase-3 activity compared with mature neutrophils, resulting in prolonged in vitro survival.
Figure 1.

Purification of immature bone marrow neutrophils. (A) After a Percoll gradient isolation, neutrophils were negatively selected. The resulting neutrophil populations were CD7 and CD36 negative. The figure also demonstrates the positively selected cells (right) that were analyzed to further control the purification process. (B) The isolated CD7−CD34−CD36− cells were MPO positive (>97%) and contained a mix of mostly immature neutrophils at different maturation stages as assessed by immunophenotyping and morphology (see Table I).
Figure 2.

Delayed spontaneous apoptosis in immature bone marrow neutrophils compared with mature blood neutrophils. (A) Viability assay in the absence of survival and death factors. Each value represents mean ± SEM calculated from 3–16 (immature cells) and 4–22 (mature cells) independent experiments, respectively. (B) Phosphatidylserine redistribution (left) and DNA fragmentation (right) assays. Mature cells (top) were analyzed after 20-h cultures and immature cells (bottom) were analyzed after 72-h cultures. The results are representative of three independent experiments performed in both cell populations. (C) Caspase-3 is rapidly processed in mature, but not in immature, neutrophils. In mature cells, a 20-kD fragment is usually seen in neutrophils cultured for 4 h. The enzymatically active 17-kD fragment is regularly detected in 8-h neutrophil cultures. In immature cells, small amounts of the 17-kD fragment are frequently seen, suggesting baseline caspase-3 activity in these cells in the absence of apoptosis. Filters were reprobed with an anti-GAPDH or anti–β-actin mAb to ensure equal loading of the gels.
Survivin Expression Is Lost During Neutrophil Differentiation.
The antiapoptotic effects of members of the IAP family have often been associated with decreased enzymatic caspase activities (1, 2). Because caspase-3 did not demonstrate evidence for high activity in immature neutrophils, we hypothesized that specific IAPs are involved in the inhibition of nascent active caspases in these cells. Expression of IAPs was investigated by immunoblotting (Fig. 3 A). XIAP levels were increased in two out of three investigated immature neutrophil populations compared with mature blood neutrophils. However, the most impressive difference was seen in the levels of survivin, which were dramatically increased in immature compared with mature cells. In contrast, levels of IAP-1, IAP-2, and NAIP did not show consistent differences between the two cell populations. Immunofluorescence and laser scan microscopic analysis confirmed that survivin was highly expressed in immature neutrophils, but not in mature neutrophils from normal individuals, and was detectable both in the nucleus and the cytoplasm (Fig. 3 B). Interestingly, caspase-3 was not expressed at early stages of neutrophil maturation (myeloblast; Fig. 3 B, arrows).
Figure 3.
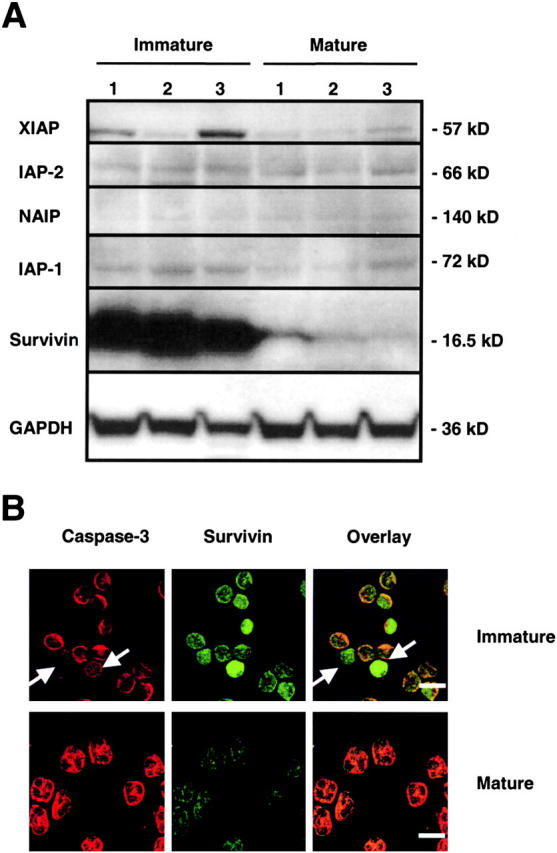
Expression of IAP family members in freshly purified immature and mature neutrophils. (A) Immunoblotting. Mature neutrophils expressed detectable levels of XIAP, IAP-1, IAP-2, and survivin. NAIP was often not detectable. Immature neutrophils expressed large amounts of survivin. Filters were reprobed with anti-GAPDH or anti–β-actin mAbs to ensure equal loading of the gels. For both immature and mature neutrophil populations, results from three different donors are shown (1–3). (B) Confocal microscopy. Survivin was readily detected in immature, but not in mature, neutrophils. Survivin was localized in both nucleus and cytoplasm of immature cells. Caspase-3 was expressed in the cytoplasm of both immature and mature neutrophils. Interestingly, myeloblasts (white arrows) did not express detectable levels of caspase-3. Bars, 10 μm. The results are representative of five independent experiments.
Survivin Expression Is Reinduced in Inflammation.
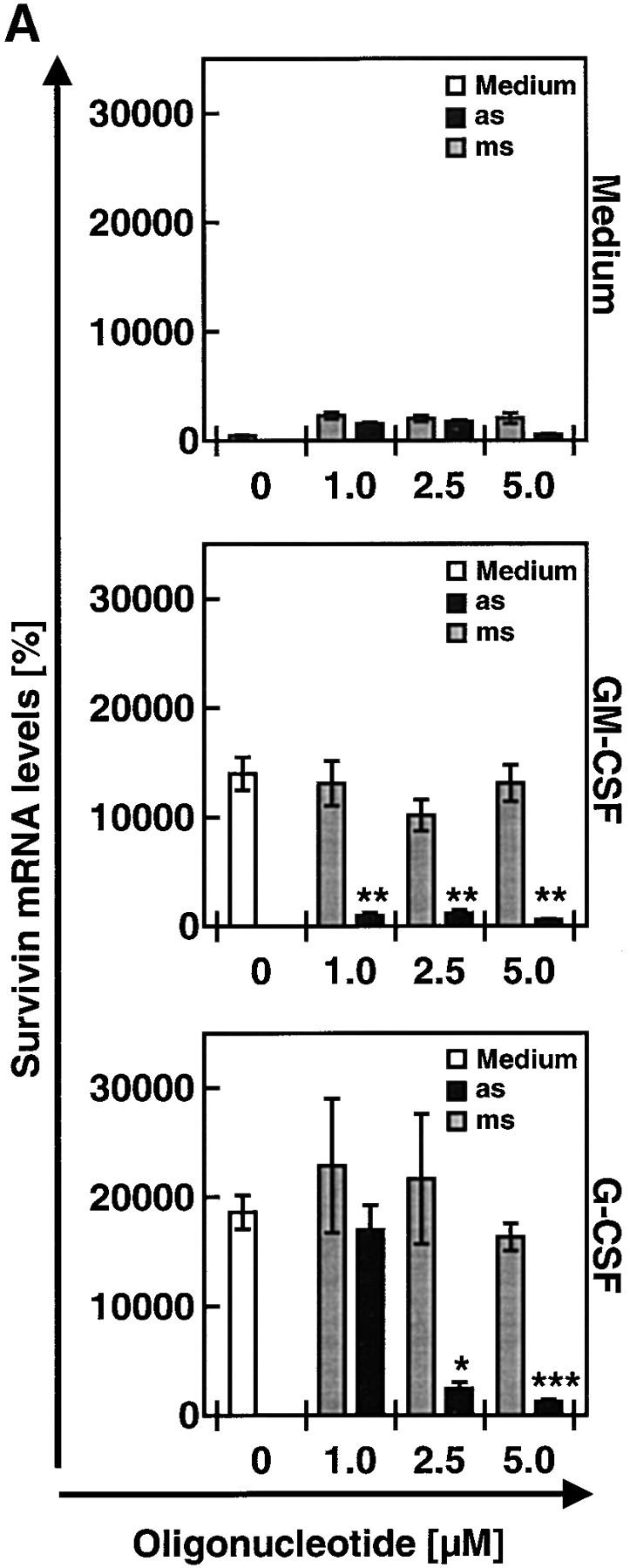
During maturation, neutrophils are exposed to high levels of cytokines in the bone marrow. Some of these cytokines, such as GM-CSF and G-CSF, are also elevated in bacterial infectious diseases (19). Interestingly, mature neutrophils derived from the blood of patients suffering from CF exhibited significantly higher levels of survivin compared with normal blood neutrophils as assessed by immunoblotting. Moreover, GM-CSF and G-CSF increased survivin expression of normal neutrophils upon in vitro stimulation for 12 h (Fig. 4 A). Elevated survivin protein levels correlated with increased mRNA expression as assessed by real-time PCR analysis. The mean mRNA levels in peripheral blood neutrophils from patients with CF were increased ∼10-fold compared with control cells. Moreover, GM-CSF and G-CSF dramatically increased survivin mRNA expression in normal mature neutrophils within 4 h (Fig. 4 B). Both cytokine-stimulated normal neutrophils and CF neutrophils did not demonstrate any evidence for cell proliferation as assessed by [3H]thymidine incorporation assays (Fig. 4 C) and DNA analysis (not depicted). In contrast with mature neutrophils, immature neutrophils demonstrated significant baseline proliferation and proliferation was inducible by both GM-CSF and G-CSF.
Figure 4.

Survivin levels are markedly increased in mature neutrophils exposed to survival factors in vitro and in neutrophils from patients with CF. (A) Immunoblotting. Levels of survivin in neutrophils were increased by culturing normal control neutrophils in the presence of GM-CSF or G-CSF for 12 h. Moreover, freshly purified neutrophils from CF patients (n = 4) showed strongly increased survivin protein levels compared with control blood neutrophils. 20 ng purified recombinant survivin served as a positive control. The filter was reprobed with an anti–β-actin mAb to ensure equal loading of the gel. (B) Real-time PCR. The lung cancer cell line A549 served as a standard (100%). Neutrophils from patients with CF contained increased amounts of survivin mRNA compared with normal neutrophils (left). The increase in survivin mRNA was mimicked by addition of GM-CSF or G-CSF to normal control neutrophils in vitro (right). Values are means ± SEM of three independent experiments. *, P < 0.05; ***, P < 0.001. (C) Proliferation assay. Freshly purified blood neutrophils from normal donors or CF patients did not demonstrate evidence for cell proliferation. The same was true for G-CSF– or GM-CSF–stimulated normal mature neutrophils. In contrast, immature neutrophils demonstrated significant proliferation, which was inducible by both GM-CSF and G-CSF. Pan–T cell–stimulated PBMCs were used as positive controls. Results of four independent experiments are shown for each cell population.
To demonstrate survivin expression in neutrophils under in vivo conditions, we analyzed neutrophils in tissue sections of patients with three distinct inflammatory diseases. As assessed by immunohistochemistry, most neutrophils in the affected intestinal mucosa from patients with acute appendicitis stained positively using an antisurvivin antibody (Fig. 5 A). To more specifically measure protein expression in neutrophils, we also performed double immunofluorescence analysis. Infiltrating neutrophils were identified using an anti-CD15 mAb. In the lungs of CF patients, practically all neutrophils expressed survivin. In the intestine of ulcerative colitis patients, we counted ∼90% survivin-expressing neutrophils. In appendicitis, ∼75% of the infiltrating neutrophils were survivin positive (Fig. 5 B). These data suggest that high levels of survivin are not restricted to immature neutrophils and that mature neutrophils are able to reinduce high levels of survivin expression upon GM-CSF and/or G-CSF exposure under in vitro and in vivo conditions.
Figure 5.
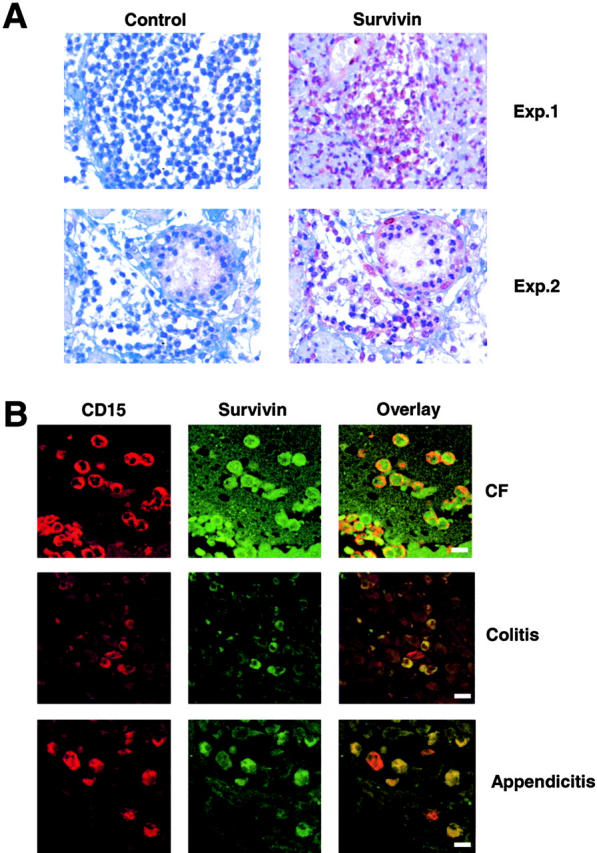
Survivin expression in mature neutrophils under in vivo inflammatory conditions. (A) Immunohistochemistry. Cells morphologically representing neutrophils expressed survivin in tissue sections from patients with appendicitis. A control antibody did not stain the cells. (B). Confocal microscopy. Tissue neutrophils from patients with CF (n = 3), ulcerative colitis (n = 3), and appendicitis (n = 3) demonstrated evidence for survivin expression. Neutrophils were specifically detected using an anti-CD15 mAb. Note that in ulcerative colitis and appendicitis, some neutrophils were survivin negative (red cells in the overlay panel). Bars, 10 μm.
Elevated levels of survivin in peripheral blood neutrophils from patients with CF were confirmed by immunofluorescence and laser scan microscopy demonstrating that almost all neutrophils expressed survivin, predominantly in the cytoplasm (Fig. 6 A). Increased survivin levels in CF neutrophils correlated with decreased caspase-3–like DEVDase in 10-h neutrophil cultures (Fig. 6 B). Moreover, caspase-3–like DEVDase activity was much less in neutrophils stimulated with G-CSF or GM-CSF in vitro.
Figure 6.
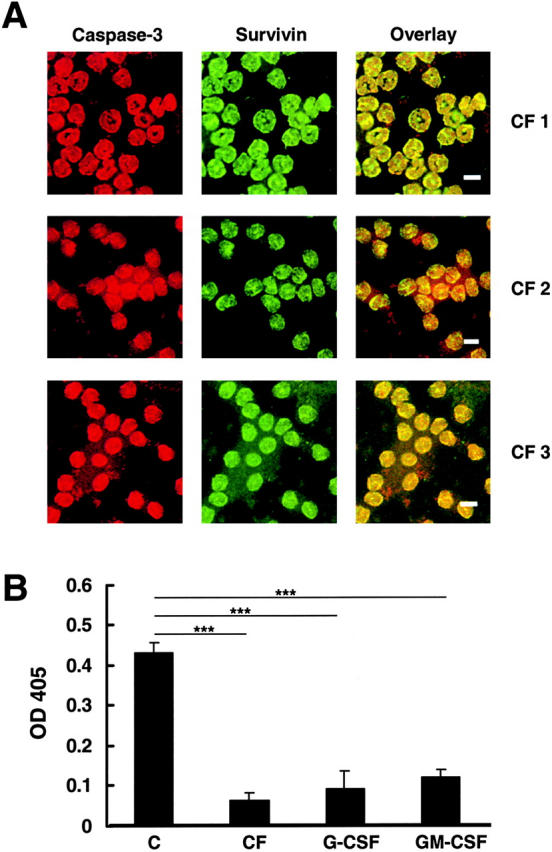
Survivin is predominantly localized in the cytoplasm of mature CF neutrophils and associated with low caspase-3 enzymatic activity. (A) Confocal microscopy. Survivin was readily detected in CF blood neutrophils where it is mostly localized in the cytoplasm. Bars, 10 μm. Three independent experiments are presented. (B) Caspase-3 activity assay. Neutrophils were analyzed after a 10-h culture period. Caspase-3 activity was much lower in CF compared with control neutrophils. In addition, when normal neutrophils were cultured in the presence of GM-CSF or G-CSF, significantly lower caspase-3 activity levels were measured. Values are means ± SEM of eleven (C), three (CF), four (G-CSF), and two (GM-CSF) independent experiments. ***, P < 0.001.
Survivin Delays Apoptosis in a Cell Cycle–independent Manner. The role of survivin as an antiapoptotic protein is still a matter of debate. Some authors concluded that survivin is less important for apoptosis regulation (30), whereas others suggested a significant role of this molecule in protecting cells from apoptosis in a cell cycle–dependent manner (3, 4). Mature neutrophils are terminally differentiated cells unable to proliferate (Fig. 4 C). Thus, this system not only represents an ideal tool to answer the question whether increased survivin levels prevent or reduce apoptosis, it also provides the unique possibility to test whether survivin exhibits antiapoptotic activity in a cell cycle–independent manner and beyond the G2-M checkpoint (5). The role of survivin in the apoptosis regulation of mature neutrophils was characterized by preventing its cytokine-mediated expression. Specific antisense but not mismatch oligonucleotides blocked the production of high levels of survivin mRNA (Fig. 7 A) and protein (Fig. 7 B) upon GM-CSF or G-CSF stimulation. Although antisense oligonucleotides partially inhibited GM-CSF–induced survivin protein expression (Fig. 7 B, top), the effects of G-CSF were completely blocked in this cell culture system at a dose of 5.0 μM (Fig. 7 B, bottom). To further prove the specificity of the survivin antisense oligonucleotides, we also analyzed XIAP and Mcl-1 protein levels. Both proteins were selected because of their potential antiapoptotic activities in neutrophils (31, 32). Although Mcl-1 levels were slightly affected at 5.0 μM antisense, no significant effects on the expression levels of these two control proteins were observed (Fig. 7 B, bottom). This suggests that reduction of survivin levels was an antisense-specific effect and not due to off-target effects mediated by nonantisense related toxicity of the oligonucleotides.
Figure 7.
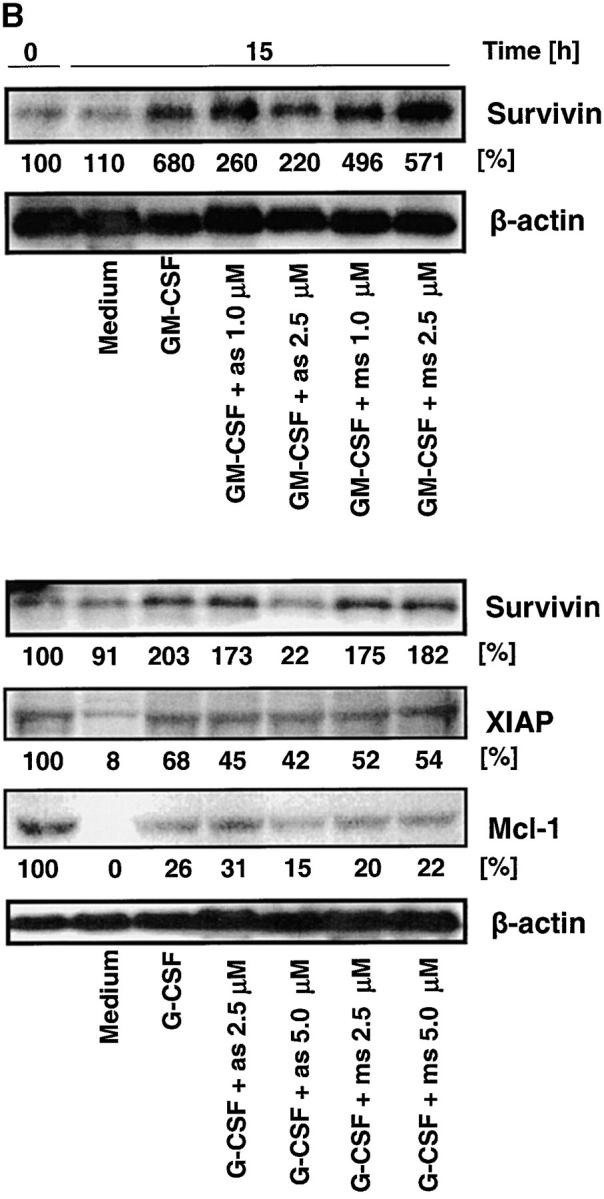

Antisense oligonucleotide treatment specifically prevents increases in survivin gene expression and antiapoptosis mediated by GM-CSF or G-CSF in mature neutrophils. (A) Real-time PCR. The lung cancer cell line A549 served as a standard (100%). Oligonucleotides (as and ms) had no effect on survivin mRNA expression in the absence of survival cytokines after a 4-h transfection period (top). Survivin-antisense (as), in contrast with mismatch control oligonucleotides (ms), prevented increases in survivin mRNA expression upon GM-CSF (middle) or G-CSF (bottom) stimulation in a dose-dependent manner. Values are means ± SEM of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) Immunoblotting. Protein expression of survivin in normal neutrophils was not affected by incubation with as or ms (not depicted). However, survivin-as, in contrast with ms, partially (GM-CSF) or completely (G-CSF) prevented cytokine-induced increases in survivin expression. To demonstrate the specificity of the survivin-as effects, we also measured XIAP and Mcl-1 levels in the experiments using G-CSF. The filters were reprobed with an anti–β-actin mAb to ensure equal loading of the gels, and results of the densitometry analysis in terms of percentage are presented below the immunoblots. Results are representative of three independent experiments. (C) DNA fragmentation assay. Targeting survivin expression by survivin-as increased spontaneous apoptosis (left). GM-CSF (middle) and G-CSF (right) prevented apoptosis, and survivin-as blocked cytokine-mediated survival in a dose-dependent manner. Neutrophils were cultured for 10 h. Values are means ± SEM of four independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D) Viability assay. Neutrophils from survivin+/− mice demonstrated reduced IL-3–mediated survival. Neutrophils were isolated from blood and cultured in the presence of IL-3 for 24 h before viability was measured. No difference in spontaneous cell death was noticed between survivin+/+ and survivin+/− mice (not depicted). Values are means ± SEM of four independent experiments. *, P < 0.05.
Survivin antisense but not mismatch oligonucleotides significantly increased spontaneous apoptosis of mature neutrophils in a dose-dependent manner as assessed by the formation of oligonucleosomal DNA fragments (Fig. 7 C). At very high concentrations (≥10 μM), mismatch oligonucleotide treatment was also associated with additional DNA fragmentation, indicating nonspecific toxicity. GM-CSF and G-CSF–treated neutrophils showed a higher content of normal diploid DNA compared with untreated cells, indicating less apoptosis. Survivin antisense oligonucleotides blocked GM-CSF and G-CSF–mediated antiapoptotic effects in a statistically significant and dose-dependent manner. Compared with the nonspecific effect of the mismatch control oligonucleotides and in agreement with the protein expression data (Fig. 7 B), antisense concentrations of 2.5 μM (for GM-CSF) and 5.0 μM (for G-CSF) were found to be optimal for neutralizing the effects of the survival factors.
To confirm the data obtained by lowering survivin protein expression using antisense oligonucleotides, we purified blood neutrophils from survivin+/− mice (20). These mice express ∼50% of normal survivin levels and demonstrated evidence for increased caspase activation in hepatocytes. Purified neutrophils from survivin+/− mice did not exhibit an accelerated cell death in vitro when compared with normal neutrophils. However, the IL-3–mediated antideath effect, observed in normal neutrophils, was significantly reduced in the genetically modified mice (Fig. 7 D).
We also monitored caspase-3 activation as a consequence of survival factor and/or survivin antisense treatment. Freshly isolated neutrophils contained procaspase-3, but no active 17-kD fragment (Fig. 8 A). Culturing of cells for 10 h resulted in decreased amounts of procaspase-3 and in the appearance of the 17-kD form. Antisense oligonucleotide treatment had no additional effects. GM-CSF delayed the proteolysis of procaspase-3. Similar effects were seen using G-CSF (reference 33 and unpublished data). This effect of the survival factors on procaspase-3 processing was completely abrogated by optimal concentrations of antisense but not mismatch oligonucleotides. Furthermore, caspase-3–like DEVDase activity was suppressed by G-CSF and GM-CSF in 10-h cultures, and this was also abolished by antisense but not mismatch oligonucleotides (Fig. 8 B). Together, to counteract G-CSF–mediated protection of procaspase-3 and inhibition of caspase-3 activity, 5.0 μM antisense oligonucleotides were required, whereas 2.5 μM were sufficient to abolish the GM-CSF effect. These data correlated well with the levels of survivin protein expression (Fig. 7 B) and the induction of apoptosis (Fig. 7 C).
Figure 8.

Survivin-antisense treatment abolishes the inhibitory effect of survival cytokines on caspase-3 activation in mature neutrophils. (A) Immunoblotting. Neutrophils cultured in the presence of GM-CSF for 10 h maintained greater amounts of the caspase-3 proform and had decreased levels of the 17-kD fragment compared with untreated or oligonucleotide-treated cells. Survivin-antisense treatment (as), but not mismatch control oligonucleotides (ms), increased caspase-3 processing in the presence of GM-CSF in a dose-dependent manner. The same results were obtained in two additional experiments. (B) Caspase-3 activity assay. Increased enzymatic activity was detectable in neutrophils undergoing spontaneous apoptosis compared with GM-CSF– or G-CSF–treated cells. Survivin-as but not ms increased caspase-3–like enzymatic activity in the presence of GM-CSF or G-CSF in a dose-dependent manner. Results of two independent experiments are shown (circles, experiment 1; triangles, experiment 2).
Discussion
In contrast with earlier studies that propagated survivin as a tumor-specific target, recent work has unveiled survivin expression also in normal adult tissues, including the proliferating fraction of CD34+ cells in the bone marrow (34). In this work, we investigated the role of survivin as a regulator of apoptosis signaling in neutrophils and report the following new findings. First, immature neutrophils express large amounts of survivin protein compared with terminally differentiated cells, which express no or little survivin. Second, elevated survivin levels in mature neutrophils are observed in active inflammatory diseases, such as acute appendicitis, ulcerative colitis, and CF. Thus, increased expression of survivin is a common phenomenon of inflammation and not restricted to cancer or other proliferating cells. Third, GM-CSF and G-CSF induce survivin gene expression in mature neutrophils in a cell cycle–independent manner. Last, survivin is able to function as an antiapoptotic protein beyond the G2-M checkpoint.
Survivin is a member of the IAP family, which seems to constitute a final false-safe mechanism to prevent intracellular damage due to premature caspase activation. For instance, it has been reported that XIAP, another member of the IAP family (1), prevents death receptor–mediated apoptosis by inhibition of caspase-3 (35). In these cells, it appears that Bid-mediated release of Smac from mitochondria is required to neutralize the IAP function to activate caspase-3 (36). Further support for the potency of IAPs to prevent premature apoptosis is provided by the observation that in some cells, mitochondria can be activated to release cytochrome c, but do not induce apoptosis (37). Thus, mitochondrial and caspase activation do not necessarily represent a “point of no return” in response to a death stimulus.
XIAP appears to be the most potent inhibitor of active caspases compared with other members of the IAP family (38). The expression of XIAP (31), as well as of IAP-1 (39) and IAP-2 (39), has been reported previously in mature neutrophils. In this work, we obtained evidence for the expression of XIAP, IAP-1, IAP-2, and survivin in these cells. XIAP and survivin were found to be inducible by G-CSF. Because XIAP expression was not always increased in immature compared with mature neutrophils, we focused our investigations in this work on the cytoprotective role of survivin.
Survivin expression seems to be a matter of active regulation in neutrophils during differentiation and inflammation. Reduced expression in mature compared with immature neutrophils was associated with caspase-3 activation and spontaneous apoptosis. Moreover, antisense-mediated inhibition of survivin expression prevented the antiapoptotic effect of GM-CSF and G-CSF in human neutrophils. A similar antiapoptosis block was observed in neutrophils from survivin+/− mice. Therefore, although survival cytokines may also increase the ratio between anti- and proapoptotic Bcl-2 family members (32, 40), their ability to increase survivin levels seems to be crucial for delayed neutrophil apoptosis.
A role of survivin in the regulation of apoptosis has previously been suggested in other cell types, particularly in tumor cells (12). The protective effect of survivin was reported to depend on its localization at the mitotic spindle and hence to be cell cycle–dependent (3–5). Moreover, survivin expression was seen at the G2-M boundary where its inhibition was accompanied by cell cycle defects. In addition, the embryonic lethal phenotype of survivin−/− mice implicated a key role for survivin in mitosis (40). The concept that survivin acts as an IAP has also been challenged by other papers in which a role in cytokinesis was suggested (41–43). Here, we demonstrate that survivin expression is not necessarily linked to the cell cycle, and that it indeed acts as an IAP without the requirement of spindle association. The notion that survivin expression is not limited to the G2-M phase is supported by earlier findings in CD34+ cells, which express survivin in all phases of the cell cycle (34, 44). Because the expression of survivin is highly regulated by survival factors and subject to rapid changes, it appears that survivin plays a key role in modulating the apoptotic threshold in neutrophils according to the biological requirements in a given physiologic or pathologic situation.
Another important finding of this work is that high survivin levels are not restricted to embryonic tissues and tumors, and particularly that inflammatory diseases are also associated with increased survivin levels. In conjunction with other papers describing its expression in hematopoietic progenitor cells (34, 44, 45) and T cells (46), this is of paramount importance and questions the value of survivin as a therapeutic target in cancer (12), due to the profound myelo- and immunosuppressive effects that might be expected from this treatment.
In summary, this paper demonstrates the importance of survivin in the regulation of neutrophil apoptosis and confirms its role as an antiapoptotic protein. Elevated survivin levels are present in neutrophils during normal differentiation in the bone marrow and represent a physiologic response in mature terminally differentiated cells under inflammatory conditions. Therefore, survivin might represent a novel target for antiinflammatory therapy. Moreover, and in contrast with previous papers, we demonstrate that survivin expression and its function as an IAP does not require cell cycle progression and mitotic spindle formation. Further analyses are needed to clarify whether additional signals exist that regulate survivin expression in primary cells and to elucidate the molecular mechanisms underlying the regulation of survivin expression in various biological systems.
Acknowledgments
We are indebted to our hematologists from the Department of Hematology at the University of Bern for diagnostic evaluation of the bone marrow aspirates. We also greatly appreciate the help in blood sampling obtained from Drs. C. Casaulta-Aebischer, S. Russmann, and D. Simon.
This work was supported by the Swiss National Science Foundation (grant nos. 31-58916.99 and 31-68449.02); the Krebsliga Schweiz (grant no. 1063-09-2000); the Bernische Krebsliga; the Kurt and Senta Herrmann Foundation; and the Edoardo R., Giovanni, Giuseppe and Chiarina Sassella Foundation. E.M. Conway was supported in part by the Fonds voor Wetenschappelijk Onderzoek (grant no. G.0382.02).
F. Altznauer and S. Martinelli contributed equally to this work.
Abbreviations used in this paper: CF, cystic fibrosis; GAPDH, glyceraldehyde-3–phosphate dehydrogenase; IAP, inhibitor of apoptosis protein; MPO, myeloperoxidase; Smac, second mitochondrial activator of caspase.
References
- 1.Salvesen, G.S., and C.S. Duckett. 2002. IAP proteins: blocking the road to death's door. Nat. Rev. 3:401–410. [DOI] [PubMed] [Google Scholar]
- 2.Tamm, I., Y. Wang, E. Sausville, D.A. Scudiero, N. Vigna, T. Oltersdorf, and J.C. Reed. 1998. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 58:5315–5320. [PubMed] [Google Scholar]
- 3.Reed, J.-C., and R.I. Reed. 1999. Survivin' cell-separation anxiety. Nat. Cell Biol. 1:E199–E200. [DOI] [PubMed] [Google Scholar]
- 4.Reed, J.-C., and J.R. Bischoff. 2000. BIRinging chromosomes through cell division--and survivin' the experience. Cell. 102:545–548. [DOI] [PubMed] [Google Scholar]
- 5.Li, F., G. Ambrosini, E.Y. Chu, J. Plescia, S. Tognin, P.C. Marchisio, and D.C. Altieri. 1998. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 396:580–583. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez, J.A., S.W. Span, C.G. Ferreira, F.A. Kruyt, and G. Giaccone. 2002. CRM1-mediated nuclear export determines the cytoplasmic localization of the antiapoptotic protein survivin. Exp. Cell Res. 275:44–53. [DOI] [PubMed] [Google Scholar]
- 7.Song, Z., X. Yao, and M. Wu. 2003. Direct interaction between survivin and Smac is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J. Biol. Chem. 278:23130–23140. [DOI] [PubMed] [Google Scholar]
- 8.Banks, D.P., J. Plescia, D.C. Altieri, J. Chen, S.H. Rosenberg, H. Zhang, and S.C. Ng. 2000. Survivin does not inhibit caspase-3 activity. Blood. 96:4002–4003. [PubMed] [Google Scholar]
- 9.Verdecia, M.A., H. Huang, E. Dutil, A. Kaiser, T. Hunter, and J.P. Noel. 2000. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat. Struct. Biol. 7:620–623. [DOI] [PubMed] [Google Scholar]
- 10.Ambrosini, G., C. Adida, and D.C. Altieri. 1997. A novel anti-apoptotic gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3:917–921. [DOI] [PubMed] [Google Scholar]
- 11.Monzo, M., R. Rosell, E. Felip, J. Astudillo, J.J. Sanchez, J. Maestre, C. Martin, A. Font, A. Barnadas, and A. Abad. 1999. A novel anti-apoptosis gene: re-expression of survivin messenger RNA as a prognostic marker in non-small cell lung cancers. J. Clin. Oncol. 17:2100–2104. [DOI] [PubMed] [Google Scholar]
- 12.Altieri, D.C. 2003. Validating survivin as a cancer therapeutic target. Nat. Rev. 4:46–54. [DOI] [PubMed] [Google Scholar]
- 13.Olie, R.A., A.P. Simoes-Wust, B. Baumann, S.H. Leech, D. Fabbro, R.A. Stahel, and U. Zangemeister-Wittke. 2000. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 60:2805–2809. [PubMed] [Google Scholar]
- 14.De Mesaeker, A., R. Häner, P. Martin, and H.E. Moser. 1995. Antisense oligonucleotides. Acc. Chem. Res. 28:366–374. [Google Scholar]
- 15.Cowland, J.B., and N. Borregaard. 1999. Isolation of neutrophil precursors from bone marrow for biochemical and transcriptional analysis. J. Immunol. Methods. 232:191–200. [DOI] [PubMed] [Google Scholar]
- 16.Borregaard, N., M. Sehested, B.S. Nielsen, H. Sengelov, and L. Kjeldsen. 1995. Biosynthesis of granule proteins in normal human bone marrow cells. Gelatinase is a marker of terminal neutrophil differentiation. Blood. 85:812–817. [PubMed] [Google Scholar]
- 17.Terstappen, L.W.M.M., M. Safford, and M.R. Loken. 1990. Flow cytometric analysis of human bone marrow. III. Neutrophil maturation. Leukemia. 4:657–663. [PubMed] [Google Scholar]
- 18.Yousefi, S., D.R. Green, K. Blaser, and H.-U. Simon. 1994. Protein tyrosine phosphorylation regulates apoptosis in human eosinophils and neutrophils. Proc. Natl. Acad. Sci. USA. 91:10868–10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dibbert, B., M. Weber, W.H. Nikolaizik, P. Vogt, M.H. Schöni, K. Blaser, and H.-U. Simon. 1999. Cytokine-mediated Bax deficiency and consequent delayed neutrophil apoptosis: a general mechanism to accumulate effector cells in inflammation. Proc. Natl. Acad. Sci. USA. 96:13330–13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conway, E.M., S. Pollefeyt, M. Steiner-Mosonyi, W. Luo, A. Devriese, F. Lupu, F. Bono, N. Leducq, F. Dol, P. Schaeffer, et al. 2002. Deficiency of survivin in transgenic mice exacerbates Fas-induced apoptosis via mitochondrial pathways. Gastroenterology. 123:619–631. [DOI] [PubMed] [Google Scholar]
- 21.Daigle, I., S. Yousefi, M. Colonna, D.R. Green, and H.-U. Simon. 2002. Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat. Med. 8:61–67. [DOI] [PubMed] [Google Scholar]
- 22.Heinisch, I.V., I. Daigle, B. Knöpfli, and H.U. Simon. 2000. CD137 activation abrogates granulocyte-macrophage colony-stimulating factor-mediated anti-apoptosis in neutrophils. Eur. J. Immunol. 30:3441–3446. [DOI] [PubMed] [Google Scholar]
- 23.Hebestreit, H., B. Dibbert, I. Balatti, D. Braun, A. Schapowal, K. Blaser, and H.-U. Simon. 1998. Disruption of Fas receptor signaling by nitric oxide in eosinophils. J. Exp. Med. 187:415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altznauer, F., S. Conus, A. Cavalli, G. Folkers, and H.-U. Simon. 2004. Calpain-1 regulates Bax and subsequent Smac-dependent caspase-3 activation in neutrophil apoptosis. J. Biol. Chem. 279:5947–5957. [DOI] [PubMed] [Google Scholar]
- 25.Simon, H.-U., S. Yousefi, C. Schranz, A. Schapowal, C. Bachert, and K. Blaser. 1997. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J. Immunol. 158:3902–3908. [PubMed] [Google Scholar]
- 26.Simon, H.-U., M. Weber, E. Becker, Y. Zilberman, K. Blaser, and F. Levi-Schaffer. 2000. Eosinophils maintain their capacity to signal and release eosinophil cationic protein upon repetitive stimulation with the same agonist. J. Immunol. 165:4069–4075. [DOI] [PubMed] [Google Scholar]
- 27.Weinmann, P., P. Gaehtgens, and B. Walzog. 1999. Bcl-xL and Bax-α-mediated regulation of apoptosis of human neutrophils via caspase-3. Blood. 93:3106–3115. [PubMed] [Google Scholar]
- 28.Daigle, I., and H.-U. Simon. 2001. Critical role for caspase-3 in neutrophil but not eosinophil apoptosis. Int. Arch. Allergy Immunol. 126:147–156. [DOI] [PubMed] [Google Scholar]
- 29.Maianski, N.A., F.P.J. Mul, J.D. van Buul, and T.W. Kuijpers. 2002. Granulocyte colony-stimulating factor inhibits the mitochondria-dependent activation of caspase-3 in neutrophils. Blood. 99:672–679. [DOI] [PubMed] [Google Scholar]
- 30.Kallio, M.J., M. Nieminen, and J.E. Eriksson. 2001. Human inhibitor of apoptosis protein (IAP) survivin participates in regulation of chromosome segregation, and mitotic exit. FASEB J. 15:2721–2723. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi, S., K. Yamashita, T. Takeoka, T. Ohtsuki, Y. Suzuki, R. Takahashi, K. Yamamoto, S.H. Kaufmann, T. Uchiyama, M. Sasada, and A. Takahashi. 2002. Calpain-mediated X-linked inhibitor of apoptosis degradation in neutrophil apoptosis and its impairment in chronic neutrophilic leukemia. J. Biol. Chem. 277:33968–33977. [DOI] [PubMed] [Google Scholar]
- 32.Moulding, D.A., C. Akgul, M. Derouet, M.R.H. White, and S.W. Edwards. 2001. Bcl-2 family expression in human neutrophils during delayed and accelerated apoptosis. J. Leukoc. Biol. 70:783–792. [PubMed] [Google Scholar]
- 33.Baumann, R., C. Casaulta, D. Simon, S. Conus, S. Yousefi, and H.-U. Simon. 2003. Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB J. 17:2221–2230. [DOI] [PubMed] [Google Scholar]
- 34.Fukuda, S., and L.M. Pelus. 2001. Regulation of the inhibitor-of-apoptosis family member survivin in normal cord blood and bone marrow CD34+ cells by hematopoietic growth factors: implication of survivin expression in normal hematopoiesis. Blood. 98:2091–2100. [DOI] [PubMed] [Google Scholar]
- 35.Devereaux, Q.L., R. Takahashi, G.S. Salvesen, and J.C. Reed. 1997. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 388:300–304. [DOI] [PubMed] [Google Scholar]
- 36.Bratton, S.B., and G.M. Cohen. 2003. Death receptors leave a caspase footprint that Smacs of XIAP. Cell Death Differ. 10:4–6. [DOI] [PubMed] [Google Scholar]
- 37.Martinou, I., S. Desagher, R. Eskes, B. Antosson, E. Andre, S. Fakan, and J.C. Martinou. 1999. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol. 144:883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holcik, M., H. Gibson, and R.G. Komeluk. 2001. XIAP: apoptotic break and promising therapeutic target. Apoptosis. 6:253–261. [DOI] [PubMed] [Google Scholar]
- 39.Hasegawa, T., K. Suzuki, C. Sakamoto, K. Ohta, S. Nishiki, M. Hino, N. Tatsumi, and S. Kitagawa. 2003. Expression of the inhibitor of apoptosis (IAP) family members in human neutrophils: up-regulation of cIAP2 by granulocyte colony-stimulating factor and overexpression of cIAP2 in chronic neutrophilic leukemia. Blood. 101:1164–1171. [DOI] [PubMed] [Google Scholar]
- 40.Simon, H.-U. 2003. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol. Rev. 193:101–110. [DOI] [PubMed] [Google Scholar]
- 41.Uren, A.G., L. Wong, M. Pakusch, K.J. Fowler, F.J. Burrows, D.L. Vaux, and K.H. Choo. 2000. Survivin, and the inner centromere protein INCENP show similar cell-cycle localization, and gene knockout phenotype. Curr. Biol. 10:1319–1328. [DOI] [PubMed] [Google Scholar]
- 42.Skoufias, D.A., C. Mollinari, F.B. Lacroix, and R.L. Margolis. 2000. Human survivin is a kinetochore-associated passenger protein. J. Cell Biol. 25:1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wheatley, S.P., A. Carvalho, P. Vagnarelli, and W.C. Earnshaw. 2001. INCENP is required for proper targeting of survivin to the centromeres, and the anaphase spindle during mitosis. Curr. Biol. 11:886–890. [DOI] [PubMed] [Google Scholar]
- 44.Fukuda, S., R.G. Foster, S.B. Porter, and L.M. Pelus. 2002. The antiapoptosis protein survivin is associated with cell cycle entry of normal cord blood CD34+ cells and modulates cell cycle and proliferation of mouse hematopoietic progenitor cells. Blood. 100:2463–2471. [DOI] [PubMed] [Google Scholar]
- 45.Fukuda, S., C.R. Mantel, and L.M. Pelus. 2004. Survivin regulates hematopoietic progenitor cell proliferation through p21WAF1/Cip1 dependent and independent pathways. Blood. 103:120–127. [DOI] [PubMed] [Google Scholar]
- 46.Xing, X., E.M. Conway, C. Kang, and A. Winoto. 2004. Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J. Exp. Med. 199:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]