

Abstract
Interleukin (IL)-18 was originally regarded to induce T helper cell (Th)1-related cytokines. In general, factors favoring interferon (IFN)-γ production are believed to abolish allergic diseases. Thus, we tested the role of IL-18 in regulation of bronchial asthma. To avoid a background response of host-derived T cells, we administered memory type Th1 or Th2 cells into unsensitized mice and examined their role in induction of bronchial asthma. Administration of antigen (Ag) induced both airway inflammation and airway hyperresponsiveness (AHR) in mice receiving memory Th2 cells. In contrast, the same treatment induced only airway inflammation but not AHR in mice receiving memory Th1 cells. However, these mice developed striking AHR when they were coadministered with IL-18. Furthermore, mice having received IFN-γ–expressing Th1 cells sorted from polarized Th1 cells developed severe airway inflammation and AHR after intranasal administration of Ag and IL-18. Thus, Th1 cells become harmful when they are stimulated with Ag and IL-18. Newly polarized Th1 cells and IFN-γ–expressing Th1 cells, both of which express IL-18 receptor α chain strongly, produce IFN-γ, IL-9, IL-13, granulocyte/macrophage colony-stimulating factor, tumor necrosis factor α, regulated on activation, normal T cell expressed and secreted, and macrophage inflammatory protein 1α upon stimulation with Ag, IL-2, and IL-18 in vitro. Thus, Ag and IL-18 stimulate memory Th1 cells to induce severe airway inflammation and AHR in the naive host.
Keywords: IL-9, IL-13, IL-18, Th1, bronchial asthma
Introduction
Asthma is a disease of respiratory tract caused by repeated immediate hypersensitivity and late phase reactions in the lungs that lead to intermittent and reversible airway obstruction (1–4). There are infiltrates of neutrophils, eosinophils, degranulated mast cells, subbasement membrane thickening, and hyperplasia of goblet cells in the airway. It is widely accepted that the Th2 cytokines (IL-4, IL-5, IL-9, and IL-13) and GM-CSF are principally responsible for inducing bronchial asthma (5–7). IL-4 is pivotal as the driving cytokine toward Th2 phenotype and induces IgE production from B cells (8). IL-5 induces maturation and activation of eosinophils (9). IL-4 and IL-9 are potent mast cell growth factors (10, 11). IL-9 and IL-13 induce mucus production by causing goblet cell hyperplasia (12–15). Furthermore, IL-13 is shown to be necessary and sufficient to induce airway hyperresponsiveness (AHR; 16–19). GM-CSF induces Th2 differentiation independently of IL-4 and also maintains the survival of eosinophils (20). In contrast, Th1 cells had been regarded to protect against bronchial asthma by damping the activity of Th2 cells (21–23). However, several studies indicated the failure of Th1 cells to attenuate Th2 cell–induced airway hypersensitivity (1, 24–28). Furthermore, Th1 cells are reported to induce AHR by recruitment and activation of neutrophils (29). These results prompted us to examine the pathological relevance of passively transferred memory-type Th1 cells in induction of bronchial asthma in unsensitized host mice. Importantly, this type of study allowed us to depict selectively the response of memory-type T cells in the naive animals.
IL-18 was originally identified as a factor that enhances IFN-γ production from Th1 cells in the presence of anti-CD3 and IL-12 (30–32). However, our recent studies and those of others demonstrated that IL-18 promotes Th2 cytokine production from T cells, NK cells, basophils, and mast cells (33–38). As IL-18 is abundantly stored in the epithelial cells of various organs (39–42), we could assume the possibility that some types of infectious agents might stimulate bronchial epithelial cells to produce IL-18. Thus, it is very important to determine the biological relevance of IL-18 in bronchial asthma. IL-18 responsiveness is determined by the expression of IL-18R, a heterodimeric complex consisting of ligand-binding α chain (IL-18Rα) and an associating β chain (IL-18Rβ; 38). Naive T cells, when stimulated with Ag and IL-12 or IL-4, develop into Th1 or Th2 cells, respectively (43). As IL-12 induces but IL-4 down-regulates IL-18Rα on T cells (44, 45), only Th1 cells become the major target cells for IL-18 and produce IFN-γ strongly when stimulated with Ag and IL-18. However, we recently found and reported that NKT cells constitutively express high levels of IL-18Rα and produce Th2 cytokines (e.g., IL-4, IL-9, IL-13, etc.), when they are stimulated with IL-2 and IL-18 without TCR engagement, although they have the potential to produce a large amount of IFN-γ in response to IL-12 and IL-18 (35). Therefore, it is important to investigate whether IL-18Rα–expressing Th1 cells also have the potential to produce Th1 or Th2 cytokines in response to IL-18 depending on cytokine milieu.
Here we have demonstrated that adoptively transferred memory-type Th1 cells when stimulated with Ag in the lung induce airway inflammation without AHR. In contrast, memory Th1 cells induce both severe airway inflammation and AHR when they are stimulated with Ag and IL-18. Surprisingly, like activated mast cells, Ag plus IL-18–stimulated Th1 cells produce IFN-γ, IL-9, IL-13, GM-CSF, TNF-α, regulated on activation, normal T cell expressed and secreted (RANTES), and macrophage inflammatory protein (MIP)-1α, suggesting their relevant role in induction of bronchial asthma.
Materials and Methods
Animals and Reagents.
Specific pathogen-free female BALB/c mice were purchased from The Jackson Laboratory. Mice transgenic (Tg) for αβ TCR recognizing OVA323–339 (DO11.10, BALB/c genetic background; reference 46) were provided by D. Loh (Washington University, St. Louis, MO). All experiments were performed on the line heterozygous for the transgene. All mice were bred under specific pathogen-free conditions at the animal facilities of Hyogo College of Medicine and were used at 6–10 wk of age. Recombinant mouse IL-4, obtained from a recombinant baculovirus (AcMNPV.IL-4), was purified by affinity chromatography and used. Recombinant mouse IL-12, IL-18, and anti–mouse IL-18Rα chain mAb (Y38; reference 47) were provided by Hayashibara Biochemical Laboratories Inc. PE-KJ1-26, which recognizes TCR from DO11.10 mice, was purchased from CALTAG Laboratories and used to specifically stain the adoptively transferred DO11.10 TCR Tg cells. Purified antibodies (anti–mouse CD28 [37.51], anti–mouse CD3 [2C11], anti–mouse IL-4 [11B11], anti–mouse IL-12p40 [C17.8], and anti–mouse IFN-γ [XMG1.2]) were prepared in our laboratory. PE anti–mouse CD4 (GK1.5) allophycocyanin (APC) anti–mouse CD4, FITC anti–rat IgG1 (RG11/39.4), and FITC anti–mouse IFN-γ (XMG1.2) were purchased from BD Biosciences.
Generation of Th1 or Th2 Cells and Isolation of IFN-γ1 Cells.
105/ml naive splenic CD4+ CD62L+ T cells from DO11.10 Tg mice were stimulated with 100 pM IL-2 and 1 μM OVA323–339 in the presence of 106/ml Ag-presenting cells (irradiated T cell–depleted BALB/c splenocytes) in six-well plates in a total 3-ml volume of RPMI 1640 supplemented with 10% FBS, 50 μM 2-ME, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin for 7 d. For induction of Th1 cells, 10 ng/ml IL-12 and 10 μg/ml anti–IL-4 (11B11) were further added to the culture. For induction of Th2 cells, 1,000 U/ml IL-4, 10 μg/ml anti–IL-12p40, and 10 μg/ml anti–IFN-γ were added. Two rounds of antigenic stimulation were performed.
For isolation of IFN-γ+ Th1 cells, 106/ml T cells cultured under Th1-inducing condition for 7 d were recultured with 100 pM IL-2, 1 μM OVA323–339, plus 106/ml APC in six-well plates in a total 3-ml volume. After 8 h, only adherent cells were collected and incubated with the anti-CD45/anti–IFN-γ bispecific antibody capture matrix (Miltenyi Biotec) for 5 min on ice. The treated cells were transferred to 50-ml conical tubes and cultured at a concentration of 5 × 104 cells/ml in 20 ml warmed medium and placed in a 37°C water bath. After 30 min, cells were washed with cold 0.5% BSA in PBS. Matrix-captured IFN-γ was detected with PE anti–IFN-γ. IFN-γ+ cells were isolated by cell sorting (Elite; Coulter Electronics).
Th Clones.
Three murine CD4+ Th clones designated 9-16 (48), 35-9D (49), and D10.G4.1 (50) were provided by Y. Asano (Ehime University, Matsuyama, Japan), T. Yoshimoto (Tokyo Medical University, Tokyo, Japan), and S. Ono (Osaka University, Osaka, Japan), respectively. 9-16 is a KLH-specific, I-Ak–restricted Th1 clone. 35-9D is an OVA-specific, I-Ab–restricted Th1 clone. D10.G4.1 is a conalbumin-specific, I-Ak–restricted Th2 clone.
In Vitro Culture.
Polarized Th1 and Th2 cells and sorted IFN-γ+ cells from Th1 cells were recultured at 105/0.2 ml/well with 100 pM IL-2, 1 μM OVA323–339, and 105/0.2 ml APC in the presence of various concentrations of IL-18 (0–100 ng/ml). Th1 clone (9-16) and Th2 clone (D10.4G.1) were cultured at 105/0.2 ml/well with immobilized anti-CD3 and anti-CD28 (each 5 μg/ml) in the presence of various concentrations of IL-18 (0–100 ng/ml). After 48 h of culture, supernatants were harvested and tested for IFN-γ, IL-4, IL-5, IL-6, IL-9, IL-13, TNF-α, RANTES, MIP-1α, and GM-CSF contents by ELISA.
Flow Cytometry.
For intracellular cytokine staining, 106/ml polarized Th1 and Th2 cells were restimulated with 100 pM IL-2, 1 μM OVA323–339, and 106/ml APC with or without 50 ng/ml IL-18 for 16 h with a pulse of 1 μg/ml Brefeldin A during the final 4 h to inhibit cytokine secretion. Such cells were first stained with PE anti-CD4 and followed by fixation with 4% (wt/vol) paraformaldehyde in PBS and permeabilization of cell membrane with ice cold PBS containing 1% FCS plus 0.1% saponin. Resultant cells were treated with 10 μg/ml anti-FcγRII/III (24G2) for 30 min and then 10 μg/ml biotinylated goat anti–mouse IL-13 or biotinylated goat anti–mouse IgG (R&D Systems) for 30 min at 4°C in staining buffer (PBS, 1% FCS). Cells were washed twice and stained with APC-labeled streptavidin plus FITC rat anti–mouse IFN-γ or isotype-matched control mAb (BD Biosciences) for 30 min. Samples were analyzed for their proportion of cytoplasmic IFN-γ+ and/or IL-13+ cells gated on CD4+ T cells by FACSCalibur™ (BD Biosciences).
For determination of IL-18Rα chain expression on Th1 and Th2 cells, after FcR blocking with anti-FcγRII/III, cells were incubated with anti–mouse IL-18Rα chain mAb or control rat IgG1 mAb (R3-34) for 30 min at 4°C, followed by FITC anti–rat IgG1 Ab and PE anti–mouse CD4 for 30 min at 4°C in staining buffer. Samples were analyzed on a FACSCalibur™.
Transfer of Cells and Administration of OVA Plus IL-18.
Preparation of Th1 or Th2 memory cells in vivo has been described (51). Equal numbers (107) of Th1 cells, Th2 cells, or sorted IFN-γ+ cells were injected i.v. into normal BALB/c mice (10–15 mice per group). At various times after adoptive transfer, peripheral blood cells were collected and stained with APC anti-CD4 and PE-KJ1-26 to ensure that all DO11.10 TCR Tg cells were included in the analysis. 30 d after transfer of cells, animals were daily exposed intranasally to PBS or 1 μg OVA and/or 0.5 μg IL-18 in 50 μl PBS for 3 d. Control mice were exposed to PBS. Mice were analyzed at 24 h after the final exposure to Ag. For the blockade of IL-13 in vivo, 20 μg sIL-13Rα2-Fc or 20 μg control human IgG (Genetics Institute Inc.) was daily administered intranasally as the mixed form with OVA and IL-18 for 3 d.
Measurement of AHR.
We measured AHR to β-methacholine (Mch) inhalation in mice by using Pulmos-I (MIPS) hardware and software. We placed a mouse in a chamber and exposed it to aerosols of saline (baseline) first and then to increased concentrations of Mch (5, 10, 15, and 20 mg/ml). After each 2-min exposure, we measured enhanced pause, a dimensionless index that reflects changes in amplitude of pressure wave form and expiratory time (52), for 3 min.
Bronchoalveolar Lavage (BAL).
BAL was performed with three aliquots of 0.5 ml PBS per mouse. Total cell counts were performed. Cytospin preparations of BAL fluid (BALF) were stained with Dif-Quik (Baxter Healthcare Corp.) and differentials were performed based on morphology and staining characteristics.
Histology.
Lungs were prepared for histology by perfusing the animal via the right ventricle with 10 ml PBS and then fixed in 10% buffered formalin, cut into 3-μm sections, and stained with hematoxylin and eosin.
Results
Coadministration of OVA and IL-18 Induces Airway Inflammation and AHR in Mice Receiving OVA-specific Memory Th1 cells.
We established Th1 or Th2 cells by two rounds of 7-d stimulation of CD4+ T cells obtained from OVA-specific TCR Tg BALB/c (DO11.10) mice with OVA peptide, IL-12, and anti–IL-4 or with OVA peptide, IL-4, and anti–IL-12/anti–IFN-γ in the presence of APC for 2 wk, respectively. We then transferred these newly polarized Th1 or Th2 cells i.v. into normal BALB/c mice (107 per mouse) and allowed them to adopt a resting memory phenotype in vivo (51). 30 d after passive cell transfer, these recipients were daily treated with intranasal administration of OVA and/or IL-18 for 3 d. Therefore in this experimental system, we could depict the capacity of the memory-type Th1 or Th2 cells to induce bronchial asthma in naive mice. Histopathological analysis of the airways of each group of mice revealed that Th2 cell–transferred mice showed a significant increase in the peribronchial and perivenular infiltration with eosinophils and lymphocytes when compared with normal mice after nasal exposure to OVA (Fig. 1, c, m, and o). Furthermore, these mice showed a dose-dependent AHR to McH challenge (Fig. 2 A). In contrast, mice having received Th1 cells 30 d earlier showed significant cell infiltration composed mainly of lymphocytes and neutrophils after OVA administration (Fig. 1, g and h) but failed to respond to Mch challenge by AHR (Fig. 2 A). However, when they were nasally exposed to both Ag and IL-18, these mice increased their pathological changes characterized by massive cell infiltration (Fig. 1, i and j) and AHR (Fig. 2 A), suggesting that Ag and IL-18 stimulate Th1 cells to produce cytokines and chemokines that induce or enhance infiltration with eosinophils, lymphocytes, and neutrophils (Fig. 1, j). In contrast, mice that received Th1 or Th2 cells 30 d earlier showed neither lung inflammation (Fig. 1, e, f, k, and l) nor AHR (Fig. 2 A) after nasal exposure to PBS or IL-18, suggesting the contribution of Ag- or Ag plus IL-18–induced recall responses of memory Th2 or Th1 cells, respectively, to induction of bronchial asthma. Thus, Ag stimulation of memory Th1 cells induced airway inflammation without AHR, whereas Ag plus IL-18 stimulation induced both airway inflammation and AHR.
Figure 1.
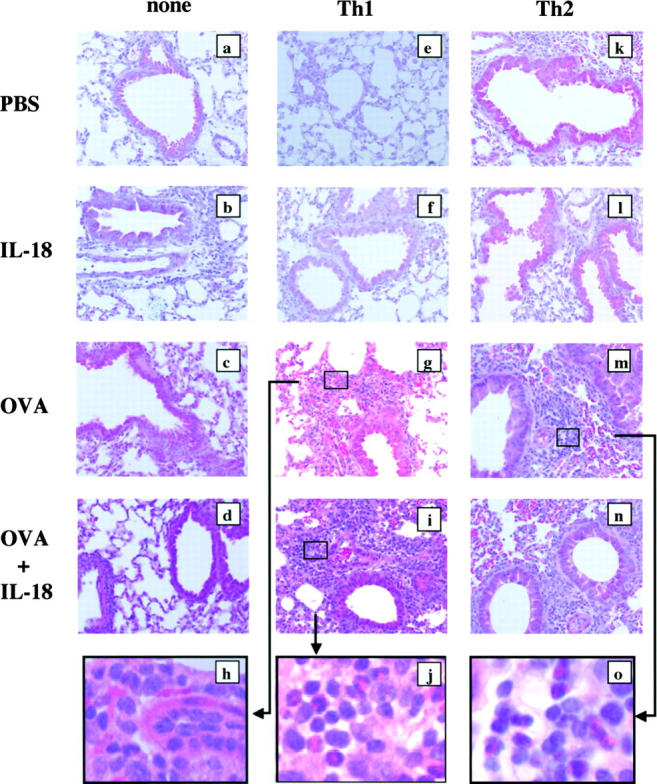
IL-18 induced airway inflammation and AHR in mice receiving memory Th1 cells. 30 d after adoptive transfer of OVA-specific Th1 (e–j) or Th2 cells (k–o) (107 cells per mouse) or none (a–d) into naive BALB/c mice intravenously, animals were daily exposed nasally to PBS alone or 1 μg OVA and/or 0.5 μg IL-18 in 50 μl PBS for 3 d. The allergic phenotype was assessed 24 h after the final exposure to antigen. Lungs were prepared for histology by perfusing the animal via the right ventricle with 10 ml PBS and then fixed in formalin, cut into 3-μm sections, and stained with hematoxylin and eosin. a–g, i, and k–n, ×50; h, j, and o, ×400. Representative results of five to seven animals are shown.
Figure 2.
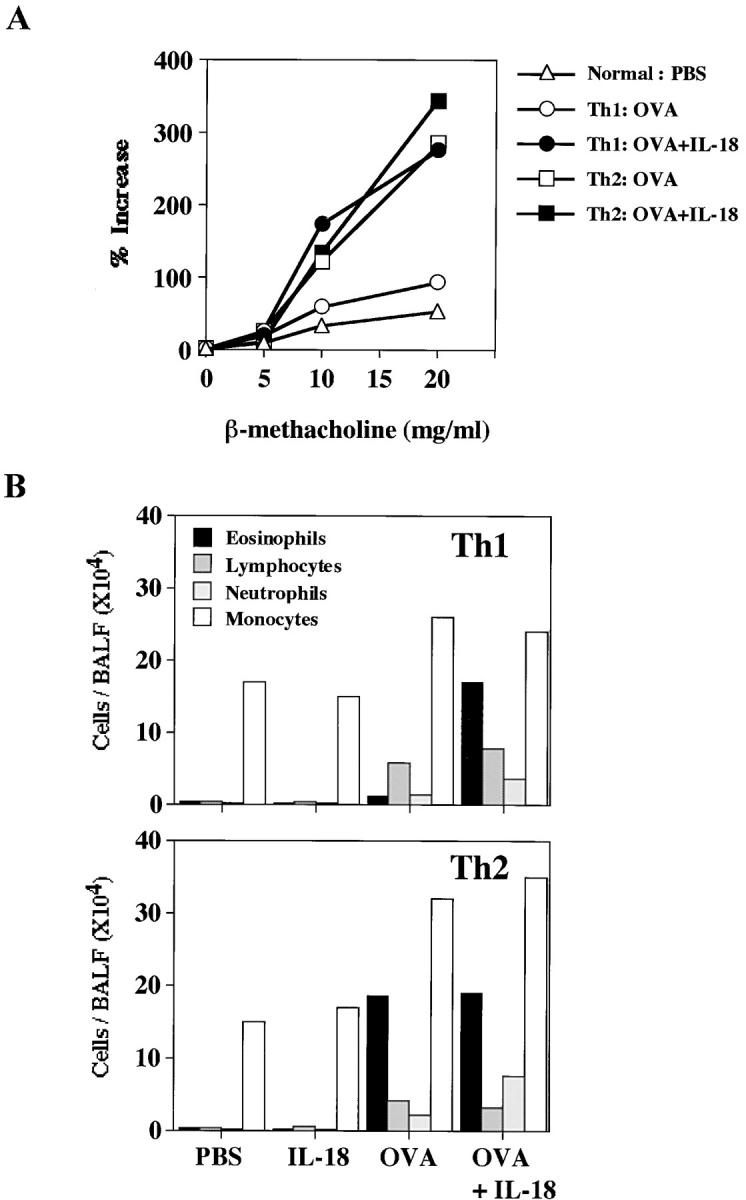
IL-18 induced AHR and eosinophilia in mice receiving memory Th1 cells. (A) AHR in response to increased concentrations of inhaled β-Mch was measured in a whole body plethysmograph. (B) Inflammatory cell composition of BALF from mice transferred with OVA-specific Th1 or Th2 cells and daily treated with intranasal administration of PBS or OVA and/or IL-18 consecutively for 3 d. Cell differential percentages were determined by light microscopic evaluation of cytospin preparation. Data are expressed as absolute numbers of cells. Representative results of five animals are shown.
It is important to know the reason why additional administration of IL-18 was required to induce bronchial asthma in Th1 cell–transferred mice. Therefore, we compared the numbers of inflammatory cells infiltrating lung tissues of Th1 cell–transferred mice after daily exposure to PBS, Ag, IL-18, or Ag plus IL-18 for 3 d (Fig. 2 B). Analysis of BALF revealed marked elevation of the eosinophil population in the lungs of Th1 cell–transferred mice after treatment with OVA plus IL-18, suggesting that Ag and IL-18 act on Th1 cells to produce soluble mediators that induce infiltration of eosinophils (Fig. 2 B). We also found that Ag plus IL-18 administration also induced increases in the number of lymphocytes, neutrophils, and monocytes (Fig. 2 B). The degree of the increase in the population of each type of cells is similar to that observed in BALF of Th2 cell–transferred mice after treatment with OVA or OVA plus IL-18 (Fig. 2 B). These results substantiated further the result of Fig. 1 that additional IL-18 stimulation changed quantitatively and qualitatively the constituents of inflammatory cells in the lungs. Thus, Ag stimulation alone only induced mild delayed-type hypersensitivity (DTH) in the lung, whereas Ag plus IL-18 stimulation induced very severe DTH, leading to induction of bronchial asthma.
IL-18 Stimulates OVA-specific Th1 Cells to Produce IFN-γ, Th2-related Cytokines, GM-CSF, and Chemokines In Vitro.
To understand how Ag and IL-18 induce airway inflammation and AHR, we next tested the types of cytokines or chemokines produced by Ag-, IL-2–, and IL-18–stimulated Th1 cells in vitro. We measured the concentrations of IL-4, IL-5, IL-6, IL-9, IL-13, TNF-α, GM-CSF, RANTES, eotaxin, MIP-1α, and IFN-γ in the culture supernatant of Th1 cells because they are reported to be deeply involved in induction of bronchial asthma (1–7). In particular, the chemokines RANTES, MIP-1α, and eotaxin are central to the delivery of eosinophils to the airway (53). Naive CD4+ T cells from OVA-specific TCR Tg mice were primed in vitro under Th1- or Th2-inducing conditions for two consecutive rounds of 7-d stimulation. Upon challenge with OVA peptide and IL-2, as reported elsewhere, Th2 cells produced significant amounts of IL-4, IL-5, IL-6, IL-9, and IL-13 but not IFN-γ, and Th1 cells produced IFN-γ but not Th2 cytokines (Fig. 3 A). Additional stimulation of Ag plus IL-2–stimulated Th1 cells with IL-18 dose dependently induced IFN-γ (Fig. 3 A; reference 44). Most surprisingly, this treatment simultaneously induced RANTES, MIP-1α, IL-9, IL-13, TNF-α (not depicted), and GM-CSF in Th1 cells (Fig. 3 A). Eotaxin was not detectable in any culture supernatant of Th1 cells. The mechanism underlying this difference in IL-18 responsiveness between Th1 and Th2 cells is principally explained by preferential expression of IL-18Rα chain on Th1 cells (44). As expected, Th1 cells express a high level of IL-18R, whereas Th2 cells express modestly (Fig. 3 B). Furthermore, they simultaneously produced a large amount of chemokines (Fig. 3 A). Thus, IL-18R–expressing Th1 cells are target cells for IL-18 and responded to it not only by increased production of IFN-γ, but also by production of some Th2 cytokines (IL-9 and IL-13), TNF-α (not depicted), GM-CSF, and chemokines. Because IL-12 plus IL-18 is known to induce IFN-γ production from Th1 cells but inhibit IL-4 and IL-13 production from Th2 cells in vivo and in vitro (30, 33, 38, 44), it is important to test the effect of IL-12 on the ability of IL-18 to induce IL-13 from Th1 cells. We found that IL-12 slightly diminished the production of IL-13 and GM-CSF from Th1 cells stimulated with IL-18 alone or IL-18 plus IL-2 in the presence of OVA (Fig. 3 C). Moreover, additional IL-12 stimulation did not affect IL-13 and GM-CSF production from Th1 cells stimulated with immobilized anti-CD3 plus anti-CD28 antibodies (not depicted). Thus, IL-12 is essential for induction of Th1 cells but not critically involved in induction of cytokine production from Th1 cells. As reported elsewhere, Th2 cells failed to respond to IL-18 by enhanced production of IL-4, although they modestly increased production of IL-5, IL-9, and IL-13 in response to IL-18 (Fig. 3 A). These results suggested the possible involvement of cytokines, particularly of IFN-γ, TNF-α, IL-9, IL-13, GM-CSF, and some chemokines from Th1 cells in induction of both severe airway inflammation and AHR.
Figure 3.

IL-18 induced IFN-γ, IL-9, IL-13, GM-CSF, RANTES, and MIP-1α production from OVA-specific Th1 cells. (A) 105/ml naive splenic CD4+ CD62L+ T cells from DO11.10 Tg mice were cultured with 100 pM IL-2 plus 1 μM OVA323–339 and 106/ml irradiated T cell–depleted BALB/c splenocytes in six-well plates in a total 3-ml volume for 7 d. For differentiation of Th1 cells, 10 ng/ml IL-12 and 10 μg/ml anti–IL-4 were added to the culture. For the differentiation of Th2 cells, 1,000 U/ml IL-4, 10 μg/ml anti–IL-12p40, and 10 μg/ml anti–IFN-γ were added. Two rounds of antigenic stimulations under polarizing conditions were performed. After priming, cells were washed and recultured at 105/0.2 ml/well with 100 pM IL-2, 1 μM OVA323–339, plus 105/0.2 ml irradiated T cell–depleted BALB/c splenocytes in the presence of various concentrations of IL-18 (0–100 ng/ml). After 48 h of culture, supernatants were harvested and tested for IFN-γ, IL-4, IL-5, IL-6, IL-9, IL-13, RANTES, MIP-1α, and GM-CSF contents by ELISA. Results are geometric means ± SEM. (B) Surface expression of IL-18Rα chain by flow cytometry. The percentages shown represent the proportion of IL-18Rα chain+ cells among CD4+ cells. (C) Polarized Th1 cells were cultured at 105/0.2 ml/well with medium alone or various combinations of 100 pM IL-2, 20 ng/ml IL-12, and 50 ng/ml IL-18 in the presence of 1 μM OVA323–339 plus 105/0.2 ml irradiated T cell–depleted BALB/c splenocytes. After 48 h of culture, supernatants were harvested and tested for IFN-γ, IL-13, and GM-CSF contents by ELISA. Results are geometric means ± SEM.
IL-18 Stimulates Ag-specific Cloned Th1 Cells to Produce IL-13 and GM-CSF.
These results still leave the question of whether some other contaminated cells, such as NK cells or NKT cells, might have responded to IL-2 and IL-18 by production of IL-9, IL-13, and GM-CSF (35, 36). Therefore, we used cloned Th1 cells (9-16; reference 48) and Th2 cells (D10.4G.1; reference 50) specific for KLH and conalbumin, respectively. We stimulated them with immobilized anti-CD3 and anti-CD28 in the presence of various doses of IL-18. As previously reported, these cloned Th2 cells produced significant amounts of IL-4 and IL-13 but not IFN-γ, and cloned Th1 cells produced IFN-γ and GM-CSF but not Th2 cytokines (Fig. 4 A). We found that like newly established Th1 cells derived from DO11.10, a large proportion of stable cloned Th1 cells express a high level of IL-18Rα chain and produce GM-CSF and IL-13 dependently of doses of IL-18 used for stimulation (Fig. 4). In contrast, cloned Th2 cells, even if a substantial fraction of Th2 cells express IL-18Rα chain, did not produce IFN-γ when stimulated with immobilized anti-CD3/anti-CD28 and IL-18 (Fig. 4). We examined another OVA-specific Th1 clone (35-9D; reference 49). This clone produced significant amounts of IFN-γ but no IL-4 and IL-13. However, like 9-16 Th1 clone, 35-9D Th1 clone expressed a high level of IL-18Rα chain (80.0%) and increased IL-13 dependently of doses of IL-18 used for stimulation (not depicted). These results strongly indicated that even stable cloned Th1 cells gained the capacity to produce Th2 cytokines and GM-CSF when they were stimulated with Ag and IL-18, suggesting the importance of IL-18 in induction of Th2 cytokines from Th1 cells.
Figure 4.
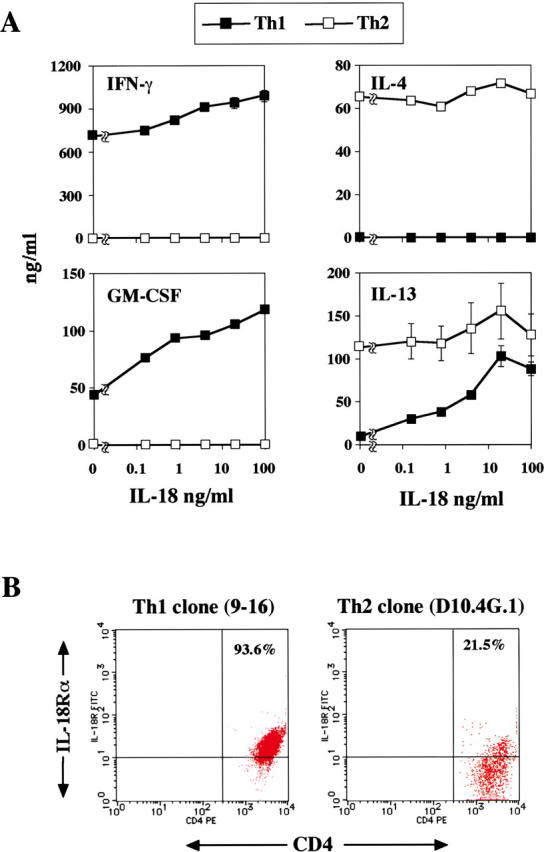
IL-18 induced IL-13 and GM-CSF production from antigen-specific Th1 clone. (A) Th1 clone (9-16), KLH-specific, I-Ak–restricted, and Th2 clone (D10.4G.1), conalbumin-splenic, I-Ak–restricted, were cultured at 105/0.2 ml/well with immobilized anti-CD3/anti-CD28 (each 5 μg/ml) in the presence of various concentrations of IL-18 (0–100 ng/ml). After 48 h of culture, supernatants were harvested and tested for IFN-γ, IL-4, GM-CSF, and IL-13 contents by ELISA. Results are geometric means ± SEM. (B) Surface expression of IL-18Rα chain by flow cytometry. The percentages shown represent the proportion of IL-18Rα chain+ cells among CD4+ cells.
Administration of IL-13 Antagonist Diminishes the Number of Eosinophils in BALF, but Fails to Abolish Ag plus IL-18–induced AHR in Th1 Cell–transferred Mice.
As IL-13 is reported to participate in airway inflammation and AHR (16–19), we next blocked the function of this cytokine by administration of IL-13 antagonist into the mice receiving Th1 cells (Fig. 5). We administered a soluble IL-13Rα2–human Fc fusion protein (IL-13R-Fc), which selectively binds to and neutralizes IL-13 but not IL-4 (34, 54), to these mice and compared them to mice that received control protein. Although this treatment significantly diminished the number of eosinophils in BALF (Fig. 5 B), little effect was seen in AHR upon Mch challenge (Fig. 5 A). In contrast, as previously reported, administration of IL-13 antagonist diminished both AHR and the number of eosinophils in BALF from Th2-transferred and OVA-stimulated mice (Fig. 5 B). Therefore, we could exclude the possibility that IL-13 is responsible for inducing AHR in mice receiving Th1 cells. Because the number of eosinophils was markedly diminished by neutralization of IL-13, these inflammatory cells were not involved in AHR in this experimental asthma model.
Figure 5.

Effect of the IL-13 blockage on IL-18 induced AHR and eosinophilia in mice receiving memory Th1 cells. 30 d after adoptive transfer of OVA-specific Th1 or Th2 cells (107 cells per mouse) into naive BALB/c mice intravenously, animals were daily exposed intranasally to PBS, 1 μg OVA alone, or OVA plus 0.5 μg IL-18 in 50 μl PBS for 3 d. For the blockade of IL-13 in vivo, 20 μg sIL-13Rα2-Fc or 20 μg control human IgG were daily administered intranasally as the mixed form with OVA and IL-18 for 3 d. 24 h after the final exposure to antigen, AHR in response to increasing concentrations of inhaled β-Mch (A) and inflammatory cell composition of BALF (B) were examined as described in Fig. 2. Representative results of four animals are shown.
IL-18 Stimulates IFN-γ+ Th1 Cells to Produce IFN-γ, IL-9, IL-13, and GM-SCF.
Although additional IL-18 stimulation dose dependently induced Ag plus IL-2–stimulated Th1 cells to produce both IFN-γ and IL-13 (Fig. 3 A), we still needed to exclude the possibility that potentially contaminated Th0 cells produce IL-13. To exclude this possibility, we examined the proportions of CD4+T cells producing IFN-γ and/or IL-13 in Th1 and Th2 cells stimulated with Ag, IL-2, and IL-18 for 16 h (Fig. 6 A). Th1 cells showed a significant increase in the proportion of Th1 cells producing IL-13 alone or both IFN-γ and IL-13. As expected, Th2 cells produced IL-13 but not IFN-γ, and responded poorly to additional IL-18 stimulation. These results strongly indicated that Th1 cells produce both IFN-γ and IL-13 when stimulated with Ag, IL-2, and IL-18.
Figure 6.

IL-18 induced IL-9, IL-13, and GM-CSF production from IFN-γ+ Th1 cells. (A) 106/ml polarized Th1 and Th2 cells, as described in Fig. 3, were restimulated with 100 pM IL-2, 1 μM OVA323–339, and 106/ml APC with or without 50 ng/ml IL-18 for 16 h. Intracellular IL-13 and IFN-γ staining was performed and analyzed as described in Materials and Methods. Numbers represent the percentage of IFN-γ+ and/or IL-13+ cells gated on CD4+ T cells. (B) 106/ml polarized Th1 cells were challenged with 106/ml OVA peptide and APC in six-well plates in a total 3-ml volume. After 8 h, cells were collected and incubated with the anti-CD45/anti–IFN-γ bispecific antibody capture matrix for 5 min on ice. The resultant cells were incubated and then washed as described in Materials and Methods. Matrix-captured IFN-γ was detected with PE anti–IFN-γ. This IFN-γ+ CD4+ T cell population was then isolated by cell sorting. Numbers represent the percentage of IFN-γ+ gated on CD4+ T cells. (C) IFN-γ+ cells were washed and recultured at 105/0.2 ml/well with 100 pM IL-2, 1 μM OVA323–339, plus 105/0.2 irradiated APC in the presence of various concentrations of IL-18 (0–100 ng/ml). After 48 h of culture, supernatants were harvested and tested for IFN-γ, IL-4, IL-9, IL-13, and GM-CSF contents by ELISA. Results are geometric means ± SEM. (D) 20 d after adoptive transfer of OVA-specific IFN-γ+ cells (107 cells per mouse) into naive BALB/c mice intravenously, animals were daily exposed intranasally to PBS alone or 1 μg OVA plus 0.5 μg IL-18 in 50 μl PBS for 3 d. 24 h after the final exposure to antigen, lungs were stained with hematoxylin and eosin. a and b, ×40; c, ×160.
We finally tested the capacity of highly purified Th1 cells to produce IFN-γ, GM-CSF, IL-9, and IL-13 in response to Ag, IL-2, and IL-18 in vitro (Fig. 6 C). For this purpose, we purified IFN-γ–expressing Th1 cells by capturing secreted IFN-γ by surface-bound anti–IFN-γ antibody. As shown in Fig. 6 B, 6.3% of Th1 cells express IFN-γ on their cell surfaces when Th1 cells stimulated with OVA peptide in the presence of APC for 8 h were examined by the affinity matrix technology, which allows detection of IFN-γ production by living cells. We then positively purified IFN-γ–producing living IFN-γ+ CD4+ Th1 cells (98.7%) by a fluorescence cell sorter. Culturing of sorted IFN-γ+ Th1 cells with OVA, IL-2, and IL-18 in the presence of APC led to an increase in IL-9, IL-13, IFN-γ, and GM-CSF but not IL-4 production (Fig. 6 C). Thus, we could formally conclude that Ag, IL-2, and IL-18 can stimulate Th1 cells to produce IFN-γ, GM-CSF, IL-9, and IL-13. To further clarify the pathological relevance of Th1 cells, we transferred FACS®-sorted OVA-specific IFN-γ+ Th1 cells into normal BALB/c mice and allowed them to adopt a resting memory phenotype. 30 d after cell transfer, these mice were nasally exposed to OVA plus IL-18 for 3 d. These animals developed peribronchial and perivenular infiltration with eosinophils and lymphocytes when examined by histopathologic analysis (Fig. 6 D) and showed a striking increase in AHR upon Mch challenge (unpublished data). Taken together, these results indicated that living IFN-γ+ Th1 cells, when stimulated with Ag and IL-18 in vivo, induce airway inflammation and bronchial asthma in unsensitized host mice. However, blockage of IL-13 again failed to inhibit airway inflammation and AHR, even though this treatment depleted eosinophils in the lung tissue (not depicted), suggesting the contribution of Ag plus IL-18–stimulated Th1 cells to induction of bronchial asthma.
Discussion
Th1-dominant immune responses have generally been considered pathologic in autoimmune disease but protective against Th2-related diseases (asthma, atopy, etc.; 21). We demonstrated that intranasal administration of Ag induced airway inflammation without AHR in mice receiving memory-type Th1 cells. Because Th1 cells induce DTH, we suspected that this Ag-induced DTH is a physiological response and is not responsible for inducing pathological manifestations such as AHR. In contrast, intranasal administration of Ag and IL-18 induced severe airway inflammation associated with AHR. Thus, Th1 cells become very harmful cells that induce pathological DTH when they are stimulated with Ag and IL-18.
Human bronchial asthma is characterized by airway hypersensitivity, eosinophilic airway inflammation, airway remodeling, mucus hypersecretion, and a high level of IgE (1–4). These findings have been traditionally explained by excited state of Th2 cells (5–7). Indeed, inactivation of GATA-3 significantly diminishes airway inflammation and hypersensitivity (55–57). As IFN-γ antagonizes Th2 response, Th1 cells have been regarded to be protective against asthma (21–23). However, recent studies revealed the contribution of Th1 cells to induction of bronchial asthma (1, 24–29). Cotransfer of allergen-specific Th1 and Th2 cells induce asthma (25). Furthermore, allergen-specific Th1 cells fail to counterbalance Th2 cell–mediated airway hypersensitivity (24). Several studies suggested that a combination of Th1 and Th2 cells instead of mutual inhibition (26, 27) augment each activity to induce bronchial asthma, suggesting the importance of both Th1 and Th2 cytokines. Indeed, IFN-γ and IL-13 induce most severe bronchial asthma (28). Furthermore, a recent study suggests another possibility that Th1 cells induce airway hypersensitivity by recruitment and activation of neutrophils (29). Therefore, Th1 cells become pathological cells under some conditions. Thus, bronchial asthma might be categorized into three types; Th1 cell–induced asthma, Th2 cell–induced asthma, and Th1/Th2 mixed cell–induced asthma.
Th2 cell–induced bronchial asthma is inhibited by neutralization of IL-13 (16, 17). Th1 cell–induced bronchial asthma is controlled by inhibition of the function of chemokines (29). It is well-known evidence that Th1 cells but not Th2 cells express IL-18Rα (44). We previously showed that IL-18 in collaboration with IL-12 strongly induces Ag or anti-CD3–activated Th1 cells to produce IFN-γ, suggesting that Th1 cells are major target cells for IL-18 (44). Therefore, it is important to compare the responsiveness of Th1 cell– or Th2 cell–administered mice to the exposure of Ag and IL-18. We established Th1- or Th2-type memory T cells. We revealed that administration of memory-type Th1 or Th2 cells induced bronchial asthma in an entirely different manner (Figs. 1 and 2). Th1 cell–administered mice require nasal exposure to both Ag and IL-18 to develop bronchial asthma, whereas Th2 cell–administered mice only required Ag. An in vitro study revealed that Th1 cells produce IFN-γ, GM-CSF, TNF-α, IL-9, IL-13, RANTES, and MIP-1α upon stimulation with Ag, IL-2, and IL-18 (Fig. 3 A). Thus, Ag plus IL-18–stimulated Th1 cells produced a similar spectrum of cytokines and chemokines to that produced by activated mast cells. Although Th1 cell–administered mice developed severe AHR, we could not detect Ag-specific IgE in their sera (not depicted), suggesting the important role of administered memory-type Th1 cells in the induction of AHR in an IgE-independent manner.
Here we could induce unique bronchial asthma by activation of Th1 cells with Ag and IL-18 to produce IFN-γ, GM-CSF, TNF-α, IL-9, IL-13, RANTES, and MIP-1α in normal BALB/c mice. We could also induce Th2-type asthma by the transfer of memory-type Th2 cells (Figs. 1 and 2). As previously reported (16, 17), we could show that neutralization of IL-13 inhibits this Th2 cell–induced AHR (Fig. 5), suggesting contribution of IL-13 to Th2-induced bronchial asthma (Fig. 5). In contrast, neutralization of IL-13 did not affect Th1 cell–induced AHR, although this treatment strongly diminished infiltration with eosinophils (Fig. 5). As Th1 cells produce IFN-γ, GM-CSF, TNF-α, IL-9, IL-13, and chemokines when they are stimulated with Ag, IL-2, and IL-18 in vitro, it might be important to point out the possibility that administration of memory-type Th1 cells could mimic the mixed asthma responses that are attainable by activation of both Th1 and Th2 cells. Eosinophils have been implicated as primary effecter cells in asthma and asthmatic AHR (58). Neutralization of IL-13 markedly diminished the number of eosinophils in the lungs of mice that were first transferred with memory-type Th1 and lately exposed to Ag and IL-18. Nevertheless, these mice developed a comparable level of AHR to that of control mice, suggesting the contribution of cytokines other than IL-13 to the development of AHR in Th1 cell–transferred mice. Thus, Ag plus IL-18–stimulated Th1 cells induce bronchial asthma independently of IL-13.
It is intriguing to speculate that IL-13 production from Th2 cells may induce accumulation and activation of eosinophils to produce eosinophil-specific basic proteins (major basic protein, eosinophil cationic protein, and eosinophil peroxidase), eosinophil-derived neurotoxins, and lipid mediators to induce the pathological process of bronchial asthma. Because Ag plus IL-18–stimulated Th1 cells produce both IL-13 and IFN-γ (Fig. 6 A), IFN-γ may partly inhibit the action of IL-13 to induce AHR. However, aside from this action, IFN-γ may directly activate accumulated neutrophils to produce chemical mediators and reactive oxygen intermediates, which in combination may strongly induce bronchial asthma. If these assumptions are right, we can understand the reason why simple depletion of eosinophils failed to attenuate bronchial asthma in human studies (59). Depletion of neutrophils may also be needed to attenuate bronchial asthma. Indeed, neutralization of some other factors including chemokines, particularly CXC chemokines, sometimes attenuates the AHR in Th1 cell–induced asthma (29). Indeed, we could detect significant amounts of RANTES and MIP-1α in BALF obtained from the mice that were adoptively transferred with Th1 cells and lately challenged with OVA and IL-18 (not depicted), even though BALF was diluted with washing fluid. In contrast, neither RANTES nor MIP-1α was detected in BALF from mice challenged with OVA alone (not depicted). We need further study to substantiate this hypothesis that IFN-γ–stimulated neutrophils contribute to induction of bronchial asthma.
In addition to IFN-γ and IL-13 (Fig. 6 A), Ag plus IL-18–stimulated Th1 cells produced a variety of cytokines including IL-9, GM-CSF, RANTES, and MIP-1α. Mast cells, when activated with allergen that cross-links IgE molecules on the cell surface, also produce various types of cytokines, chemokines, and chemical mediators (1, 60). However, we recently found that IL-18 directly stimulates mast cells and basophils to produce Th2 cytokines and chemical mediators (33). However, as simple administration of IL-18 failed to induce airway inflammation (Fig. 1) and bronchial asthma in normal mice, we suspected the importance of activation of Th1 cells with Ag and IL-18 instead of IL-18–dependent activation of mast cells to induce asthma in the naive animals that are equipped with memory-type Th1 cells but lack both Ag-specific IgE and activated mast cells. We found that like activated mast cells (1, 60), Ag plus IL-18–stimulated Th1 cells produce Th1 cytokines, Th2 cytokines, and chemokines. As IL-18 is abundantly stored in the epithelial cells of various organs (39–42), we could assume that some types of infectious agents induce IL-18 production from bronchial epithelial cells. Ag from the infectious agent and IL-18 from epithelial cells then stimulate memory-type Th1 cells, which were induced by previous infection, to produce cytokines including IFN-γ, IL-9, IL-13, GM-CSF, TNF-α, RANTES, and MIP-1α. Thus, Th1 cells may play a relevant role in induction of bronchial asthma under some infectious conditions.
Here we demonstrated that Th1 cells are critically involved in the induction of airway hypersensitivity and histopathological changes. Th1 cells have the striking capacity to produce IFN-γ, IL-9, IL-13, GM-CSF, RANTES, and MIP-1α when they are stimulated with Ag, IL-2, and IL-18 in vitro (Figs. 3 A and 6 C). Moreover, passive transfer of Th1 cells causes normal mice to develop severe airway inflammation and AHR when they are nasally exposed to Ag and IL-18 (Figs. 1 and 2). Thus, memory-type Th1 cells become pathological cells when they are stimulated with Ag and IL-18 in vivo. Although we could depict the asthma-associated cytokines in the products of Ag-, IL-2–, and IL-18-stimulated Th1 cells, we still need to identify the definitive causative effecter cells or factors in this experimental bronchial asthma model. However, most importantly, we could reveal that IL-18 is an upstream cytokine of IFN-γ, IL-9, IL-13, GM-CSF, TNF-α, RANTES, and MIP-1α, providing a potential therapeutic target molecule for the establishment of the treatment for allergic disorders.
Acknowledgments
We are grateful to Dr. William E. Paul (Laboratory of Immunology, National Institute of Allergy and Infectious Diseases, and National Institutes of Health) for his valuable comments on this manuscript. We express sincere thanks to Hayashibara Biochemical Laboratories Inc. for providing us with recombinant mouse IL-12, IL-18, and anti–IL-18Rα chain mAb.
This study is supported by Grant-in-Aid for Scientific Research on Priority Areas and Hitech Research Center grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
T. Sugimoto and Y. Ishikawa contributed equally to this work.
Abbreviations used in this paper: AHR, airway hyperresponsiveness; APC, allophycocyanin; BAL, bronchoalveolar lavage; BALF, BAL fluid; DTH, delayed-type hypersensitivity; Mch, methacholine; MIP, macrophage inflammatory protein; RANTES, regulated on activation, normal T cell expressed and secreted; Tg, transgenic.
References
- 1.Busse, W.W., and R.J. Lemanske. 2001. Asthma. N. Engl. J. Med. 344:350–362. [DOI] [PubMed] [Google Scholar]
- 2.Davies, D.E., J. Wicks, R.M. Powell, S.M. Puddicombe, and S.T. Holgate. 2003. Airway remodeling in a asthma: new insights. J. Allergy Clin. Immunol. 111:215-225. [DOI] [PubMed] [Google Scholar]
- 3.Wills-Karp, M. 1999. Immunologic basis of antigen-induced airway hyper-responsiveness. Annu. Rev. Immunol. 17:255–281. [DOI] [PubMed] [Google Scholar]
- 4.Bochner, B.S., B.J. Undem, and L.M. Lichtenstein. 1994. Immunological aspects of allergic asthma. Annu. Rev. Immunol. 12:295–335. [DOI] [PubMed] [Google Scholar]
- 5.Renauld, J.C. 2001. New insights into the role of cytokines in asthma. J. Clin. Pathol. 54:577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Umetsu, D.T., J.J. McIntire, O. Akbari, C. Macaubas, and R.H. DeKruyff. 2002. Asthma: an epidemic of dysregulated immunity. Nat. Immunol. 3:715–720. [DOI] [PubMed] [Google Scholar]
- 7.Elias, J.A., C.G. Lee, T. Zheng, B. Ma, R.J. Homer, and Z. Zhu. 2003. New insights into the pathogenesis of asthma. J. Clin. Invest. 111:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seder, R.A., and W.E. Paul. 1994. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12:635–673. [DOI] [PubMed] [Google Scholar]
- 9.Hamelmann, E., and E.W. Gelfand. 2001. IL-5-induced airway eosinophilia–the key to asthma? Immunol. Rev. 179:182–191. [DOI] [PubMed] [Google Scholar]
- 10.Mosmann, T.R., M.W. Bond, R.L. Coffman, J. Ohara, and W.E. Paul. 1986. T-cell and mast cell lines respond to B-cell stimulatory factor 1. Proc. Natl. Acad. Sci. USA. 83:5654–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hultner, L., C. Druez, J. Moeller, C. Uyttenhove, E. Schmitt, E. Rude, P. Dormer, and S.J. Van. 1990. Mast cell growth-enhancing activity (MEA) is structurally related and functionally identical to the novel mouse T cell growth factor P40/TCGFIII (interleukin 9). Eur. J. Immunol. 20:1413–1416. [DOI] [PubMed] [Google Scholar]
- 12.Temann, U.A., G.P. Geba, J.A. Rankin, and R.A. Flavell. 1998. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J. Exp. Med. 188:1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Louahed, J., M. Toda, J. Jen, Q. Hamid, J.C. Renauld, R.C. Levitt, and N.C. Nicolaides. 2000. Interleukin-9 upregulates mucus expression in the airways. Am. J. Respir. Cell. Mol. Biol. 22:649–656. [DOI] [PubMed] [Google Scholar]
- 14.Vermeer, P.D., R. Harson, L.A. Einwalter, T. Moninger, and J. Zabner. 2003. Interleukin-9 induces goblet cell hyperplasia during repair of human airway epithelia. Am. J. Respir. Cell. Mol. Biol. 28:286–295. [DOI] [PubMed] [Google Scholar]
- 15.McKenzie, G.J., A. Bancroft, R.K. Grencis, and A.N. McKenzie. 1998. A distinct role for interleukin-13 in Th2-cell-mediated immune responses. Curr. Biol. 8:339–342. [DOI] [PubMed] [Google Scholar]
- 16.Wills-Karp, M., J. Luyimbazi, X. Xu, B. Schofield, T.Y. Neben, C.L. Karp, and D.D. Donaldson. 1998. Interleukin-13: central mediator of allergic asthma. Science. 282:2258–2261. [DOI] [PubMed] [Google Scholar]
- 17.Grunig, G., M. Warnock, A.E. Wakil, R. Venkayya, F. Brombacher, D.M. Rennick, D. Sheppard, M. Mohrs, D.D. Donaldson, R.M. Locksley, et al. 1998. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 282:2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuperman, D.A., X. Huang, L.L. Koth, G.H. Chang, G.M. Dolganov, Z. Zhu, J.A. Elias, D. Sheppard, and D.J. Erle. 2002. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8:885–889. [DOI] [PubMed] [Google Scholar]
- 19.Wills-Karp, M., and M. Chiaramonte. 2003. Interleukin-13 in asthma. Curr. Opin. Pulm. Med. 9:21–27. [DOI] [PubMed] [Google Scholar]
- 20.Ritz, S.A., M.R. Stampfli, D.E. Davies, S.T. Holgate, and M. Jordana. 2002. On the generation of allergic airway diseases: from GM-CSF to Kyoto. Trends Immunol. 23:396–402. [DOI] [PubMed] [Google Scholar]
- 21.Del Prete, A. 1998. The concept of type-1 and type-2 helper T cells and their cytokines in humans. Int. Rev. Immunol. 16:427–455. [DOI] [PubMed] [Google Scholar]
- 22.Barnes, P.J. 1999. Therapeutic strategies for allergic diseases. Nature. 402:B31–B38. [DOI] [PubMed] [Google Scholar]
- 23.Huang, T.J., P.A. MacAry, P. Eynott, A. Moussavi, K.C. Daniel, P.W. Askenase, D.M. Kemeny, and K.F. Chung. 2001. Allergen-specific Th1 cells counteract efferent Th2 cell-dependent bronchial hyperresponsiveness and eosinophilic inflammation partly via IFN-γ. J. Immunol. 166:207–217. [DOI] [PubMed] [Google Scholar]
- 24.Hansen, G., G. Berry, R.H. DeKruyff, and D.T. Umetsu. 1999. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J. Clin. Invest. 103:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Randolph, D.A., C.J. Carruthers, S.J. Szabo, K.M. Murphy, and D.D. Chaplin. 1999. Modulation of airway inflammation by passive transfer of allergen-specific Th1 and Th2 cells in a mouse model of asthma. J. Immunol. 162:2375–2383. [PubMed] [Google Scholar]
- 26.Randolph, D.A., R. Stephens, C.J. Carruthers, and D.D. Chaplin. 1999. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J. Clin. Invest. 104:1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, L., Y. Xia, A. Nguyen, L. Feng, and D. Lo. 1998. Th2-induced eotaxin expression and eosinophilia coexist with Th1 responses at the effector stage of lung inflammation. J. Immunol. 161:3128–3135. [PubMed] [Google Scholar]
- 28.Ford, J.G., D. Rennick, D.D. Donaldson, R. Venkayya, C. McArthur, E. Hansell, V.P. Kurup, M. Warnock, and G. Grunig. 2001. IL-13 and IFN-γ: interactions in lung inflammation. J. Immunol. 167:1769–1777. [DOI] [PubMed] [Google Scholar]
- 29.Takaoka, A., Y. Tanaka, T. Tsuji, T. Jinushi, A. Hoshino, Y. Asakura, Y. Mita, K. Watanabe, S. Nakaike, Y. Togashi, et al. 2001. A critical role for mouse CXC chemokine(s) in pulmonary neutrophilia during Th type 1-dependent airway inflammation. J. Immunol. 167:2349–2353. [DOI] [PubMed] [Google Scholar]
- 30.Okamura, H., H. Tsutsi, T. Komatsu, M. Yutsudo, A. Hakura, T. Tanimoto, K. Torigoe, T. Okura, Y. Nukada, K. Hattori, et al. 1995. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature. 378:88–91. [DOI] [PubMed] [Google Scholar]
- 31.Kohno, K., J. Kataoka, T. Ohtsuki, Y. Suemoto, I. Okamoto, M. Usui, M. Ikeda, and M. Kurimoto. 1997. IFN-gamma-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J. Immunol. 158:1541–1550. [PubMed] [Google Scholar]
- 32.Okamura, H., H. Tsutsui, S. Kashiwamura, T. Yoshimoto, and K. Nakanishi. 1998. Interleukin-18: a novel cytokine that augments both innate and acquired immunity. Adv. Immunol. 70:281–312. [DOI] [PubMed] [Google Scholar]
- 33.Yoshimoto, T., H. Tsutsui, K. Tominaga, K. Hoshino, H. Okamura, S. Akira, W.E. Paul, and K. Nakanishi. 1999. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc. Natl. Acad. Sci. USA. 96:13962–13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshimoto, T., H. Mizutani, H. Tsutsui, T.N. Noben, K. Yamanaka, M. Tanaka, S. Izumi, H. Okamura, W.E. Paul, and K. Nakanishi. 2000. IL-18 induction of IgE: dependence on CD4+ T cells, IL-4 and STAT6. Nat. Immunol. 1:132–137. [DOI] [PubMed] [Google Scholar]
- 35.Yoshimoto, T., B. Min, T. Sugimoto, N. Hayashi, Y. Ishikawa, Y. Sasaki, H. Hata, K. Takeda, K. Okumura, K.L. Van, et al. 2003. Nonredundant roles for CD1d-restricted natural killer T cells and conventional CD4+ T cells in the induction of immunoglobulin E antibodies in response to interleukin 18 treatment of mice. J. Exp. Med. 197:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoshino, T., R.H. Wiltrout, and H.A. Young. 1999. IL-18 is a potent coinducer of IL-13 in NK and T cells: a new potential role for IL-18 in modulating the immune response. J. Immunol. 162:5070–5077. [PubMed] [Google Scholar]
- 37.Wild, J.S., A. Sigounas, N, Sur, M.S. Siddiqui, R. Alam, M. Kurimoto, and S. Sur. 2000. IFN-γ-inducing factor (IL-18) increases allergic sensitization, serum IgE, Th2 cytokines, and airway eosinophilia in a mouse model of allergic asthma. J. Immunol. 164:2701–2710. [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi, K., T. Yoshimoto, H. Tsutsui, and H. Okamura. 2001. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 19:423–474. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi, M., Y. Nishizaki, O. Sano, T. Ohta, M. Ikeda, and M. Kurimoto. 1997. Immunohistochemical and immuno-electron-microscopic detection of interferon-γ-inducing factor (“interleukin-18”) in mouse intestinal epithelial cells. Cell Tissue Res. 289:499–503. [DOI] [PubMed] [Google Scholar]
- 40.Pizarro, T.T., M.H. Michie, M. Bentz, J. Woraratanadharm, M.J. Smith, E. Foley, C.A. Moskaluk, S.J. Bickston, and F. Cominelli. 1999. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn's disease: expression and localization in intestinal mucosal cells. J. Immunol. 162:6829–6835. [PubMed] [Google Scholar]
- 41.Cameron, L.A., R.A. Taha, A. Tsicopoulos, M. Kurimoto, R. Olivenstein, B. Wallaert, E.M. Minshall, and Q.A. Hamid. 1999. Airway epithelium expresses interleukin-18. Eur. Respir. J. 14:553–559. [DOI] [PubMed] [Google Scholar]
- 42.Sugawara, S., A. Uehara, T. Nochi, T. Yamaguchi, H. Ueda, A. Sugiyama, K. Hanzawa, K. Kumagai, H. Okamura, and H. Takada. 2001. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J. Immunol. 167:6568–6575. [DOI] [PubMed] [Google Scholar]
- 43.Abbas, A.K., K.M. Murphy, and A. Sher. 1996. Functional diversity of helper T lymphocytes. Nature. 383:787–793. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimoto, T., K. Takeda, T. Tanaka, K. Ohkusu, S. Kashiwamura, H. Okamura, S. Akira, and K. Nakanishi. 1998. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-γ production. J. Immunol. 161:3400–3407. [PubMed] [Google Scholar]
- 45.Tominaga, K., T. Yoshimoto, K. Torigoe, M. Kurimoto, K. Matsui, T. Hada, H. Okamura, and K. Nakanishi. 2000. IL-12 synergizes with IL-18 or IL-1β for IFN-γ production from human T cells. Int. Immunol. 12:151–160. [DOI] [PubMed] [Google Scholar]
- 46.Murphy, K.M., A.B. Heimberger, and D.Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+CD8+ TCRlo thymocytes in vivo. Science. 250:1720–1723. [DOI] [PubMed] [Google Scholar]
- 47.Hyodo, Y., K. Matsui, N. Hayashi, H. Tsutsui, S. Kashiwamura, H. Yamauchi, K. Hiroishi, K. Takeda, Y. Tagawa, Y. Iwakura, et al. 1999. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 162:1662–1668. [PubMed] [Google Scholar]
- 48.Asano, Y., and R.J. Hodes. 1985. T cell regulation of B cell activation. Lyt-1+, 2− T cells modify the MHC-restricted function of heterogeneous and cloned T suppressor cells. J. Exp. Med. 162:413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamura, T., and H. Nariuchi. 1992. T cell activation through TCR/-CD3 complex. IL-2 production of T cell clones stimulated with anti-CD3 without cross-linkage. J. Immunol. 148:2370–2377. [PubMed] [Google Scholar]
- 50.Kaye, J., S. Porcelli, J. Tite, B. Jones, and C.J. Janeway. 1983. Both a monoclonal antibody and antisera specific for determinants unique to individual cloned helper T cell lines can substitute for antigen and antigen-presenting cells in the activation of T cells. J. Exp. Med. 158:836–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayashi, N., D. Liu, B. Min, S.S. Ben, and W.E. Paul. 2002. Antigen challenge leads to in vivo activation and elimination of highly polarized TH1 memory T cells. Proc. Natl. Acad. Sci. USA. 99:6187–6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hamelmann, E., J. Schwarze, K. Takeda, A. Oshiba, G.L. Larsen, C.G. Irvin, and E.W. Gelfand. 1997. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care. Med. 156:766–775. [DOI] [PubMed] [Google Scholar]
- 53.Luster, A.D. 1998. Chemokines—chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 12:436–445. [DOI] [PubMed] [Google Scholar]
- 54.Urban, J.J., T.N. Noben, D.D. Donaldson, K.B. Madden, S.C. Morris, M. Collins, and F.D. Finkelman. 1998. IL-13, IL-4Rα, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity. 8:255–264. [DOI] [PubMed] [Google Scholar]
- 55.Ray, A., and L. Cohn. 1999. Th2 cells and GATA-3 in asthma: new insights into the regulation of airway inflammation. J. Clin. Invest. 104:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang, D.H., L. Yang, L. Cohn, L. Parkyn, R. Homer, P. Ray, and A. Ray. 1999. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity. 11:473–482. [DOI] [PubMed] [Google Scholar]
- 57.Finotto, S., S.G. De, H.A. Lehr, U. Herz, M. Buerke, M. Schipp, B. Bartsch, R. Atreya, E. Schmitt, P.R. Galle, et al. 2001. Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J. Exp. Med. 193:1247–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gleich, G.J. 1990. The eosinophil and bronchial asthma: current understanding. J. Allergy. Clin. Immunol. 85:422–436. [DOI] [PubMed] [Google Scholar]
- 59.Leckie, M.J., A. ten Brinke, J. Khan, Z. Diamant, B.J. O'Connor, C.M. Walls, A.K. Mathur, H.C. Cowley, K.F. Chung, R. Djukanovic, et al. 2000. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 356:2144–2148. [DOI] [PubMed] [Google Scholar]
- 60.Kawakami, T., and S.J. Galli. 2002. Regulation of mast-cell and basophil function and survival by IgE. Nat. Rev. Immunol. 2:773–786. [DOI] [PubMed] [Google Scholar]