

Abstract
The Epstein-Barr virus (EBV)-encoded nuclear antigen 1 (EBNA1) is expressed in all EBV-associated tumors, making it an important target for immunotherapy. However, evidence for major histocompatibility complex (MHC) class I–restricted EBNA1 peptides endogenously presented by EBV-transformed B and tumor cells remains elusive. Here we describe for the first time the identification of an endogenously processed human histocompatibility leukocyte antigen (HLA)-B8–restricted EBNA1 peptide that is recognized by CD8+ T cells. T cell recognition could be inhibited by the treatment of target cells with proteasome inhibitors that block the MHC class I antigen processing pathway, but not by an inhibitor (chloroquine) of MHC class II antigen processing. We also demonstrate that new protein synthesis is required for the generation of the HLA-B8 epitope for T cell recognition, suggesting that defective ribosomal products (DRiPs) are the major source of T cell epitopes. Experiments with protease inhibitors indicate that some serine proteases may participate in the degradation of EBNA1 DRiPs before they are further processed by proteasomes. These findings not only provide the first evidence of the presentation of an MHC class I–restricted EBNA1 epitope to CD8+ T cells, but also offer new insight into the molecular mechanisms involved in the processing and presentation of EBNA1.
Keywords: cancer vaccines, immunotherapy, MHC class I–restricted peptides, antigen presentation, CD8+ T cells
Introduction
EBV, a human γ herpesvirus with tropism for B cells, has been implicated in the pathogenesis of a variety of human tumors, including Burkitt's lymphoma (BL), posttransplant lymphoproliferative disorder, nasopharyngeal carcinoma (NPC), and Hodgkin's disease (HD; 1, 2). Although a subset of genes is responsible for the growth-transforming function of EBV, EBV-encoded nuclear antigen 1 (EBNA1) is the only viral gene that is detected in all EBV-associated tumors (BL, NPC, posttransplant lymphoproliferative disorder, and HD), and is critical for the maintenance of the viral episome and the pathogenesis of EBV-associated tumors (3, 4). Although two additional viral antigens, the latent membrane proteins 1 and 2, are expressed in NPC and HD (type II latency tumors) but not BL (type I latency tumor), expression of the remaining viral genes is undetectable in type I or II latency tumors. Thus, EBNA1 is a potentially important target for immunotherapy of EBV-associated malignancies.
Although much effort has been directed toward the identification of MHC class I–restricted endogenously processed EBNA1 peptides, no such epitopes have been reported (4, 5). For example, EBNA1-specific CD8+ T cells could be generated from human PBMCs after in vitro stimulation with EBV-transformed B lymphoblastoid cell lines (LCLs) pulsed with the exogenous EBNA1 protein, but they failed to recognize virally infected target cells such as LCLs or BL tumor cells (5–7). The failure of T cells to recognize EBV-infected cells has been largely attributed to the Gly-Ala repeat (GAr) domain within the EBNA1 protein, which blocks endogenous antigen processing and presentation by MHC class I molecules (6–8). However, several MHC class II–restricted T cell epitopes have recently been identified (9–11), suggesting that the processing and presentation of EBNA1 through MHC class II pathway are not inhibited by the GAr domain.
In this study we established EBNA1-reactive CD8+ T cell lines/clones from human PBMCs stimulated with EBNA1 peptides. We show for the first time that these peptide-induced CD8+ T cells can recognize LCLs as well as target cells transfected with the EBNA1 and HLA-B8 cDNAs based on cytokine release and cytotoxic assays. T cell recognition of EBNA1 can be blocked by proteasome inhibitors, but not by an inhibitor of MHC class II pathway, suggesting that the EBNA1 peptide is generated through the MHC class I pathway. We show further that treatment of target cells with an irreversible protein synthesis inhibitor, emetine, results in significant inhibition of the presentation of the HLA-B8 T cell epitope for T cell recognition, suggesting that T cell epitopes are derived primarily from the defective ribosomal products (DRiPs). We also identified two protease inhibitors (AEBSF and TPCK) that block antigen processing and presentation of the HLA-B8–restricted epitope, suggesting that some serine proteases may participate in the degradation of DRiPs of EBNA1 before further processing by proteasomes.
Materials and Methods
Cell Lines and Antibodies.
EBV-transformed LCLs 1, 2, 8, 111, 888, and 1088, melanoma cell lines 102mel, 586mel, and 1359mel, 1359 fibroblasts, and HEK 293 cell lines were maintained in RPMI 1640 (Sigma-Aldrich) supplemented with 10% FCS (Gemini) growth medium. Antibodies from hybridoma HB55 (anti-DR), HB 95 (anti–class I), HB 144 (anti-DQ), and HB 145 (anti–class II; American Type Culture Collection) were purified from culture supernatants of hybridoma. FITC-conjugated anti-CD4 and anti-CD8 were purchased from BD Biosciences.
Chemicals.
Proteasome inhibitors Z-Ile-Glu(OtBu)-Ala-Leucinal (ZAL) and lactacystin were purchased from Calbiochem. MHC class II pathway inhibitor chloroquine was purchased from Sigma-Aldrich. Protein synthesis inhibitors emetine, cycloheximide, and puromycin were purchased from Calbiochem. Protease inhibitors, including AEBSF, E-64, EST, leupeptin, pepstatin A, TLCK, and TPCK were purchased from Calbiochem. All chemicals were dissolved in solvents recommended by the manufacturers.
HLA Typing of Donor PBMCs.
HLA serotypes and DNA genotypes of human PBMCs were determined by the National Institutes of Health HLA Laboratory. The HLA genotype of PBMCs from donor M was HLA-A*01, 0201, B*08, DRβ1*0301, 0401, DQβ1*0201, 0301, DRβ3*0101, DRβ4*01. The genotype for donor Q was HLA-A*01, 6802, B*15, 53, DRβ1*0401, 1302, DQβ1*0301, 0501, DRβ3*0301, DRβ4*0101. The genotype for donor S was HLA-A*0301, 29, B*44, 4501, DRβ1*0401, 0701, DQβ1*0201, 0301, DRβ4*01. The genotype for 1359mel was HLA-A*01, B*8, 40, CW*03, 07, DRβ1*0401, 17, DQβ1*02, 03, DRβ3*0101, β4*0101. The genotype for donors 1, 2, and 4 was HLA-B*8 and for donor 3 it was HLA-A*01, 30, B*8, 13, CW*06, 07, DRβ1*3, 7, DQβ1*02, DRβ3*01, β4*01.
Synthetic EBNA1-Peptides.
10 13–15-mer peptides were made and purified as previously described (11). The purity and molecular masses of peptides were determined by HPLC and mass spectrometry.
Generation of Human CD8+ T Cell Lines and Clones.
Human PBMCs from three HLA-DR4–expressing donors (M, Q, and S) were used for peptide stimulation in vitro in lymphocyte culture medium at 1.5 × 105 cells per well in a flat-bottom 96-well plate as previously described (11). One T cell line from donor M was generated that showed specific T cell reactivity against peptide-pulsed 1359mel cells and was blocked by anti–MHC class I monoclonal antibody. T cell line M was further cloned using limiting dilution methods as previously described (12).
Cytokine Release, Cytotoxicity, and ELISPOT Assays.
Human EBNA1-specific T cell clones were identified on the basis of their ability to release IFN-γ. Cytotoxicity and cold target inhibition assays were performed as previously described (13). In brief, LCL 111 hot target cells were labeled with 100 μCi Na2 51 CrO4, either alone or in the presence of 1 μM EBNA1-P518–526 peptide for 8 h at 37°C. Target cells were washed three times with RPMI 1640, counted, and then mixed with CD8+ T cells at the indicated effector to target (E/T) ratios. Chromium release was measured after 16 h of incubation. In cold target inhibition assays, LCL 111 target cells were pulsed with 1 μM EBNA1-P518–526 or EBNA1-P572–584 peptides for 90 min, washed three times with RPMI, counted, and incubated with CD8+ T cells for 30 min before the addition of hot target cells. An E/T ratio of 40:1 and a cold to hot target ratio of 4:104 were used in these assays. The percentage of specific lysis was determined from the equation [(cpm experimental well-cpm spontaneous release)/(cpm maximum release-cpm spontaneous release)] × 100%. We used the ELISPOT assay to detect antigen-specific T cells in fresh PBMCs as previously described (14).
Transfection of EBNA1 Expression Constructs.
The EBNA1 full-length, EBNA1–green fluorescent protein (GFP), and EBNA1-GAr-del-GFP constructs have been described (15). A retroviral construct encoding EBNA1-GFP was generated and retroviral supernatants of retrovirus were made as previously described (16). Ii-EBNA1 (invariant chain, first 80 amino acids, fused to EBNA1) was constructed by subcloning full-length EBNA1 into a pTSX expression vector to express as an Ii fusion protein (17). HEK293 and 1359mel cells were transfected with LipofectAMINE reagent (Invitrogen) according to the manufacturer's instructions.
Effects of MHC Class I and II Antigen Presentation Inhibitors on CD8+ T Cell Recognition.
The effects of various inhibitors, including proteasome inhibitors, MHC class II antigen-processing inhibitor, protein synthesis inhibitors, and protease inhibitors on CD8+ T cell activity were examined in HEK293, 1359mel, and LCL 111 target cells expressing HLA-B8 plus EBNA1-GFP or EBNA1 genes. Target cells were incubated in the absence or presence of various concentrations of inhibitors for different periods of time depending on the type of inhibitors used. The cells were then washed and counted and cocultured with T cells overnight for IFN-γ release assays. The solvents used to dissolve the inhibitors, such as DMSO, methanol, and ethanol were also used as controls. Melanoma-derived TIL 102 CD4+ T cell recognition of 102mel tumor cells was used to demonstrate the specificity of lactacystin and chloroquine inhibitors of MHC class I and II antigen presentation, respectively.
Results
Generation of MHC Class I–restricted T Cells Specific for EBNA1.
Recent studies demonstrated that human CD4+ T cells consistently and predominantly respond to MHC class II–restricted peptides derived from EBNA1 (9–11, 18), suggesting that EBNA1 can be processed through the MHC class II pathway for T cell recognition. During the course of our work on the identification of MHC class II–restricted EBNA1 peptides, we generated several T cell lines that are capable of stimulating T cells after coculturing with EBNA1-P518–530 (YNLRRGTALAIPQ) peptide-pulsed 1359mel cells. Representative data from one of these cell lines, designated M3-W1, is shown in Fig. 1 A. To determine the restriction element for T cell recognition, we tested M3-W1 T cell activity in response to the peptide-pulsed target cells in the presence of anti–MHC class I (HLA-A, HLA-B, and HLA-C), anti–HLA-DR, anti–HLA-DP, and anti–HLA-DQ or isotype control antibodies. To our surprise, T cell recognition of the EBNA1-P518–530 peptide by T cell line M3-W1 was specifically blocked by an anti–MHC class I monoclonal antibody, but not by anti–HLA-DP, anti-DQ, anti-DR, or isotype control antibodies (Fig. 1 B). These results suggest that human M3-W1 T cells recognize a peptide derived from EBNA1 presented by HLA class I molecules.
Figure 1.
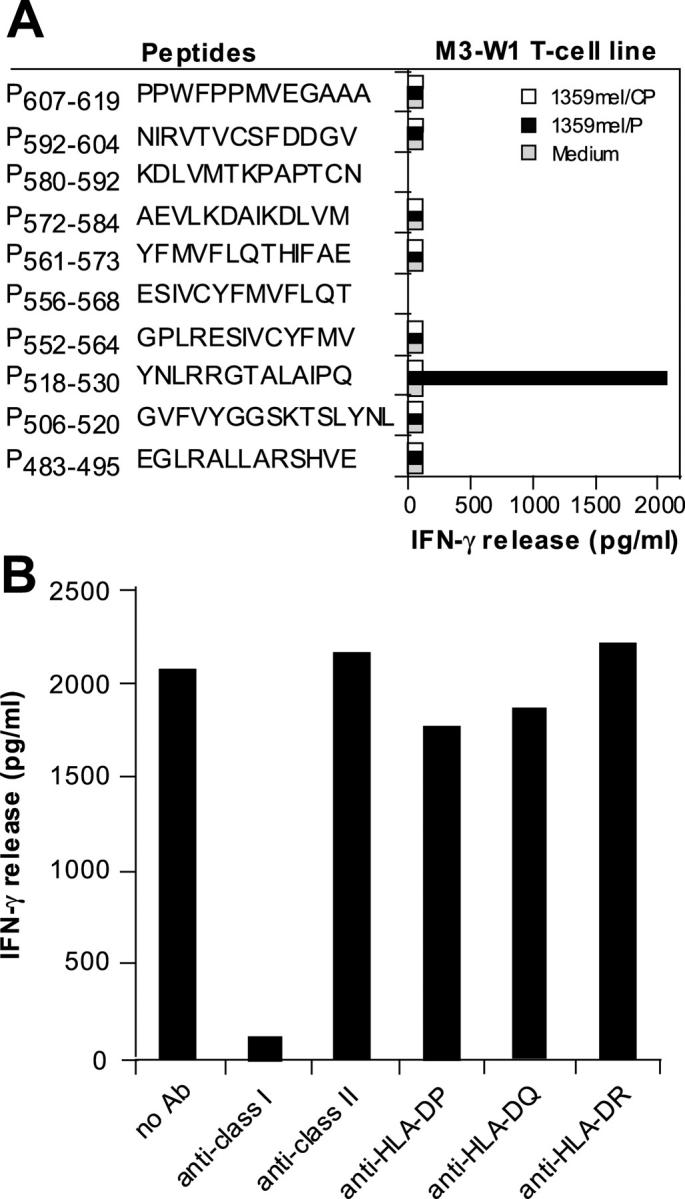
Generation of EBNA1-specific T cells. (A) T cells were generated from HLA-B8–expressing PBMCs of donor M after in vitro stimulation with synthetic peptides from EBNA1. 1.5 × 105 cells per well of PBMCs were used to generate EBNA1-P518–530 peptide–specific T cells. Peptides other than those used for repeated stimulations served as negative controls. For T cell recognition assays, peptides were pulsed onto 1359mel target cells and cocultured with T cells overnight. Cytokine release assays were performed as previously described (reference 11). (B) T cell recognition (T cell line M3-W1 from donor M) of 1359mel cells pulsed with EBNA1-P518–530 peptide was specifically inhibited by antibody against MHC class I molecules. T cell recognition assays were performed at an E/T ratio of 1:1. All results are expressed as IFN-γ release in picogram/milliliter and are the averages of duplicate values. All antibodies were used at a final concentration of 20 μg/ml each.
Characterization of T Cell Clones and Their Antigenic Peptides.
To further characterize the T cell line M3-W1, we generated 18 T cell clones by limiting dilution methods. Recognition of LCL 111 by different T cell clones is presented in Fig. 2 A. One of the T cell clones (designated M3W1-B9) with strong T cell reactivity was chosen for further study. FACS® analysis revealed that the M3W1-B9 T cells were CD8+ (Fig. 2 B). Recognition of the EBNA1-P518–530 peptide by M3W1-B9 CD8+ T cells was also blocked by antibody against MHC class I, but not by MHC class II or control antibodies, suggesting that these T cell clones resemble the bulk CD8+ T cell line (not depicted).
Figure 2.

Characterization of EBNA1-specific T cells. (A) Recognition of LCL 111 by T cell clones derived from the M3-W1 T cell line. (B) FACS® analysis of M3-W1-B9 T cells for CD8 expression. T cells were stained with anti–CD4-PE or anti–CD8-FITC. Positive staining for CD8 T cells is shown as an open histogram and control antibody staining is represented as a shaded histogram. (C) Identification of minimal EBNA1 T cell epitope for MHC class I binding. Four different peptides were made and pulsed onto 1359mel cells at 10 μM concentration. After washing, the peptide-pulsed cells were cocultured with T cells overnight. IFN-γ release was determined from culture supernatants. (D) EBNA1-P518–526 peptide titration experiment for M3W1-B9 T cell recognition. EBNA1-P518–526 peptide at various concentrations were pulsed on 1359mel cells and used as target cells to stimulate T cells. A control peptide EBNA1-P 572–584 was also used at various concentrations. Experiments were repeated twice with similar results.
Because the optimal peptide lengths for MHC class I molecules are generally 9–10 amino acids, we made three additional peptides: one 9-mer EBNA1-P518–526 (YNLRRGTAL) containing the HLA-B8 peptide binding motif and two 10-mer peptides (EBNA1-P518–527 and EBNA1-P519–528) from the parental EBNA1-P518–530 peptide. These peptides were tested for their ability to stimulate M3W1-B9 CD8+ T cells. As shown in Fig. 2 C, the EBNA1-P518–526 peptide was recognized more readily by the M3W1-B9 CD8+ T cells than the parental 13-mer peptide. By contrast, both EBNA1-P518–527 (YNLRRGTALA) and EBNA1-P519–528 (NLRRGTALAI) peptides exhibited lower or no activity for T cell recognition. Peptide titration experiments showed that T cell reactivity of the 9-mer EBNA1-P518–526 peptide could be detected at a concentration of 63 nM (Fig. 2 D). Thus, the 9-mer EBNA1-P518–526 peptide is optimal for recognition by the M3W1-B9 CD8+ T cells.
EBNA1 Peptides Are Naturally Processed and Presented to M3-W1-B9 CD8+ T Cells.
To determine if the T cell peptide derived from the EBNA1 antigen can be endogenously processed and presented to CD8+ T cells, we transfected plasmid DNAs carrying full-length EBNA1-GFP or EBNA1-GAr-del-GFP into 1359mel and 1359 fibroblast cells as targets for T cell recognition. As shown in Fig. 3 A, M3W1-B9 CD8+ T cells recognized 1359mel/EBNA1-GFP and 1359mel/EBNA1-GAr-del-GFP target cells equally well, whereas no T cell reactivity was detected with 1359mel/GFP cells. The expression of EBNA1 as a fusion protein with GFP allowed us to monitor gene expression and transfection efficiency throughout the course of the experiments. CD8+ T cells also recognized 1359 fibroblast cells transfected with full-length EBNA1-GFP cDNA (Fig. 3 B). The transfection efficiency of 1359mel and fibroblasts is ∼10–15%. T cell recognition of 1359mel/EBNA1-GFP was also blocked by antibody against MHC class I, confirming that the recognition is MHC class I restricted (not depicted). To evaluate endogenous processing of EBNA1 in human PBMCs, we constructed a retrovirus-encoding EBNA1-GFP for introducing genes into PBMCs. Although the transduction efficiency of the retrovirus-encoding EBNA1-GFP into PBMCs from donor M was <10%, we found that T cell recognition of PBMCs infected with recombinant retrovirus increased threefold in terms of IFN-γ release from T cells compared with that of uninfected PBMCs (Fig. 3 C). These results indicate that the MHC class I–restricted EBNA1 peptides are endogenously processed and presented to T cells.
Figure 3.
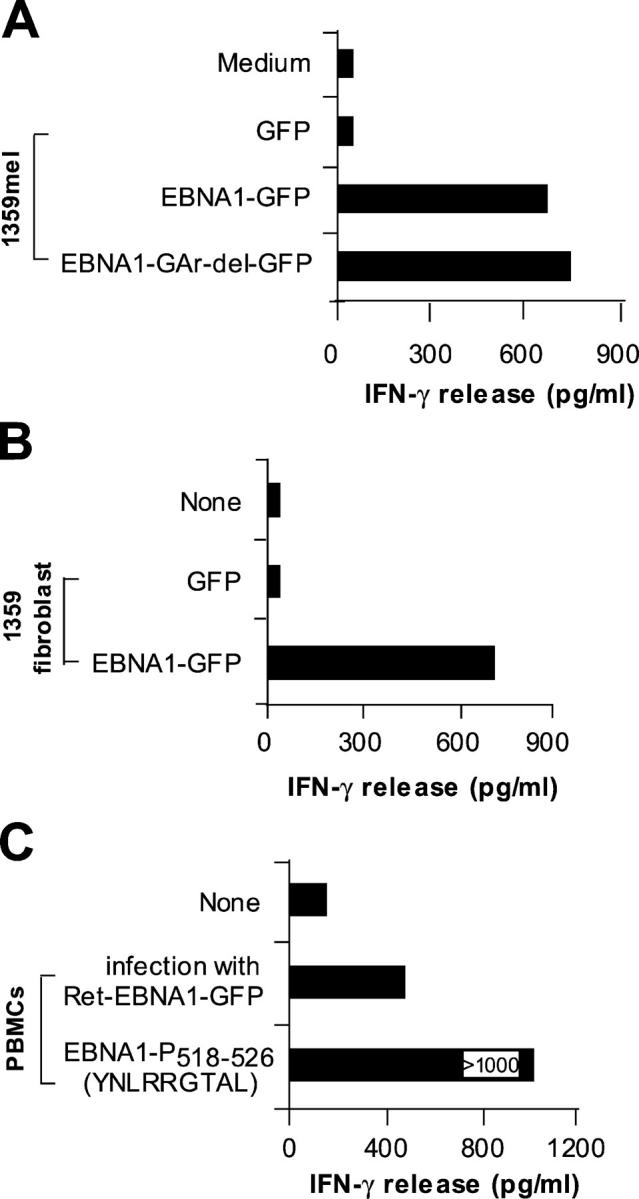
Natural processing and presentation of EBNA1 to M3-W1-B9 CD8+ T cells. (A) Recognition of full-length EBNA1-transfected 1359mel cells by M3-W1-B9 T cells. 1359mel cells were transfected with 200 ng EBNA1-GFP or EBNA1-GAr-del-GFP plasmid DNAs using LipofectAMINE. IFN-γ release was determined as described in Fig. 1. (B) Recognition of 1359 fibroblasts transfected with EBNA1-GFP by M3-W1-B9 T cells. 1359 fibroblasts were transfected with EBNA1 plasmid DNA by electroporation. T cell assays were performed at an E/T ratio of 2:1. (C) T cell recognition of autologous PBMCs infected with retroviral/EBNA1-GFP.
M3W1-B9 CD8+ T Cells Recognize EBNA1 Peptides Presented by HLA-B8 Molecules.
To determine the restriction element for M3W1-B9 CD8+ T cells, we pulsed the 9-mer EBNA1-P518–526 peptide onto various MHC class I+ melanoma cell lines and tested for T cell recognition. We found that CD8+ T cells recognized 1359mel cells pulsed with the EBNA1-P518–526 peptide, but not other cells pulsed with the same peptide (Fig. 4 A), suggesting that HLA-B8 is a putative restriction element for T cell recognition. To test this possibility, we cloned the HLA-B8 cDNA and transfected it into HEK293 cells along with EBNA1-GFP or EBNA1-GAr-del-GFP and assessed their ability to present the antigenic peptide to T cells. Although EBNA1-GFP, EBNA1-GAr-del-GFP, or HLA-B8 expressed alone in HEK293 cell did not stimulate T cell responses, HEK293 cells transfected with EBNA1-GFP plus HLA-B8 or EBNA1-GAr-del-GFP plus HLA-B8 cDNAs strongly stimulated IFN-γ release from CD8+ T cells (Fig. 4 B). Although T cell recognition of HEK293 cells transfected with EBNA1-GAr-del-GFP plus HLA-B8 cDNAs was slightly higher than that of HEK293 cells transfected with EBNA1-GFP and HLA-B8 cDNAs, we did not observe any significant inhibitory effect of the GAr domain on T cell responses (Fig. 4 B). These studies indicate that HLA-B8 is an antigen-presenting molecule for M3W1-B9 CD8+ T cells.
Figure 4.

HLA-B8 molecule functions as a restriction element for M3-W1-B9 CD8+ T cells. (A) T cell recognition of peptide-pulsed HLA-B8–expressing cell lines. (B) Identification of HLA-B8 molecule as a restriction element for T cell recognition. HEK293 cells cotransfected with HLA-B8 plus full-length EBNA1-GFP or EBNA1-GAr-del-GFP cDNAs (with GAr domain deleted) were tested for recognition by M3-W1-B9 CD8+ T cells. Positive and negative signs indicate cotransfection of target cells in the presence or absence of HLA-B8 cDNA, respectively. (C) Natural processing and presentation of the native form of EBNA1 for T cell recognition. HEK293 cells cotransfected with full-length EBNA1 and HLA-B8 cDNAs were cocultured with M3-W1-B9 CD8+ T cells overnight. IFN-γ secretion from T cells was determined by ELISA. (D) Endogenous generation of HLA-B8–restricted EBNA1 peptide for T cell recognition. HEK293 cells transfected with HLA-B8 cDNA were mixed with HEK293 cells transfected with EBNA1-GFP cDNA at a 1:1 ratio. The mixed cells were then cocultured with M3-W1-B9 CD8+ T cells overnight. IFN-γ release from CD8+ T cells was measured from culture supernatants.
To exclude the possibility that GFP fusion to EBNA1 alters antigen processing and presentation, we tested whether HEK293 cells cotransfected with HLA-B8 and the native form of EBNA1 cDNA can stimulate T cells. As shown in Fig. 4 C, HEK293 cells transfected with EBNA1 plus HLA-B8 cDNA strongly stimulated IFN-γ release from CD8+ T cells, whereas HEK293 cells transfected with either one alone failed to stimulate T cell response. These studies suggest that the native form of EBNA1 protein, like the GFP-tagged EBNA1, can be processed and presented to CD8+ T cells by the HLA-B8 molecules.
It is well known that dendritic cells have the capacity to capture and deliver exogenous antigens into the MHC class I processing pathway (19, 20). To determine whether the recognition of EBNA1 by M3W1-B9 CD8+ T cells requires the cross-presentation pathway, we transfected HEK293 with HLA-B8 cDNA or EBNA1-GFP cDNA separately, and then mixed them (1:1) together as target cells in a T cell assay. As shown in Fig. 4 D, M3W1-B9 CD8+ T cells did not respond to the mixed cells of transfected HEK293/HLA-B8 and HEK293/EBNA1-GFP, but they actively recognized HEK293 cells cotransfected with HLA-B8 and EBNA1-GFP cDNAs. These studies suggest that the coexpression of HLA-B8 and EBNA1 in the same HEK293 cells is required for T cell recognition by M3W1-B9 CD8+ T cells and that HEK293 cells are not capable of cross-presenting EBNA1 antigen to T cells.
Recognition and Lysis of EBV+ LCL Cells by M3W1-B9 T Cells.
Although peptide-specific CD8+ T cells against putative tumor antigens or peptides can often be generated from human PBMCs, they show no reactivity with tumor cells. Possible explanations include the low affinity of the T cells for the MHC–peptide complexes or the failure of presentation of naturally processed peptides on the surface of tumor cells (21). Indeed, EBNA1 peptide-specific CD8+ T cells generated from human PBMCs after in vitro stimulation failed to recognize autologous EBV+ LCLs (5). T cell reactivity was only found when autologous EBV+ LCL cells were pulsed with exogenous protein or peptides (5). To test whether the CD8+ T cells generated in this study were capable of recognizing naturally processed peptides on EBV-infected cells, we used several EBV B cell lines as target cells. As shown in Fig. 5 A, a cytokine release assay demonstrated that M3W1-B9 CD8+ T cells strongly recognized HLA-B8+ LCL 8, 111, and 1088 cells, but did not respond to HLA-mismatched LCL 1 and 2 cells. To further test whether M3W1-B9 CD8+ T cells can lyse tumor target cells, we performed chromium release assays. CD8+ T cells specifically lysed LCL 111 cells but not the HLA-mismatched LCL1 cells (Fig. 5 B). Cold target inhibition experiments showed that the specific killing of chromium-labeled (hot) LCL 111 target cells by the M3W1-B9 CD8+ T cells could be inhibited by the 9-mer EBNA1-P518–526 peptide–pulsed unlabeled (cold) LCL 111 cells, but not by control peptide–pulsed cold LCL 111 cells (Fig. 5 C). Taken together, these results indicate that the 9-mer EBNA1-P518–526 peptide can specifically block the recognition and lysis of EBV+ LCL 111 cells by the M3W1-B9 CD8+ T cells, implying that a similar or identical EBNA1 peptide is endogenously processed and presented to T cells by HLA-B8 molecules on the surface of EBV+ LCL cells by a mechanism that overrides the inhibitory effect of the GAr domain on processing and presentation of EBNA1.
Figure 5.
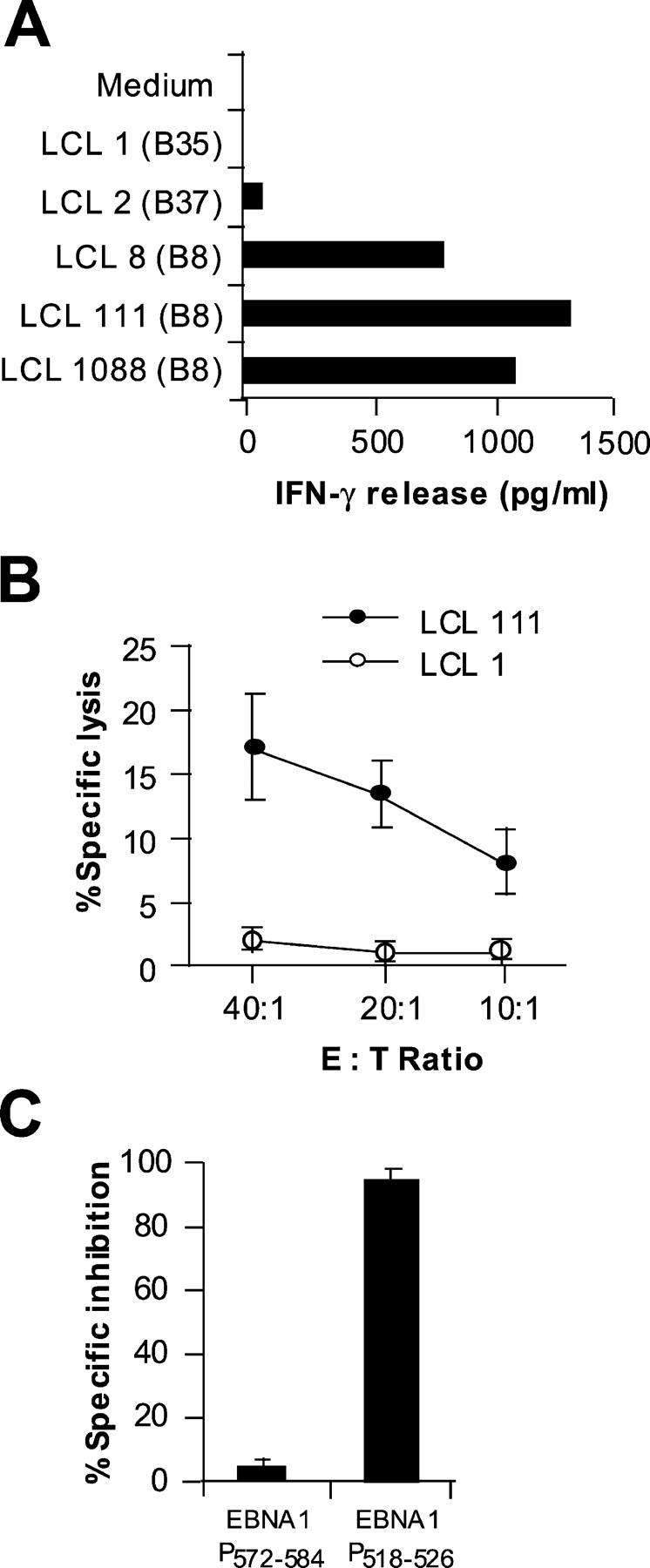
Specific lysis of HLA-B8–matched EBV-transformed LCLs by M3-W1-B9 CD8+ T cells. (A) Recognition of HLA-B8–matched LCLs by M3-W1-B9 CD8+ T cells. LCLs were cocultured with M3-W1-B9 CD8+ T cells at an E/T ratio of 1:1. Mismatched LCLs were used as negative controls. (B) Specific lysis of HLA-B8–matched LCL 111 cells by CD8+ T cells at different E/T ratios. LCL 1 was used as a negative control. LCL cells were labeled with 51chromium. Cytolysis by CD8+ T cells was determined in a 16-h chromium release assay. (C) Cold target inhibition of recognition of LCL 111 cells by M3-W1-B9 CD8+ T cells. Lysis of LCLs by M3-W1-B9 CD8+ T cells was specifically inhibited when EBNA1-P518–526–pulsed cold LCL 111 targets were used. Lysis was tested with an effector to hot target ratio of 40:1. Cold LCL 111 target cells were pulsed with EBNA1-P518–526 or EBNA1-P572–584 peptide at a concentration of 1 μM and were mixed with hot targets at a ratio of 4:1. All experiments were repeated twice with similar results.
Recognition of EBNA1-P518–530 Peptide by EBNA1-specific CD4+ and CD8+ T Cells.
Because the HLA-B8–restricted EBNA1-P518–526 peptide overlaps peptides presented by both HLA-DR1 and HLA-DP3 molecules (Fig. 6 A), we sought to determine if the same peptide could be recognized by EBNA1-specific CD4+ and CD8+ T cell clones. Fig. 6 B shows that the EBNA1-P518–530 peptide could be recognized by HLA-DR1– and HLA-DP3–restricted CD4+ as well as M3W1-B9 CD8+ T cells when pulsed on their corresponding HLA-matched target cells. However, the 9-mer EBNA1-P518–526 peptide was recognized only by CD8+ T cells and not by HLA-DR1– and HLA-DP3–restricted CD4+ T cells, suggesting that the short peptide is specific for CD8+ T cells. These results imply that the 13-mer EBNA1-P518–530 peptide has a dual function as it can stimulate both CD4+ and CD8+ T cell responses.
Figure 6.
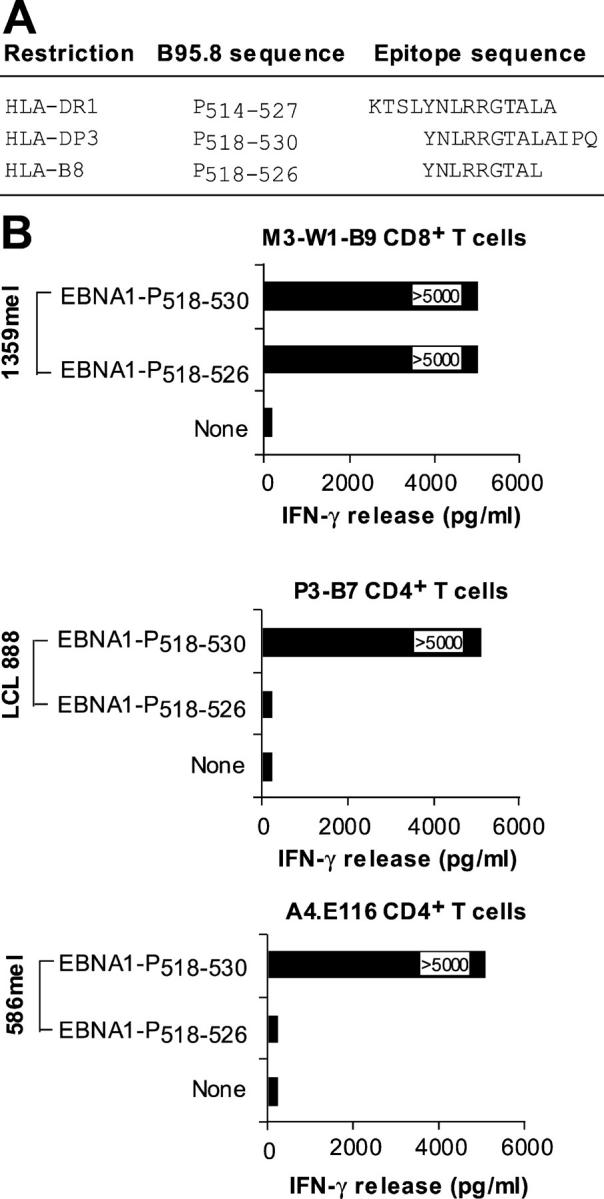
Recognition of the EBNA1-P518–530 peptide by CD4+ and CD8+ T cells. (A) Alignment of HLA-DR1–, HLA-DP3–, and HLA-B8–restricted peptides. (B) Recognition of peptide-pulsed target cells by three different HLA-B8–, HLA-DR1–, and HLA-DP3–restricted T cell lines/clones. The HLA-DP3–restricted P3-B7 CD4+ T cells are described in a previous study (reference 11). The HLA-DR1–restricted A4.E116 CD4+ T cells, also previously described (reference 10), recognized an HLA-DR1–restricted EBNA1 peptide.
Determination of EBNA1-specific HLA-B8–restricted CD8+ T Cells in Other Donor PBMCs.
Next, we tested whether EBNA1-specific HLA-B8–restricted CD8+ T cells are present in the PBMCs of other donors expressing HLA-B8. Five donor PBMCs were obtained for ELISPOT assays using EBNA1-P518–526 peptide–pulsed target cells. NY-ESO-1 peptide–pulsed target cells served as a specific control. Three of the five donor PBMCs (donors 1, 2, and 3) express HLA-B8 molecules and are serum positive for EBV, whereas donor 4 is positive for HLA-B8 expression but is serum negative for EBV. A mismatched donor PBMC (donor 5) served as a negative control. We found that three of the four HLA-B8+ donor PBMCs specifically responded to the EBNA1-P518–526 peptide (Fig. 7 A). By contrast, neither PBMCs from HLA-B8+ but seronegative nor HLA-B8− donors responded to the EBNA1-P518–526 peptide (Fig. 7 A). These results suggest that EBNA1-specific, HLA-B8–restricted CD8+ T cells are commonly present in HLA-B8+ EBV-infected individuals. To further test whether these EBNA1-specific, HLA-B8–restricted CD8+ T cells are capable of recognizing EBV+ LCL cells, we established T cell clones from the PBMCs of donor 3 by limiting dilution methods. Recognition of HLA-B8–expressing LCL 1088 by these T cell clones is shown in Fig. 7 B. One T clone, D1-B11, was selected for further testing of its ability to recognize HLA-B8+ EBV+ LCL cells. These T cells exhibited strong T cell reactivity against HLA-B8+ LCLs 8 and 1088, but did not respond to HLA-B8− LCL 1 and 2 cells (Fig. 7 C). Taken together, these studies suggest that the PBMCs of EBV-infected donors expressing HLA-B8 molecules contain EBNA1-specific CD8+ T cells that are capable of recognizing HLA-B8+ LCL targets.
Figure 7.
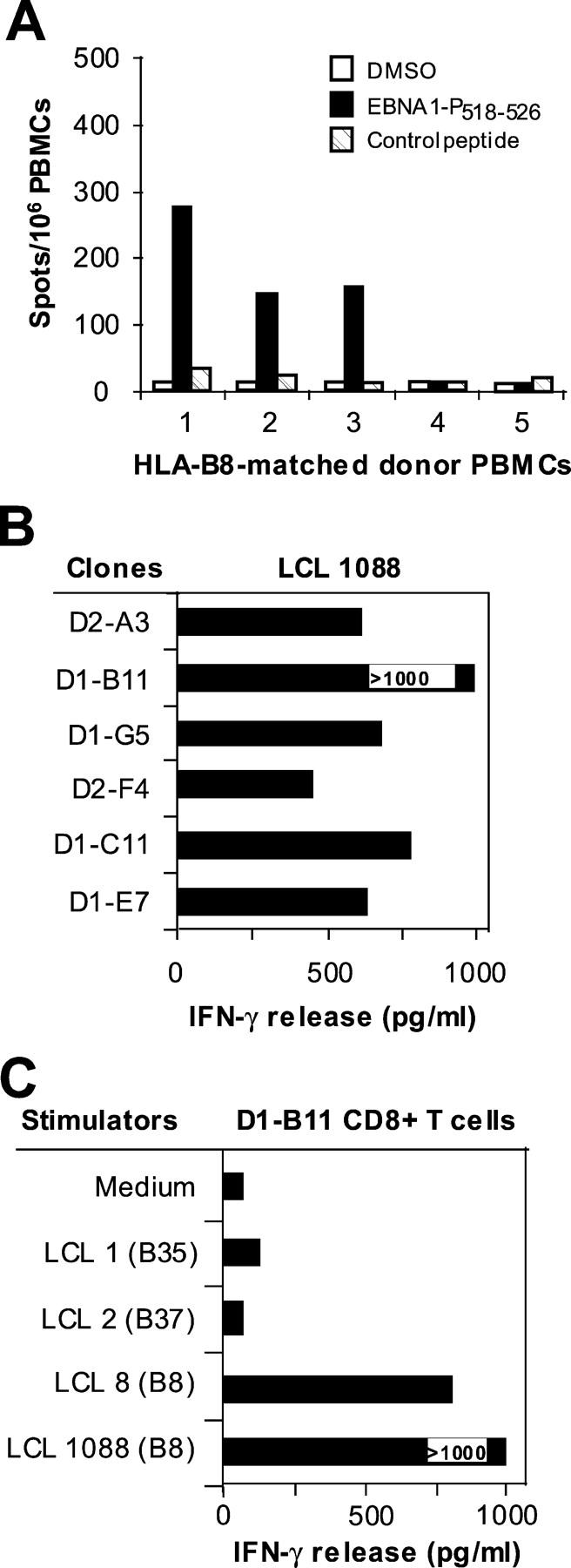
Generation of EBNA1-P518–526 peptide–specific T cells from HLA-B8–expressing PBMCs. (A) Detection of EBNA1-P518–526 peptide–reactive T cells from HLA-B8+ donor PBMCs. 105 PBMCs were seeded per well and experiments were performed in quadruplicate wells. An HLA-A2–restricted NY-ESO-1 peptide served as a control. HLA-mismatched donor 5 and an HLA-B8+ PMBC donor 4 that is seronegative for EBV were also included. (B) Recognition of LCL 1088 by CD8+ T cell clones from the PBMCs of donor 3. T cells generated from PBMCs were stimulated with EBNA1-P518–526 peptide as described in Fig. 1. Six CD8+ T cell clones were generated from two T cell lines and were capable of recognizing HLA-B8–expressing LCL 1088 target cells. (C) D1-B11 CD8+ T cell recognition of HLA-B8–matched LCLs. LCLs were cocultured with T cells at an E/T ratio of 1:1. All experiments were repeated twice with similar results.
Inhibition of T Cell Recognition by Proteasome Inhibitors.
Next, we sought to elucidate the mechanism(s) by which HLA-B8–restricted peptides are generated. One possibility is that HLA-B8 peptides are generated through the conventional MHC class I pathway. If so, specific inhibitors of proteasomes should inhibit the degradation of proteins and thus the presentation of peptides by MHC class I molecules for T cell recognition (22). Alternatively, MHC class I molecules bind and present peptides in MHC class II compartments, one of which is transported to the plasma membrane for T cell recognition (23). Because the EBNA1-P518–526 peptide overlaps with HLA-DR1– and HLA-DP3–restricted peptides that are shown to be endogenously processed and presented to CD4+ T cells (10, 11), MHC class I molecules might acquire and present a peptide generated through the MHC class II pathway. To test this possibility, we transfected HEK293 cells with EBNA1-GFP and HLA-B8 cDNAs, and then treated them with different concentrations of ZAL and lactacystin, specific inhibitors of proteasomes (22). When antigen-specific CD8+ T cells were then cocultured with target cells to evaluate T cell responses, we found that T cell reactivity of HEK293 transfected with HLA-B8 plus EBNA1-GFP cDNAs by the M3W1-B9 CD8+ T cells decreased with increasing concentrations of proteasome inhibitor ZAL, but not in the presence of control DMSO (Fig. 8 A). Similarly, T cell reactivity against LCL 111 cells was inhibited with increasing concentration of ZAL inhibitor. To exclude the potential nonspecific effect of ZAL on T cell recognition, we tested the effects of ZAL inhibitor on melanoma-reactive TIL102-CD4+ T cells, which recognize an MHC class II–restricted epitope on the cell surface of 102mel tumor cells, and of P3-B7 CD4+ T cells, which recognize HLA-DP3–expressing HEK293 cells transfected with Ii-EBNA1 as target cells. The ZAL inhibitor did not have any inhibitory effect on recognition of target cells by TIL102 CD4+ T cells or P3-B7 T CD4+ cells (Fig. 8 A). We also tested the effects of lactacystin, another specific proteasome inhibitor, on T cell recognition. As shown in Fig. 8 B, recognition of 1359mel/EBNA1-GFP cells by M3-W1-B9 CD8+ T cell was decreased with increasing concentrations of lactacystin, whereas no inhibitory effect was observed with TIL102 CD4+ T cell recognition of 102mel target cells. These results suggest that the inhibitory effect of ZAL and lactacystin is specific for the presentation of HLA-B8–restricted epitope to CD8+ T cells, but not for MHC class II antigen processing and presentation.
Figure 8.

Specific inhibition of T cell recognition of EBNA1 by proteasomes inhibitors. (A) Blocking of T cell recognition of EBNA1 by a ZAL proteasome inhibitor. HEK 293 cells cotransfected with EBNA1-GFP were treated with various concentrations of ZAL inhibitor for 10 h. After washing, cells were incubated with T cells overnight for IFN-γ release assays. Various dilutions of DMSO were used as controls. T cell activity in the absence of inhibitor was used at 100% activity. Two CD4+ T cells were used to demonstrate the specificity of ZAL inhibitor. TIL102 and P3-B7 CD4+ T cells able to recognize 102mel and HEK293/DP3/Ii-EBNA1 target cells, respectively, were not inhibited by ZAL. (B) Inhibition of M3-W1-B9 CD8+ T cell recognition of 1359mel target cells stably expressing EBNA1-GFP by lactacystin proteasome inhibitor. The lactacystin inhibitor did not affect recognition of 102mel tumor cells by TIL102 CD4+ cells. (C) Blocking of MHC class II antigen processing by chloroquine. Inhibition of T cell recognition of 102mel cells by TIL102 CD4+ was observed after treatment with chloroquine in a dose-dependent fashion. By contrast, T cell recognition of LCL 111 and HEK293 transfected with HLA-B8 and EBNA1-GFP cDNAs was not significantly affected after the treatment of chloroquine.
To further exclude the possibility that the EBNA1 peptide recognized by the M3W1-B9 CD8+ T cells might be processed through MHC class II pathway and then recycled back to MHC class I pathway to be presented by HLA-B8 molecules, we tested this possibility in the presence of chloroquine, a lysosomotropic agent that inhibits MHC class II antigen processing (24). T cell recognition of HEK293 cells transfected with HLA-B8 plus EBNA1-GFP cDNAs or LCL 111 by M3W1-B9 CD8+ T cells was not significantly affected after the target cells were treated with different concentrations of chloroquine (Fig. 8 C). By contrast, T cell activity of TIL102-CD4+ T cells against 102mel cells was significantly inhibited by chloroquine, suggesting that chloroquine can specifically inhibit MHC class II but not class I pathway for antigen presentation. Taken together, these results suggest that the processing and presentation of the HLA-B8–restricted EBNA1 peptides require the participation of proteasomes in the MHC class I pathway.
HLA-B8–restricted EBNA1 Epitope Is Derived from DRiPs.
The GAr domain has been demonstrated to efficiently block its proteasomal degradation of full-length EBNA1, thus inhibiting the generation of MHC class I–restricted peptide for T cell recognition. However, this inhibition may not be absolute, because some T cell epitopes are still processed and presented to T cells. Alternatively, the HLA-B8–restricted T cell epitope might be derived through the rapid degradation of DRiPs (25, 26), which have been estimated to constitute upwards of 30% of newly synthesized proteins and are considered an important source of peptide for presentation by MHC class I molecules (25–27). We reasoned that if the HLA-B8–restricted epitope is generated from proteasomal degradation of full-length EBNA1, the treatment of target cells with protein synthesis inhibitors would not affect T cell recognition because the full-length EBNA1 protein is still present after treatment. Otherwise, it would suggest that the HLA-B8–restricted epitope is derived from DRiPs rather than EBNA1. To test these possibilities, we treated HLA-B8–expressing HEK293/EBNA1-GFP cells with different concentrations of irreversible (emetine) and reversible (puromycin and cycloheximide) protein synthesis inhibitors for 1 h. After washing to remove inhibitors, the treated cells were cocultured with antigen-specific CD8+ T cells. Fig. 9 A shows that recognition of target cells by M3W1-B9 CD8+ T cells was significantly (94%) inhibited at a low concentration (1 μM) and completely inhibited at a 5 μM concentration of the irreversible inhibitor emetine. By contrast, no inhibitory effect was observed when HLA-B8–expressing 293/EBNA1-GFP cells were treated with reversible inhibitors puromycin and cycloheximide, respectively. Because blocking protein synthesis should rapidly decrease the peptide supply required for the export of MHC class I molecules from the ER to the cell surface as well as synthesis of MHC class I molecules (25–27), the treatment of cells with emetine would decrease the overall antigen presentation by MHC class I molecules. To exclude the possibility that inhibition of T cell recognition of the HLA-B8–restricted EBNA1 peptide was due to the expression and export of MHC class I molecules, we pulsed HLA-B8+ 1359mel cells with the EBNA1-P518–526 peptide after the cells were treated with different concentrations of emetine and washed. T cell recognition of peptide-pulsed target cells was slightly inhibited (Fig. 9 A), suggesting that effect of emetine on MHC class I molecules could not account for the inhibition of T cell recognition. To further test the sensitivity of antigen processing of other tumor antigens, such as tyrosinase-related protein 2 (TRP2), to protein synthesis inhibition, we treated 1363mel cells (TRP2+ and HLA-A2+) with different concentrations of emetine and then tested for their ability to stimulate TRP2-specific CD8+ T cells. As shown in Fig. 9 B, the inhibition of recognition of 1363mel target cells by TRP2-specific CD8+ T cells increased with the increasing concentrations of emetine. However, there was only 30% inhibition of T cell recognition at 1 μM emetine treatment compared with >90% inhibition of EBNA1-specific CD8+ T cells at the same concentration of emetine (Fig. 9 A). As expected, T cell recognition of the TRP2 peptide–pulsed target cells after the emetine treatment was slightly inhibited (Fig. 9 B). We also found that the treatment of target cells with emetine at a 1 μM concentration resulted in 20% inhibition of EBNA1-specific P3-B7 CD4+ T cell recognition (not depicted). Taken together, these results indicate that new protein synthesis is necessary and required for the generation of the HLA-B8–restricted epitope for T cell recognition, thus implying that DRiPs are the primary source of CD8+ T cell peptides. On the other hand, the processing and presentation of the TRP2 and CD4+ T cell EBNA1 epitopes are less dependent on the production of short-lived DRiPs.
Figure 9.
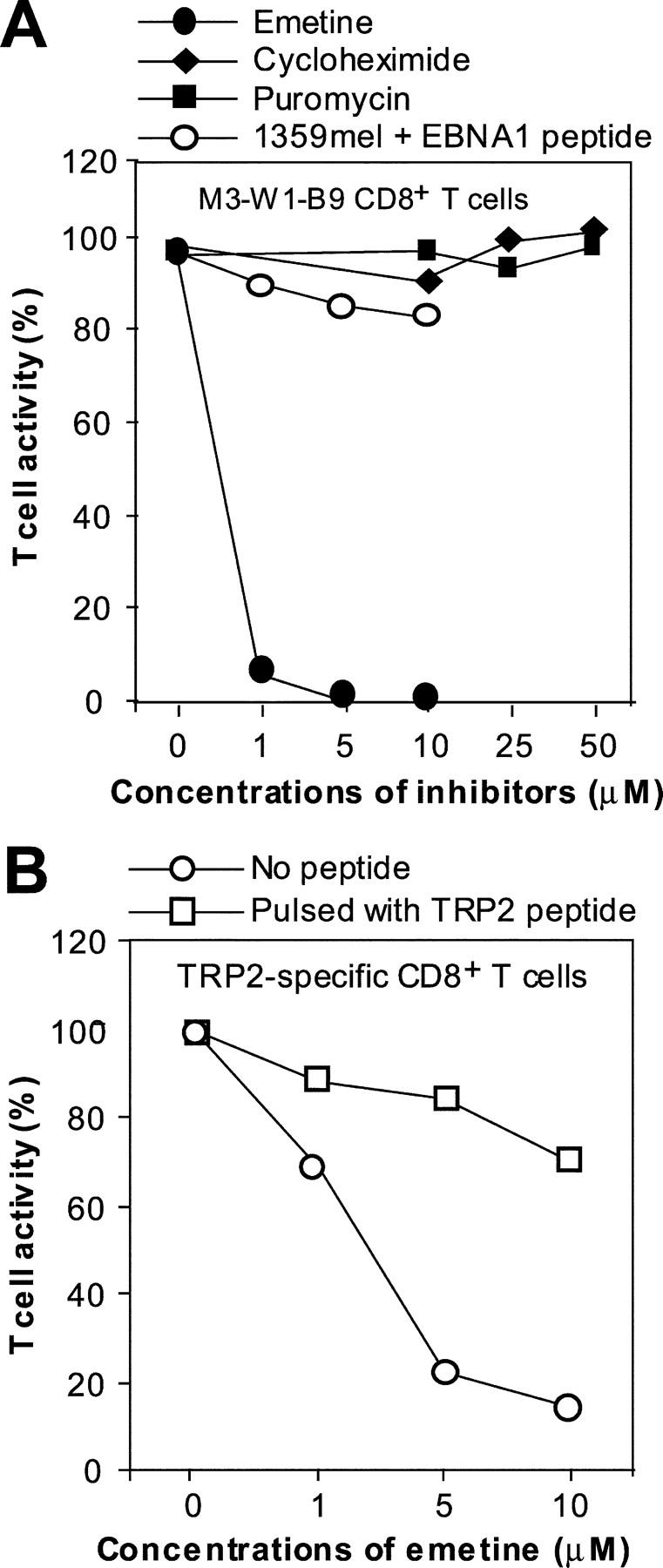
Inhibition of T cell recognition of EBNA1 by protein synthesis inhibitors. (A) Specific inhibition of M3-W1-B9 CD8+ T cell recognition of HEK293/B8/EBNA1-GFP target cells by an irreversible protein synthesis inhibitor emetine. HLA-B8–expressing HEK293/EBNA1-GFP target cells were treated with an emetine inhibitor at three concentrations for 1 h. After three washes, cells were cocultured with M3-W1-B9 CD8+ T cells overnight for IFN-γ release assays. Similar experiments were performed for the treatment of cells with cycloheximide or puromycin. To determine effect of emetine on recognition of MHC class I/EBNA1 peptide on the cell surface, we also pulsed HLA-B8+ 1359 cells with the EBNA1-P 518–526 peptide after the treatment of 1359mel cells with three different concentration of emetine. (B) Determination of the sensitivity of recognition of TRP2-specific CD8+ T cells to the treatment with emetine. 1363mel cells were treated with three different concentrations of emetine. After three washes, the cells were cocultured with TRP2-specific CD8+ T cells. The treated cells pulsed with a TRP2 peptide were used to examine the effect of emetine on recognition of MHC class I–TRP2 complexes on the cell surface.
Serine Proteases Are Involved in the Generation of T Cell Epitopes.
Next, we sought to determine if other proteases are involved in the degradation of short-lived DRiPs of EBNA1 because the GAr domain may need to be removed before they are further degraded by proteasomes. To identify individual protease inhibitors that might inhibit the presentation of the HLA-B8–restricted epitope to T cells, we selected seven protease inhibitors for further testing. Fig. 10 A shows that two such protease inhibitors, TPCK and AEBSF, significantly blocked CD8+ T cell recognition of the HLA-B8–expressing HEK293/EBNA1-GFP target cells when treated for 2 h at the lowest effective concentrations suggested by the manufacturer. None of the other protease inhibitors were effective, even after treatment for 2 or 8 h. To test whether TPCK or AEBSF affected T cell recognition of other antigens, we treated 1359mel with the same concentrations of TPCK or AEBSF as shown in Fig. 10 A. No inhibition was observed for recognition of 1359mel cells by 1359mel-specific CD8+ T cells (Fig. 10 B), suggesting that both inhibitors are specific for processing and presentation of the HLA-B8–restricted EBNA1 epitope. TPCK or AEBSF inhibited T cell recognition of target cells in a dose-dependent manner (Fig. 10 C), whereas TLCK produced a partial inhibitory effect when used at a high concentration. Although TLCK and TPCK have similar specificities, TLCK is unstable in solution according to the manufacturer. AEBSF is an irreversible, specific inhibitor of serine proteases, whereas TPCK is an irreversible inhibitor of chymotrypsin and many other serine and cysteine proteases. These results suggest that serine proteases are also required for the processing of the HLA-B8–restricted T cell epitope.
Figure 10.

Requirement of serine proteases for the generation of EBNA1 T cell epitope. (A) Specific inhibition of M3-W1-B9 CD8+ T cell recognition of HEK293/B8/EBNA1-GFP target cells by protease inhibitors. Target cells were incubated with various protease inhibitors for 2 h, washed, and cocultured with CD8+ T cells overnight for IFN-γ release assays. Solvents used to solubilize inhibitors were also used as controls. Experiments were performed in triplicate wells. CD8+ T cell recognition of target cells was inhibited by treatment with TPCK and AEBSF inhibitors. (B) Effect of protease inhibitors on recognition of 1359mel cells by 1359mel-specific CD8+ T cells. (C) Dose-dependent inhibition of M3-W1-B9 CD8+ T cell recognition of HEK293/B8/EBNA1-GFP target cells by protease inhibitors. Target cells were treated with different concentrations of protease inhibitors. After washes, the treated cells were cocultured with M3-W1-B9 CD8+ T cells overnight for IFN-γ release assays.
Discussion
Because of its important role in many EBV-associated cancers, EBNA1 has been suggested as an important target for immunotherapy (4, 18). However, efforts by several groups to identify MHC class I–restricted peptides from EBNA1 have proved disappointing. CD8+ T cells were generated that could recognize the peptide-pulsed target cells, but failed to recognize EBNA-1–expressing B cell lines and tumor cells (5). The results presented here demonstrate that CD8+ T cells generated from in vitro peptide stimulation are capable of recognizing 13 and 9-mer EBNA1 peptides, and that the 9-mer EBNA1 peptide is the more effectively recognized of the two (Fig. 2 C). More importantly, our data showed that EBNA1-specific CD8+ T cells can recognize EBNA1/HLA-B8 expressed by HEK293, 1359mel, and LCL 111 cells (Figs. 3–5), suggesting that EBNA1 could be endogenously processed and presented by HLA-B8 molecules for T cell recognition.
Although LCLs or HEK293 cells can take up exogenous EBNA1 protein from dead cells and then process and present derivative peptides to T cells through cross-presentation (28), our coculture experiments demonstrated that mixing HLA-B8–expressing HEK293 cells with HEK293 cells transfected with EBNA1 did not confer T cell recognition (Fig. 4 D). Hence, HLA-B8 and EBNA1 must be expressed in the same HEK293 cells for EBNA1 to be processed and presented by HLA-B8 molecules. Like HEK293 cells, LCLs differ from dendritic cells in having a poor capacity to cross-present antigen to T cells (18–20).
The discrepancy between previous studies and ours with regard to the recognition of EBV+ LCL cells by CD8+ T cells may have several explanations. First, the avidity of T cells generated from PBMCs stimulated in vitro is a critical factor. T cells with a low avidity may recognize only the peptide-pulsed target cells, whereas T cells with a high avidity would recognize both the peptide-pulsed target cells, EBV+ LCL, and tumor cells. For example, the EBNA1-specific CD4+ T cells described previously by two groups recognized HLA-DR1–restricted EBNA1 peptide, but failed to recognize EBV+ tumor cells (9, 29). However, a recent study showed that CD4+ T cells recognizing the same HLA-DR1–restricted peptide also recognized EBV+ BL tumor cells (10). Second, it has been reported that some CD8+ T cells exhibit poor cytolytic activity, but secrete high levels of cytokines such as IFN-γ (30). In another study, antiviral CD8+ T cells were not cytolytic due to their low level of perforin (31). Our strategy to generate EBNA1-specific T cells was based on measurement of IFN-γ release, but not on cytolytic activity. We generated T cell clones by a limiting dilution method and screened them for high IFN-γ release, showing that our CD8+ T cell clones could recognize peptide-pulsed, EBNA1-transfected target cells as well as EBV+ B cells based on IFN-γ release assays. Although our T cells clearly lyse EBV+ LCLs, their cytolytic activity is moderate compared with cytokine release. These issues warrant further investigation in the near future.
The identification of the HLA-B8–restricted EBNA1 peptide may provide a new opportunity for the development of immunotherapy against BL. However, due to MHC class I down-regulation in BL (4), the usefulness of the HLA-B8–restricted epitope requires further investigation. Our study demonstrates that the EBNA1-P518–526 peptide-specific CD8+ T cells are readily detectable in the HLA-B8–expressing donor PBMCs, suggesting that such CD8+ T cell responses to this peptide are commonplace in HLA-B8+ EBV-infected individuals. HLA-B8–restricted CD8+ T cells established from these HLA-B8+ donors responded functionally to the peptide-pulsed target cells as well as HLA-B8–matched LCLs (Fig. 7), further suggesting that the EBNA1 T cell peptides are naturally processed and presented to CD8+ T cells. Interestingly, this EBNA1 peptide overlaps with those presented by both DR1 (P514–527) and DP3 (P518–530) molecules (10, 11). Because our CD8+ T cells recognized HLA-DR1– and HLA-DP3–restricted peptides (Fig. 6), immunization with this dual peptide might stimulate both CD4+ and CD8+ T cell responses (32). Thus, the findings presented here could contribute to the development of vaccines for the prevention and treatment of EBV-associated tumors.
The failure to generate CD8+ T cells capable of recognizing naturally processed EBNA1 peptide on EBV+ cells has been attributed to the inhibitory effect of the GAr domain within EBNA1 on MHC class I–restricted antigen processing and presentation (6–8). In this study, we showed that all forms of EBNA1 (native, EBNA1-GFP, and EBNA1-GAr-del-GFP) conferred T cell recognition when cotransfected into HEK293 cells with the HLA-B8 cDNA (Fig. 4, B and C). Hence, it appears that the GAr domain does not play a significant role in inhibiting the processing and presentation of the HLA-B8–restricted EBNA1 peptide through the MHC class I pathway. This notion is further supported by our data showing that proteasome inhibitors can block the processing and presentation of HLA-B8–restricted EBNA1 peptide for T cell recognition through the MHC class I pathway (Fig. 8). The failure of chloroquine, an MHC class II pathway inhibitor, to inhibit T cell recognition of EBNA1 and HLA-B8–expressing HEK293 cells and LCL 111 cells further suggests that the MHC class II pathway does not play an important role in generating HLA-B8–restricted EBNA1 peptide (Fig. 8). Our data further indicate that newly synthesized DRiPs, rather than the full-length EBNA1 antigen, are the major source of the HLA-B8–restricted epitope (Fig. 9). These results are consistent with a recent study showing that inhibition of proteasomal degradation by the GAr domain is not sufficient to prevent presentation of epitopes from the GAr-containing proteins to the MHC class I pathway (33). Importantly, the increase in antigenic peptide production from the GAr-containing proteins correlates with the rate of protein synthesis (33). Taken together, these data support a mechanism by which the MHC class I peptides are derived through the degradation of short-lived DRiPs. The molecular basis for this mechanism was investigated by treating target cells with AEBSF and TPCK at a low effective concentration that resulted in significant inhibition of T cell recognition. Other protease inhibitors failed to inhibit the presentation of the HLA-B8–restricted T cell epitope to T cells (Fig. 10). Because both AEBSF and TPCK inhibit serine proteases, we suggest that at least one or more serine proteases are required for the initial cleavage of the GAr domain from the DRiPs of EBNA1 before further digestion by proteasomes. Thus, our results demonstrate for the first time that the short-lived DRiPs of EBNA1 may serve as the primary source of the HLA-B8–restricted epitope and that their degradation requires participation of both serine proteases and proteasomes.
Acknowledgments
We thank Drs. R. Khanna at Division of Infectious Diseases and Immunology, Queensland Institute of Medical Research, University of Queensland, Queensland, Australia for kindly providing plasmids encoding EBNA1-GFP and EBNA1-GAr-del-GFP, and for helpful discussions and suggestions, and Christian Münz at the Rockefeller University, NY, for kindly providing the A4.E116 CD4+ T cells.
This work was supported in part by the Startup funds of Baylor College of Medicine and grants from the National Institutes of Health (to R.F. Wang).
Abbreviations used in this paper: BL, Burkitt's lymphoma; DRiPs, defective ribosomal products; EBNA1, EBV-encoded nuclear antigen 1; E/T, effector to target; GAr, Gly-Ala repeat; GFP, green fluorescent protein; HD, Hodgkin's disease; LCL, EBV-transformed B lymphoblastoid cell line; NPC, nasopharyngeal carcinoma; TRP2, tyrosinase-related protein 2; ZAL, Z-Ile-Glu(OtBu)-Ala-Leucinal.
References
- 1.Kieff, E. 1995. Epstein-Barr virus–increasing evidence of a link to carcinoma. N. Engl. J. Med. 333:724–726. [DOI] [PubMed] [Google Scholar]
- 2.Rickinson, A.B., and D.J. Moss. 1997. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annu. Rev. Immunol. 15:405–431. [DOI] [PubMed] [Google Scholar]
- 3.Rowe, M., D.T. Rowe, C.D. Gregory, L.S. Young, P.J. Farrell, H. Rupani, and A.B. Rickinson. 1987. Differences in B cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt's lymphoma cells. EMBO J. 6:2743–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khanna, R., D.J. Moss, and S.R. Burrows. 1999. Vaccine strategies against Epstein-Barr virus-associated diseases: lessons from studies on cytotoxic T-cell-mediated immune regulation. Immunol. Rev. 170:49–64. [DOI] [PubMed] [Google Scholar]
- 5.Blake, N., T. Haigh, G. Shaka'a, D. Croom-Carter, and A. Rickinson. 2000. The importance of exogenous antigen in priming the human CD8+ T cell response: lessons from the EBV nuclear antigen EBNA1. J. Immunol. 165:7078–7087. [DOI] [PubMed] [Google Scholar]
- 6.Levitskaya, J., M. Coram, V. Levitsky, S. Imreh, P.M. Steigerwald-Mullen, G. Klein, M.G. Kurilla, and M.G. Masucci. 1995. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature. 375:685–688. [DOI] [PubMed] [Google Scholar]
- 7.Blake, N., S. Lee, I. Redchenko, W. Thomas, N. Steven, A. Leese, P. Steigerwald-Mullen, M.G. Kurilla, L. Frappier, and A. Rickinson. 1997. Human CD8+ T cell responses to EBV EBNA1:HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity. 7:791–802. [DOI] [PubMed] [Google Scholar]
- 8.Levitskaya, J., A. Sharipo, A. Leonchiks, A. Ciechanover, and M.G. Masucci. 1997. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA. 94:12616–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leen, A., P. Meij, I. Redchenko, J. Middeldorp, E. Bloemena, A. Rickinson, and N. Blake. 2001. Differential immunogenicity of Epstein-Barr virus latent-cycle proteins for human CD4(+) T-helper 1 responses. J. Virol. 75:8649–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paludan, C., K. Bickham, S. Nikiforow, M.L. Tsang, K. Goodman, W.A. Hanekom, J.F. Fonteneau, S. Stevanovic, and C. Munz. 2002. Epstein-Barr nuclear antigen 1-specific CD4(+) Th1 cells kill Burkitt's lymphoma cells. J. Immunol. 169:1593–1603. [DOI] [PubMed] [Google Scholar]
- 11.Voo, K.S., T. Fu, H.E. Heslop, M.K. Brenner, C.M. Rooney, and R.-F. Wang. 2002. Identification of HLA-DP3-restricted peptides from EBNA1 recognized by CD4+ T cells. Cancer Res. 62:7195–7199. [PubMed] [Google Scholar]
- 12.Wang, R.-F., S.L. Johnston, G. Zeng, D.J. Schwartzentruber, and S.A. Rosenberg. 1998. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J. Immunol. 161:3596–3606. [PubMed] [Google Scholar]
- 13.Wang, R.-F., M.R. Parkhurst, Y. Kawakami, P.F. Robbins, and S.A. Rosenberg. 1996. Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J. Exp. Med. 183:1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan, L.C., N. Gudgeon, N.E. Annels, P. Hansasuta, C.A. O'Callaghan, S. Rowland-Jones, A.J. McMichael, A.B. Rickinson, and M.F. Callan. 1999. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J. Immunol. 162:1827–1835. [PubMed] [Google Scholar]
- 15.Tellam, J., M. Sherritt, S. Thomson, R. Tellam, D.J. Moss, S.R. Burrows, E. Wiertz, and R. Khanna. 2001. Targeting of EBNA1 for rapid intracellular degradation overrides the inhibitory effects of the Gly-Ala repeat domain and restores CD8+ T cell recognition. J. Biol. Chem. 276:33353–33360. [DOI] [PubMed] [Google Scholar]
- 16.Wang, R.-F., X. Wang, S.L. Johnston, G. Zeng, P.F. Robbins, and S.A. Rosenberg. 1998. Development of a retrovirus-based complementary DNA expression system for the cloning of tumor antigens. Cancer Res. 58:3519–3525. [PubMed] [Google Scholar]
- 17.Wang, R.-F., X. Wang, A.C. Atwood, S.L. Topalian, and S.A. Rosenberg. 1999. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science. 284:1351–1354. [DOI] [PubMed] [Google Scholar]
- 18.Munz, C., K.L. Bickham, M. Subklewe, M.L. Tsang, A. Chahroudi, M.G. Kurilla, D. Zhang, M. O'Donnell, and R.M. Steinman. 2000. Human CD4+ T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J. Exp. Med. 191:1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinman, R.M., and M. Dhodapkar. 2001. Active immunization against cancer with dendritic cells: the near future. Int. J. Cancer. 94:459–473. [DOI] [PubMed] [Google Scholar]
- 20.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19:47–64. [DOI] [PubMed] [Google Scholar]
- 21.Wang, R.F., and S.A. Rosenberg. 1999. Human tumor antigens for cancer vaccine development. Immunol. Rev. 170:85–100. [DOI] [PubMed] [Google Scholar]
- 22.Rock, K.L., I.A. York, T. Saric, and A.L. Goldberg. 2002. Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 80:1–70. [DOI] [PubMed] [Google Scholar]
- 23.Gromme, M., F.G. Uytdehaag, H. Janssen, J. Calafat, R.S. van Binnendijk, M.J. Kenter, A. Tulp, D. Verwoerd, and J. Neefjes. 1999. Recycling MHC class I molecules and endosomal peptide loading. Proc. Natl. Acad. Sci. USA. 96:10326–10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Germain, R.N. 1994. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 76:287–299. [DOI] [PubMed] [Google Scholar]
- 25.Schubert, U., L.C. Anton, J. Gibbs, C.C. Norbury, J.W. Yewdell, and J.R. Bennink. 2000. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 404:770–774. [DOI] [PubMed] [Google Scholar]
- 26.Reits, E.A., J.C. Vos, M. Gromme, and J. Neefjes. 2000. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature. 404:774–778. [DOI] [PubMed] [Google Scholar]
- 27.Yewdell, J.W., U. Schubert, and J.R. Bennink. 2001. At the crossroads of cell biology and immunology: DRiPs and other sources of peptide ligands for MHC class I molecules. J. Cell Sci. 114:845–851. [DOI] [PubMed] [Google Scholar]
- 28.Larsson, M., J.F. Fonteneau, and N. Bhardwaj. 2001. Dendritic cells resurrect antigens from dead cells. Trends Immunol. 22:141–148. [DOI] [PubMed] [Google Scholar]
- 29.Khanna, R., S.R. Burrows, P.M. Steigerwald-Mullen, S.A. Thomson, M.G. Kurilla, and D.J. Moss. 1995. Isolation of cytotoxic T lymphocytes from healthy seropositive individuals specific for peptide epitopes from Epstein-Barr virus nuclear antigen 1: implications for viral persistence and tumor surveillance. Virology. 214:633–637. [DOI] [PubMed] [Google Scholar]
- 30.Shi, Y., and C.T. Lutz. 2002. Interferon-gamma control of EBV-transformed B cells: a role for CD8+ T cells that poorly kill EBV-infected cells. Viral Immunol. 15:213–225. [DOI] [PubMed] [Google Scholar]
- 31.Zhang, D., P. Shankar, Z. Xu, B. Harnisch, G. Chen, C. Lange, S.J. Lee, H. Valdez, M.M. Lederman, and J. Lieberman. 2003. Most antiviral CD8 T cells during chronic viral infection do not express high levels of perforin and are not directly cytotoxic. Blood. 101:226–235. [DOI] [PubMed] [Google Scholar]
- 32.Wang, R.-F. 2001. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol. 22:269–276. [DOI] [PubMed] [Google Scholar]
- 33.Yin, Y., B. Manoury, and R. Fahraeus. 2003. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science. 301:1371–1374. [DOI] [PubMed] [Google Scholar]