

Abstract
Carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1) is a cell surface molecule that has been proposed to negatively regulate T cell function. We have shown that CEACAM1 is associated with specific regulation of T helper cell (Th)1 pathways, T-bet–mediated Th1 cytokine signaling, and Th1-mediated immunopathology in vivo. Mice treated with anti–mouse CEACAM1-specific monoclonal antibody (mAb) CC1 during the effector phase exhibited a reduced severity of trinitrobenzene sulfonic acid colitis in association with decreased interferon (IFN)-γ production. Although oxazolone colitis has been reported as Th2 mediated, mice treated with the CC1 mAb or a CEACAM1-Fc chimeric protein exhibited a reduced severity of colitis in association with a significant reduction of IFN-γ and T-bet activation, whereas signal transducer and activator of antigen 4 activation was unaffected. Both interleukin-4 and IFN-γ gene–deficient mice exhibited less severe colitis induction by oxazolone. Direct ligation of T cells in vitro with the murine hepatitis virus spike protein, a natural ligand for the N-domain of CEACAM1, inhibited the differentiation of naive cells into Th1 but not Th2 cells and activation of Th1 but not Th2 cytokine production. These results indicate that CEACAM1 isoforms are a novel class of activation-induced cell surface molecules on T cells that function in the specific regulation of Th1-mediated inflammation such as that associated with inflammatory bowel disease.
Keywords: CEACAM1, inflammatory bowel disease, hapten-induced colitis, T cell immunity, Th1 cytokine
Introduction
Carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1; also known as CD66a or biliary glycoprotein) is a member of the carcinoembryonic antigen family of glycoproteins with homologues in rodents (mice and rats) and humans (1, 2). CEACAM1 is expressed in a wide variety of organs and cell types including a variety of epithelial and hematopoietic cell lineages (NK cells, B cells, polymorphonuclear leukocytes, monocytes, and DCs; 2–7). The extracellular domain of CEACAM1 isoforms consists of a membrane distal IgV-like N-domain followed by variable numbers of alternating IgI1 and IgI2 set domains with either a long or short cytoplasmic tail (1, 8). In mice, two alleles of CEACAM1 exist (a and b) based upon differences in the N-domain. Most mouse strains express the “a” allele (1). The long, 73–amino acid cytoplasmic tail of CEACAM1 contains two immunoreceptor tyrosine-based inhibitory motifs (ITIMs) that have been shown to negatively regulate cellular activation in epithelial cells (2, 9). This inhibitory property of CEACAM1 is triggered by phosphorylation of tyrosine residues within the ITIMs resulting in recruitment of the Src homology 2 domain–containing tyrosine phosphatases SHP-1 and SHP-2 and the Src homology 2–containing inositol phosphatase (10). CEACAM1 is characterized by homophilic adhesion between N-domains of CEACAM1 and likely other CEACAM family members (11) and heterophilic interactions with several types of bacteria and viruses (8, 12–14). These interactions are likely linked to cell signaling leading to regulation of cell growth and differentiation (15–17).
CEACAM1 is also expressed on human and mouse T cells (18) early after activation (18–22). Ligation of the N-domain of CEACAM1 with either CEACAM1-specific mAbs or bacterial products either significantly suppresses or activates T cells in vitro (18, 20–22). The explanation for their discrepant functional effect remains unknown. It has been hypothesized that the inhibition observed might be related to expression of long cytoplasmic tail isoforms of CEACAM1, which contain two ITIMs in mouse and human T cells (3, 21). However, there is no available in vivo evidence that either supports a role for CEACAM1 in regulating T cell–mediated inflammatory processes and the consequences (inhibition or activation) of ligating CEACAM1 in the setting of inflammation, or the ligands involved.
Therefore, we examined the effects of CEACAM1a ligation either in vivo with a CEACAM1a-specific mAb and a homophilic ligand for CEACAM1a (CEACAM1a-Fc fusion protein) in the trinitrobenzene sulfonic acid (TNBS) and oxazolone colitis models (23–25) or in vitro with a heterophilic ligand for CEACAM1a, the mouse hepatitis virus (MHV) spike glycoprotein. Through these studies, we have linked CEACAM1 to specific regulation of Th1 pathways and their immunopathologic consequences.
Materials and Methods
Mice.
5–8-wk-old female C57BL/6 and BALB/c mice were obtained from Charles River Laboratories or the central breeding facility at the University of Mainz, respectively. IFN-γ and IL-4 gene–deficient mice on the C57BL/6 background were purchased from The Jackson Laboratory. Mice were provided sterile food and water ad libitum, kept in microisolator cages, and maintained in the animal facility of Harvard Medical School. All studies were performed under approval of the Harvard Medical School Standing Committee on Animals or the ethical committee of Rheinland-Pfalz.
Induction of Hapten-induced Colitis.
To generate a more chronic T cell–mediated inflammation as previously described (26, 27), mice were sensitized with 150 μl of the haptenating agent TNBS (2, 4, 6-TNBS; Sigma-Aldrich) at a concentration of 2.5% in 50% ethanol by skin painting on day 1. On day 8, 150 μl of 1% TNBS in 50% ethanol was administered intrarectally via a 3.5F catheter under anesthesia with tribromoethanol. To ensure distribution of the TNBS within the entire colon and cecum, mice were held in a vertical position for 1 min after the instillation. Oxazolone colitis was induced by sensitizing mice with oxazolone (4-ethoxymethylene-2-phenyl-2-oxazolin-5-one; Sigma-Aldrich) at a concentration of 3% in 100% ethanol by skin painting on day 1 followed by intrarectal administration at a concentration of 1% in 50% ethanol on day 8 as described above. On day 12, mice were killed and immunopathologic characterization was performed as previously described (23, 26, 27).
In Vivo Treatment with CC1 mAb, Anti–IL-4 mAb, and CEACAM1-Fc Fusion Protein.
Anti-CEACAM1a mAb, CC1, was purified from a hybridoma derived by fusion of SP20 cells with spleen cells from SJL/J mice immunized with purified BALB/c intestinal brush border membranes. Control mouse IgG1 mAbs directed against irrelevant antigen (the B subunit of cholera toxin) were purified by standard methods (28–31). Either 1 mg of the CC1 or control mouse IgG1 mAb was administered 24 h before skin painting and/or rectal challenge with the haptenating reagent. Anti–IL-4 mAb was produced from the 11B11 hybridoma cell line (25) and a control rat IgG antibody was obtained from Sigma-Aldrich. C57BL/6 mice with oxazolone colitis were injected with 200 μg/dosage N-CEACAM1-Fc (32) every other day starting 1 d before skin painting and ending 1 d before killing the mice.
Flow Cytometry.
Single cell suspensions of mononuclear cells prepared from mesenteric lymph nodes and colonic lamina propria were stained with either the CC1 or murine IgG1 isotype control mAb for the primary Ab and FITC-conjugated rat anti–mouse IgG1 (BD Biosciences) for the secondary Ab. The cells were costained with PE anti-CD3 mAb (BD Biosciences) and histograms of CD3+ cells were analyzed by FACSort™ (Becton Dickinson) with CELLQuest™ software (Becton Dickinson).
Grading of Histologic Changes.
Colonic tissues were removed on day 12 and embedded in paraffin for staining with hematoxylin and eosin. The degree of inflammation on microscopic cross sections of the colon was graded semiquantitatively. For the histopathologic grading of TNBS-induced colitis, the severity of colitis was assessed based on two criteria, inflammation and injury. Inflammation was graded from 0 to 4 (0, none; 1, low; 2, moderate; 3, high; 4, severe with transmural leukocyte infiltration, loss of goblet cells) and severity of injury was graded from 0 to 3 (0, none; 1, occasional epithelial lesion; 2, clear-cut ulceration; 3, extensive ulceration). For histopathologic grading of oxazolone-induced colitis, five criteria (hypervascularization, presence of mononuclear cells, epithelial hyperplasia, epithelial injury, and presence of granulocytes) were scored from 0 to 3 in each with an additive score between 0 (no colitis) and 15 (maximal colitis activity). Grading was performed in a blinded fashion by the same pathologist (J.N. Glickman).
Cytokine Assays.
Lamina propria mononuclear cells (5 × 105 cells/well) isolated on day 12 were stimulated with 5 μg/ml plate-bound hamster anti–mouse CD3ɛ mAb (BD Biosciences) and 2 μg/ml soluble hamster anti–mouse CD28 mAb (BD Biosciences). Culture supernatants were harvested 48 h after the cell culture and concentrations of IFN-γ, IL-2, IL-4, and TNF-α were measured with OptEIA ELISA sets (BD Biosciences).
Isolation of Nuclear Proteins.
Spleen mononuclear cells were washed twice in cold PBS and resuspended in 500 μl buffer A (10 mM Hepes, 1.5 mM MgCl2, 19 mM KCl, 0.5 mM PMSF, 1 mM DTT) followed by the addition of 20 μl Triton X-100 (Sigma-Aldrich) and incubation on ice for 5 min. Cells were centrifuged for 10 min at 4°C followed by the resuspension of the nuclear pellet in buffer C (20 mM Hepes, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 0.5 mM PMSF, 1 mM DTT). Finally, the nuclei were homogenized using mini-stir bars for 1 h at 4°C followed by centrifugation at 15,000 RPM for 15 min at 4°C. The concentration of nuclear proteins in supernatants was assessed using the Bio-Rad Protein Assay.
Western Blots.
Equal amounts of extract were added to 8 μl electrophoresis sample buffer (Firma Roth). After boiling, the extracts were loaded on 15% SDS-PAGE gels and electrophoretically separated. Proteins were transferred to nitrocellulose membranes and detection was performed with the ECL Western blotting analysis system (Amersham Biosciences). Western blots were made using the following antibodies: rabbit anti–T-bet antibody (1:1,000 dilution; provided by L.H. Glimcher, Harvard School of Public Health, Boston, MA; reference 33), anti–human signal transducer and activator of antigen 4 (STAT-4; 1:500 dilution; Santa Cruz Biotechnology, Inc.), and horseradish peroxidase–linked secondary antibodies (1:5,000 dilution; Amersham Biosciences).
Preparation of CD11c+ Cells and IL-12 p70 Cytokine Assay.
CD11c+ cells were isolated from the spleens of C57BL/6 mice injected with Flt-3 ligand–bearing B16-F10 melanoma cell line by using the MACS CD11c microbeads (Miltenyi Biotec) as previously described (34). The percentage of CD11c+ cells was >95% after the purification. For the analysis of IL-12 p70 production, cells were stimulated in vitro with 1 mg/ml LPS (Sigma-Aldrich) and 10 U/ml recombinant mouse IFN-γ. Production of IL-12 p70 was measured with OptEIA ELISA sets (BD Biosciences).
In Vitro Differentiation Assay.
CD4+ CD62L+ naive T cells were isolated from spleens and mesenteric lymphocytes by naive T cell column kit (R&D Systems). For generation of Th1 cells, CD4+ CD62L+ cells were incubated with 1 μg/ml anti-CD3 mAb, 1 μg/ml anti-CD28 mAb, 5 ng/ml recombinant IL-12 (R&D Systems), and 5 μg/ml anti–IL-4 mAb (BD Biosciences) for 72 h. To generate Th2 cells, CD4+ CD62L+ cells were incubated with 1 μg/ml anti-CD3 mAb, 1 μg/ml anti-CD28 mAb, 50 ng/ml recombinant IL-4 (R&D Systems), 1 μg/ml anti–IL-12 mAb (BD Biosciences), and 10 μg/ml anti–IFN-γ mAb (BD Biosciences) for 72 h. After 72 h of incubation, the cells were extensively washed to remove the recombinant cytokines added in culture and restimulated in vitro with 5 μg/ml anti-CD3 and 5 μg/ml anti-CD28 for 48 h. The culture supernatants were subjected to cytokine analysis by ELISA. Several concentrations of MHV spike glycoprotein (A59S1) or bovine serum albumin (0, 0.1, and 1.0 μg/ml) together with or without antiserum to 100 μg/ml of the spike glycoprotein were added during or after the differentiation to Th1- or Th2-type cells.
Statistical Analysis.
Statistical analysis was performed using the Student's t test and the program Microsoft Excel.
Online Supplemental Material.
Fig. S1 shows induction of oxazolone colitis in IFN-γ– and IL-4–deficient mice. Fig. S2 shows histograms of CEACAM1a expression on CD3+ cells in mesenteric lymph node and colonic lamina propria isolated before and immediately after skin painting, and 24 and 72 h after rectal administration with TNBS. Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20030437/DC1.
Results
Anti-CEACAM1a mAb, CC1, Protects Mice from TNBS Colitis When Administered Before the Effector Phase in Association with Specific Reduction in IFN-γ Production.
CEACAM1 is known to be expressed constitutively by murine and human DCs and to be an early activation antigen on human and mouse T cells that may persist for up to 1 wk after activation (3, 18–22). However, little is known about ligation of CEACAM1 on these cell types in vivo. Therefore, we assessed whether ligation of CEACAM1 with an anti-CEACAM1–specific mAb CC1, which is directed against the NH2-terminal domain of CEACAM1a, affected the course of TNBS colitis, a model mediated primarily by Th1 cytokines in certain strains of mice (23). To test whether CEACAM1a is involved in T cell–mediated colonic inflammation at the time of T cell priming by DCs or at the time of the effector response by intestinal T cells, we assessed the effects of CC1 mAb at the time of either T cell priming (before skin painting) and/or the effector phase (before rectal challenge) in the TNBS colitis model. Animals that received TNBS in association with either a control mAb (Fig. 1 a, □) or the CC1 mAb before skin painting (Fig. 1 a, •) experienced severe weight loss. In contrast, mice that received the CC1 mAb either before rectal challenge (Fig. 1 a, ○) or both skin painting and rectal challenge twice (Fig. 1 a, ▪) experienced less weight loss. This was directly reflected in the levels of macroscopic injury observed in that mice treated with the CC1 mAb either before rectal challenge or twice did not exhibit significant shortening and thickening of the colon (Fig. 1 b). Consistent with these macroscopic changes, the control mAb–treated group and groups receiving the CC1 mAb only before skin painting exhibited marked, transmural infiltration with inflammatory cells and injury with ulceration (Fig. 1 c, A and C, respectively). In contrast, mice treated with the CC1 mAb either twice or before rectal challenge exhibited less severe histologic features of colitis (Fig. 1 c, B and D, respectively). When quantified by a histologic scoring system for evidence of inflammation and injury, these histologic challenges were highly significant (Fig. 1 d).
Figure 1.
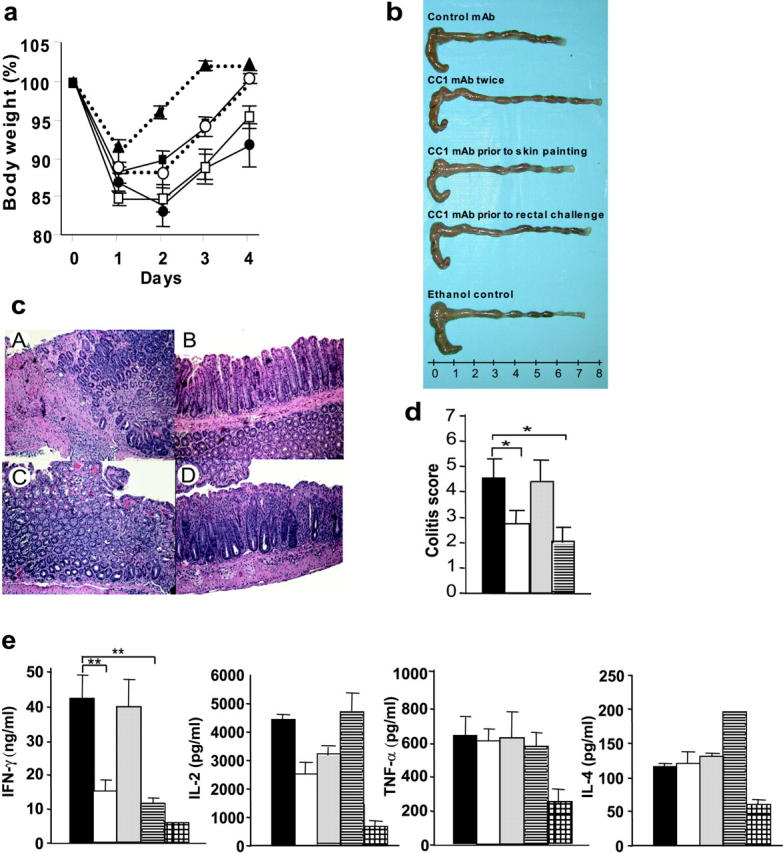
Effect of CC1 mAb injection on the induction of TNBS colitis and cytokine production. (a) Body weight of mice subjected to TNBS colitis treated either with control IgG1 mAb (□), with CC1 mAb before skin painting and before rectal challenge twice (▪), before skin painting (•), or before rectal challenge (○) in C57BL/6 mice are shown. One group was injected with 50% ethanol (▴) instead of TNBS. Data are shown as mean values ± SEM and represent eight mice per group. (b) Macroscopic pictures of colons from mice induced with TNBS colitis treated with or without CC1 mAb are shown. (c) Hematoxylin and eosin–stained pictures from TNBS colitis treated with or without CC1 mAb are shown (×100). One representative picture from each group of eight is shown. A, control mAb; B, CC1 mAb administered twice; C, CC1 mAb administered before skin painting; D, CC1 mAb administered before rectal challenge. (d) Quantitative histopathologic assessment of TNBS colitis activity shows a significant (*, P < 0.05 by t test) suppression in mice treated with CC1 mAb either twice or before rectal challenge when compared with the control mAb–treated group. Samples were collected from mice with TNBS colitis treated either with control mAb (solid bar) or CC1 mAb twice (open bar) before skin painting (shaded bar) or before rectal challenge (striped bar). Data are shown as mean values ± SEM and represent eight mice per group. (e) Th1 and Th2 cytokine production from LPLs was analyzed by ELISA. Samples were collected from mice with TNBS colitis treated either with control mAb (solid bars) or CC1 mAb twice (open bars) before skin painting (shaded bars) or before rectal challenge (striped bars). One group of mice was administered ethanol without TNBS for the skin sensitization and rectal challenge (hatched bars). CC1 mAb–treated group treated either twice or before rectal challenge exhibited significant suppression of IFN-γ production when compared with the control mAb–treated group (**, P < 0.01). Data are shown as mean values ± SEM and represent pooled values from eight independent experiments.
To determine whether this mAb CC1-mediated protection from colitis was associated with alterations in cytokine production, Th1 (IFN-γ, IL-2, TNF-α)– and Th2 (IL-4)–associated cytokine production by lamina propria lymphocytes (LPLs) was assessed in the various treatment groups. These studies revealed a significant reduction in IFN-γ production by LPLs from mice that received the CC1 mAb either before rectal challenge or twice, but not under the other conditions examined (Fig. 1 e). Interestingly, no effects were observed on IL-2, TNF-α, and Th2 cytokine production as defined by assessment of IL-4. A similar inhibition of IFN-γ but not IL-4 as a consequence of CC1 mAb treatment was observed with splenocytes stimulated with anti-CD3 plus anti-CD28 mAbs (unpublished data). Moreover, the quantities of CD4+ T cells within the lamina propria did not differ between CC1-treated and untreated animals, indicating that the effects observed were not simply due to decreased T cells in this compartment (unpublished data). These results indicate that suppression of TNBS colitis by administration of the CC1 mAb is associated with a specific reduction in production of IFN-γ but not IL-4, which is consistent with the role of IFN-γ in this model of colitis (23).
Anti-CEACAM1a mAb CC1 Also Protects Mice from Oxazolone Colitis When Administered Before the Effector Phase in Association with Reduction in IFN-γ.
To determine whether these effects of CEACAM1 on the development of T cell–mediated immunopathology in the colon could be extended to a Th2-associated colitis, we next analyzed the effect of the CC1 mAb in the oxazolone colitis model, which has been previously shown to be primarily Th2 mediated in the SJL strain of mice (25). The same general effects of CC1 mAb on TNBS colitis on the body weight were also evident in oxazolone colitis (Fig. 2 a). Specifically, mice treated with the CC1 mAb either before rectal challenge (Fig. 2 a, ○) or twice (Fig. 2 a, ▪) and, to a lesser extent, before skin painting (Fig. 2 a, •) exhibited less weight loss than mice treated with the control mAb (Fig. 2 a, □). An assessment for macroscopic evidence of colon shortening confirmed these clinical findings (Fig. 2 b). Histologic analysis also revealed that colon tissues from mice treated with the CC1 mAb either twice or before rectal challenge exhibited significantly less severe histologic evidence of colitis (Fig. 2 c, B and D, respectively). In contrast, the control mAb–treated mice or mice treated with CC1 mAb before skin sensitization developed severe colitis (Fig. 2 c, A and C, respectively). When analyzed quantitatively with a scoring system previously applied to this model, these differences were highly significant (Fig. 2 d).
Figure 2.
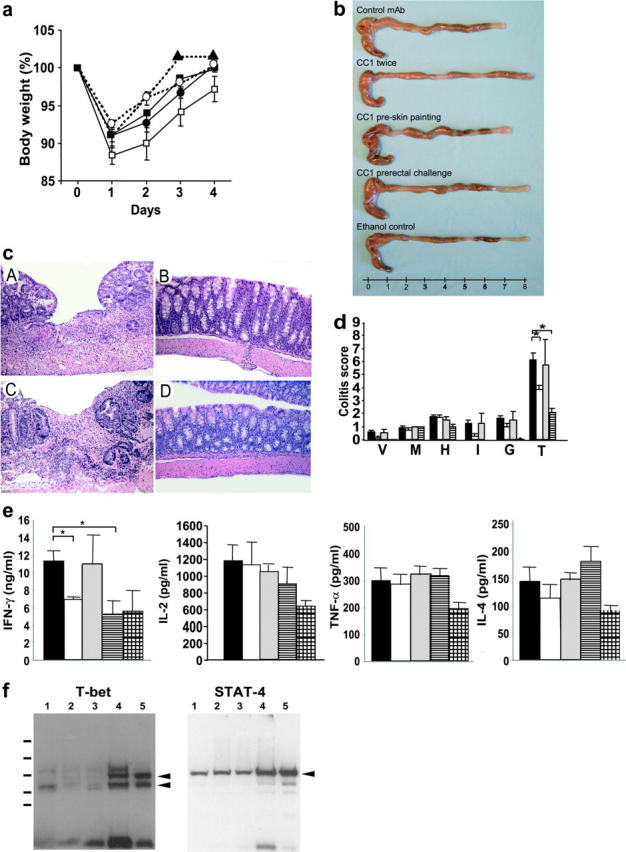
Effect of CC1 mAb injection on the induction of oxazolone colitis and cytokine production. (a) Oxazolone colitis was induced in C57BL/6 mice and mice were monitored for body weight after various combinations of treatments with CC1 or control mAb. Mice were treated either with control IgG1 mAb (□), with CC1 mAb twice (▪), before skin painting (•), or before rectal challenge (○). One group was subjected to intrarectal administration with 50% ethanol (▴) instead of oxazolone. Data are shown as mean values ± SEM and represent eight mice per group. (b) Macroscopic pictures of oxazolone colitis treated with or without CC1 mAb. (c) Histologic hematoxylin and eosin–stained pictures from oxazolone colitis treated with or without CC1 mAb are shown (×100). One representative picture from each group of eight is shown. A, control mAb; B, CC1 mAb administered twice; C, CC1 mAb administered before skin painting; D, CC1 mAb administered before rectal challenge. (d) Quantitative histopathologic assessment of oxazolone colitis activity shows a significant suppression in mice treated with CC1 mAb either twice or before rectal challenge in comparison to the control mAb–treated group (*, P < 0.01). Samples were collected from mice with oxazolone colitis that were treated either with control mAb (solid bar) or CC1 mAb either twice (open bar), before skin painting (shaded bar), or before rectal challenge (striped bar). Data are provided as mean values ± SEM and represent eight mice per group. V, hyper-vascularization; M, presence of mononuclear cells; H, epithelial cell hyperplasia; I, epithelial injury; G, presence of granulocytes; T, total score. (e) Th1 and Th2 cytokine production from LPLs was analyzed by ELISA. Samples were collected from mice with oxazolone colitis that were treated either with control mAb (solid bars) or CC1 mAb either twice (open bars), before skin painting (shaded bars), or before rectal challenge (striped bars). One group of mice was administered ethanol instead of oxazolone (hatched bars). Mice treated with CC1 mAb either twice or before rectal challenge exhibited significant suppression of IFN-γ production when compared with the control mAb–treated group (*, P < 0.01). Data shown represent pooled values from eight independent experiments. Data are shown as mean values ± SEM. (f) Analysis of T-bet and STAT-4 levels in splenic mononuclear cells from mice with oxazolone-induced colitis upon CEACAM1 ligation. Mice with oxazolone-induced colitis were treated with the CC1 mAb as described above followed by isolation of splenic cells and extraction of nuclear proteins. Spleen mononuclear cells were stimulated in a T cell–specific fashion with antibodies to CD3 and CD28 for 24 h. Equal amounts of proteins from two to three mice per group were then subjected to Western blot analysis for expression of STAT-4 and T-bet as indicated. Samples from 1 through 3 were obtained from mice treated with CC1 mAb and samples from 4 and 5 were obtained from mice treated with control mAb. T-bet gave two bands by Western blot analysis as previously described (references 27 and 33).
The oxazolone colitis model was originally reported to be a predominantly Th2-mediated model in SJL mice (25). These results were therefore interesting in that they would have predicted that CEACAM1 was regulating Th2 pathways. Quite surprisingly, however, when the cytokines produced by LPLs after anti-CD3 plus anti-CD28 stimulation were analyzed in this model, IFN-γ but not the Th2 cytokine (IL-4) was significantly decreased in C57BL/6 mice that received the CC1 mAb either before rectal challenge or twice (Fig. 2 e). Although TNF-α is crucial in the induction of Th1-type immune pathways and the pathogenesis of murine model of inflammatory bowel disease (35), production of TNF-α was not affected by the CC1 mAb treatment in either TNBS or oxazolone colitis (Figs. 1 e and 2 e). Other regulatory cytokines (e.g., TGF-β, IL-10) and Th1/Th2 cytokines (e.g., IL-2, IL-5, IL-10, IL-13) were not affected by CC1 mAb treatment in both the TNBS and oxazolone colitis models (unpublished data). Thus, as observed in the TNBS colitis model, ligation of CEACAM1a by the CC1 mAb at the time of the effector phase of oxazolone-induced colitis caused a decrease in colonic immunopathology that was associated with specific inhibition of the Th1 cytokine, IFN-γ, but not Th2 or regulatory cytokines.
Ligation of CEACAM1 Suppresses IFN-γ Signaling via T-bet but Not STAT-4 Signaling.
To determine the effects of CEACAM1 ligation on Th1 cytokine signaling, we next determined the expression of signature transcription factors in spleen cells from mice with oxazolone-induced colitis upon antibody treatment. Specifically, we analyzed the nuclear levels of the transcription factors T-bet and STAT-4 because these transcription factors are essential for IFN-γ (33) and IL-12 signaling (36) in T lymphocytes, respectively. Accordingly, we isolated nuclear proteins from the spleen cells of oxazolone-treated mice upon administration of anti-CEACAM1 and control antibodies. As shown in Fig. 2 f, ligation of CEACAM1 by the CC1 mAb led to a profound down-regulation of nuclear T-bet expression as determined by Western blot analysis. In contrast, nuclear levels of STAT-4 were nearly unaffected suggesting that CEACAM1 strongly controls IFN-γ but not IL-12 signaling in T cells via T-bet.
Deficiency of IFN-γ or IL-4 Is Associated with Less Severe Colon Injury in Response to Oxazolone.
Because our results suggested a novel role for Th1 cytokines in oxazolone colitis in C57BL/6 mice, we investigated further the involvement of Th1 and Th2 cytokines in this model. Given previous reports that oxazolone colitis was predominantly Th2 mediated, we investigated this issue further by inducing oxazolone colitis in IFN-γ–deficient (IFN-γ−/−) and IL-4–deficient (IL-4−/−) mice on a C57BL/6 background. Both IFN-γ−/− and IL-4−/− mice were significantly protected from body weight loss and shortening of the colon when compared with wild-type mice that were treated similarly (Fig. S1, a and b, available at http://www.jem.org/cgi/content/full/jem.20030437/DC1). Consistent with these effects, histologic analyses showed that both IFN-γ−/− and IL-4−/− mice exhibited less severe but still mild colitis compared with wild-type mice (Fig. S1 c, available at http://www.jem.org/cgi/content/full/jem.20030437/DC1). The average colitis scores of wild-type, IFN-γ−/−, and IL-4−/− mice were 7.2, 2.4, and 4.0, respectively. Notably, although the colitis scores of the IFN-γ−/− and IL-4−/− mice were significantly lower than that of the wild-type mice (P < 0.01 and P < 0.05, respectively), colitis was still present. An analysis of cytokines produced by LPLs showed that there was no difference in the levels of cytokines (IFN-γ, IL-2, IL-4, and IL-5) except those depleted by the gene targeting (unpublished data). A similar reduction in the severity of colitis was observed in IFN-γ−/− mice on a BALB/c background (unpublished data). These results indicate that both IFN-γ and IL-4 play independent and additive roles in the induction of oxazolone colitis in both the C57BL/6 and BALB/c backgrounds.
Administration of CC1 mAb Together with Anti–IL-4 mAb Enhances the Protection of Mice from Oxazolone Colitis.
The studies described above suggested that CEACAM1a ligation specifically impacts upon Th1 but not Th2 pathways in these models. Given that oxazolone colitis represents a mixed Th1/Th2 colitis in C57BL/6 mice as shown above, we examined the effects of administering the CC1 mAb and/or anti–IL-4 mAb to inhibit both Th1 and Th2 pathways, respectively. If oxazolone colitis was indeed due to both Th1 and Th2 cytokines and CEACAM1a was specific for the Th1 pathway, it would be predicted that more complete clinicopathologic protection would be provided by blocking both pathways. Protection as defined by body weight loss was nearly complete when mice received treatment with an anti–IL-4 mAb in combination with the CC1 mAb (Fig. 3 a, ♦) in comparison to either the control Ab–treated (Fig. 3 a, ▴) or anti–IL-4– and control Ab–treated (Fig. 3 a, □) groups. Notably, an essential role for IL-4 in this model was suggested by the fact that blockade of IL-4 either alone or together with CEACAM1a ligation via treatment with the CC1 mAb afforded significant macroscopic protection in comparison to the control rat IgG and mouse IgG1–treated group (Fig. 3 b). Microscopic analysis clearly supported a role for both IL-4 and IFN-γ in the pathogenesis of oxazolone colitis and a specific role for CEACAM1a in IFN-γ regulation. Specifically, treatment with the anti–IL-4 mAb with or without the CC1 mAb resulted in less severe histologic features of colitis compared with the control group (Fig. 3 c). However, the greatest protection from histologic injury was provided by a combination of the CC1 and IL-4 mAbs because the mean colitis score of this group was 3.8 in comparison to scores of 5.0 and 11.3 for the anti–IL-4 plus control and control Ab–treated groups, respectively (P < 0.01, anti–IL-4 and CC1 mAb vs. control mAbs; P < 0.05, anti–IL-4 plus mouse IgG1 mAb vs. control mAbs). In addition, anti–IL-4 and CC1 mAb treatment, but not either alone, suppressed the levels of both IFN-γ and IL-4 to baseline levels in the colonic lamina propria in the context of oxazolone colitis (unpublished data). These results confirm that oxazolone colitis is caused by both Th1 and Th2 cytokine pathways such that blockade of both provides optimal protection from clinical evidence of colitis and immunopathology, and that CEACAM1a is specifically linked to Th1 pathways allowing for an enhancement of the protective effect provided by anti–IL-4 mAb treatment.
Figure 3.

Effect of anti–IL-4 mAb and CC1 mAb injection on the induction of oxazolone colitis. (a) Body weight of mice treated with anti–IL-4 mAb together with CC1 mAb (♦), anti–IL-4 mAb and control mouse IgG1 (□), or control rat IgG and mouse IgG1 (▴). Prevention of body weight loss was significant (*, P < 0.05; **, P < 0.01) in mice administered anti–IL-4 and CC1 mAb when compared with the control Ab–treated group. Data shown are expressed as mean values ± SEM from six mice per group. Representative macroscopic (b) and microscopic (c; ×100) pictures of mice treated with or without anti–IL-4 and CC1 mAbs are shown.
Neither In Vivo nor In Vitro Treatment with the CC1 mAb Affects IL-12 p70 Production.
These studies indicate that CEACAM1a ligation by the CC1 mAb in vivo specifically inhibits IFN-γ during the course of T cell–mediated intestinal inflammation. One possible mechanism to explain this result is through CEACAM1a regulation of IL-12 production given that IFN-γ production from Th1 cells is regulated by IL-12 p70 produced by APCs (e.g., DCs, macrophages; references 37 and 38). In addition, it has been previously reported that CEACAM1a ligation on mouse DCs in vitro results in IL-12 release and thus promotion of IFN-γ production (3). Thus, we assessed IL-12 production in the TNBS colitis model. IL-12 p70 production by spleen (unpublished data) and lamina propria cells from mice exposed to TNBS was not affected by in vivo treatment with the CC1 mAb (control mAb, 317.5 ± 59.3 pg/ml; CC1 mAb twice, 400.3 ± 82.1 pg/ml; CC1 mAb pre-skin, 280.3 ± 57.5 pg/ml; CC1 mAb pre-rectal, 415.3 ± 86.3 pg/ml; not significant). In addition, IL-12 p70 production by CD11c+ DCs stimulated with LPS and IFN-γ in vitro was not significantly altered by treatment with the CC1 mAb in comparison to an isotype control mAb (unpublished data). Thus, inhibition of IFN-γ production by treatment with the CC1 mAb was not likely to effect IL-12 p70 production by APCs including DCs.
CEACAM1a Chimeric Protein Prevents Oxazolone Colitis and Suppresses IFN-γ Production.
Our results, as shown here, indicate that ligation of CEACAM1a with the CC1 mAb significantly suppresses colitis in association with inhibition of T cell production of IFN-γ by LPLs. However, these results do not define either the nature of the natural ligand for CEACAM1a in vivo and whether inhibition of Th1-mediated immunopathology can be caused by ligation of CEACAM1a with its natural ligand. To define the ligand for CEACAM1a in vivo, we examined the effects of a CEACAM1a-Fc fusion protein (sMHVR-Ig) encoding the extracellular portion of the mCEACAM1a-4L isoform that has been shown to homophilically ligate the CEACAM1a molecule in vitro (32). Therefore, mice were treated with either the CEACAM1a-Fc fusion protein or a control isotype–matched Fc fragment at a dose of 200 μg administered every other day for seven doses beginning at the time of skin sensitization in the oxazolone colitis model. Macroscopic assessment revealed that shortening of the colon was less severe in the mice treated with CEACAM1a-Fc compared with the mice administered the control-Fc fragment (Fig. 4 a). Similarly, histologic evidence of colitis was less pronounced in the CEACAM1a-Fc–treated group compared with the control-Fc–treated group (Fig. 4, b and c). These pathologic findings were associated with a significant decrease in IFN-γ production by LPLs from mice treated with the CEACAM1a-Fc fusion protein (Fig. 4 d). The fact that the decrease in immunopathologic injury observed occurred in association with suppression of IFN-γ production indicated that these were causally related.
Figure 4.

Effect of CEACAM1-Fc chimeric protein on the induction of oxazolone colitis. Macroscopic (a) and histologic pictures (b; ×100) of colon isolated from mice induced to develop oxazolone colitis treated with CEACAM1-Fc or a control-Fc fragment. (c) Colitis scores of colon-induced oxazolone colitis treated with CEACAM1-Fc (solid bar) or a control-Fc fragment (open bar). Mice treated with CEACAM1-Fc exhibited significant reduction in colitis scores (*, P < 0.005; **, P < 0.01). V, hyper-vascularization; M, presence of mononuclear cells; H, epithelial cell hyperplasia; I, epithelial injury; G, presence of granulocytes; T, total score. (d) Th1 and Th2 cytokine production from LPLs was analyzed by ELISA of mice with oxazolone colitis treated either with CEACAM1-Fc (solid bars) or control-Fc fragment (open bars). Suppression of IFN-γ was significant (**, P < 0.01). Data are shown as mean values ± SEM from four independent experiments.
Ligation of CEACAM1a on T Cells Specifically Inhibits the Differentiation and Activation of Th1 Cells In Vitro.
The observations described above suggest that homophilic ligation of CEACAM1a in vivo during the effector phase of colitis induces an inhibitory pathway associated with down-regulation of IFN-γ production. Given the absence of an effect of the CC1 mAb on IL-12 production, we reasoned that ligation of CEACAM1a on a T cell might mediate this response. To determine this, we first examined the expression of CEACAM1a on CD3+ cells during the course of TNBS colitis as defined by flow cytometry. As predicted by in vitro studies showing that CEACAM1a is an activation antigen on mouse and human T cells (18, 19, 21, 22), we observed that a small proportion (∼5%) of T cells expressed CEACAM1a within mesenteric lymph nodes and LPLs in the normal intestine, presumably consistent with the physiologic inflammation that is normally ascribed to this organ system (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20030437/DC1). By 4 d after induction of colitis induced by TNBS administration, CEACAM1a continued to be detectable on a similar but unchanged proportion of CD3+ LPLs (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20030437/DC1). Similar observations were made at various other time points after TNBS administration and after induction of colitis with oxazolone administration (unpublished data), indicating that CEACAM1a is expressed in vivo in T cells during intestinal inflammation and amenable to ligation with CEACAM1a-related ligands.
Next, we sought to directly address whether ligation of CEACAM1a regulates IFN-γ production by T cells. Naive T cells were differentiated and activated into either Th1 or Th2 cells in the presence of a natural CEACAM1a ligand that binds within the homophilic binding site of the N-domain. For this purpose, a his-tagged version of the MHV spike glycoprotein was added during the differentiation and activation of either Th1 or Th2 cells (39). We used the MHV spike glycoprotein due to nonspecific effects of either control-Fc fragments or IgG antibodies in the model system used that prevented interpretation of similar studies with either the CEACAM1a-Fc fusion protein or the CC1 mAb (unpublished data). IFN-γ production was significantly suppressed in a dose-dependent fashion by addition of the spike glycoprotein during the differentiation of naive T cells into Th1 cells (Fig. 5 a). Addition of the spike glycoprotein also inhibited the production of IFN-γ by differentiated Th1 cells (Fig. 5 b). The inhibition of IFN-γ production was completely reversed by addition of a blocking antibody specific for the viral spike glycoprotein and was not observed when similar concentrations of bovine serum albumin were added as controls (Fig. 5, a and b). In contrast, ligation of CEACAM1a by the spike glycoprotein had no effect on the differentiation and activation of IL-4–producing Th2-type cells (Fig. 5, c and d). These results show that ligation of CEACAM1a on the cell surface of T cells by a ligand that naturally binds to the homophilic interaction site of the N-domain causes specific inhibition of Th1 differentiation and cytokine production.
Figure 5.

CEACAM1a-mediated inhibition of T cells. Naive T cells isolated from C57BL/6 mice were stimulated in vitro with anti-CD3 mAb and anti-CD28 mAb under Th1-inducing conditions (a and b) or Th2-inducing (c and d) conditions. Different concentrations of the MHV spike glycoprotein with or without antisera to the anti-MHV spike glycoprotein were added in the culture during (a and c) or after (b and d) the differentiation to Th1/Th2 cells. Production of IFN-γ was significantly suppressed by the spike glycoprotein when added during and after the differentiation to Th1 (*, P < 0.05) but not Th2 cells. ▪, BSA; □, spike protein plus control serum; ○, spike protein plus antisera to spike glycoprotein. Data are shown as mean ± SEM and are representative of three separate experiments.
Discussion
These studies identify a novel role for CEACAM1a in regulating T cell–dependent immunopathology in vivo based upon an analysis of two different types of murine colitis models, i.e., TNBS and oxazolone colitis, and link this directly with specific inhibition of Th1 pathways and Th1 cytokine signaling. Both of these colitis models are induced by haptenating reagents that induce activation of haptenated antigen-specific T cells (23, 40). The TNBS colitis model has been reported to be specifically mediated by Th1 cytokines in most mouse strains (23, 41). Thus, our studies that have revealed that ligation of CEACAM1a with an anti-CEACAM1a–specific mAb (CC1) could protect against the development of TNBS colitis in association with specific reduction in IFN-γ production by LPLs strongly suggest that the diminution in immunopathology caused by the CC1 mAb is due to inhibition of Th1 pathways.
The reason for the specific down-regulation of Th1 cytokines and in particular IFN-γ by CEACAM1a ligation was determined in subsequent studies. Although it has been previously reported that cross-linking of CEACAM1a on mouse DCs by another mouse CEACAM1a-specific mAb AgB10 induces Th1 cytokine production via the release of IL-12 from DCs (3), we did not observe any effects of the CC1 mAbs on IL-12 production by CD11c+ cells stimulated in vitro. In addition, we observed no difference in the levels of IL-12 produced by spleen and lamina propria mononuclear cells from mice stimulated ex vivo in which TNBS colitis had been induced in the presence of the CC1 mAb. Moreover, treatment of mice with the CC1 mAb before skin sensitization, when T cell priming by DCs occurs and IL-12 production is likely to be maximally induced by DC–T cell interactions involving CD40 and CD40L for example (42), did not significantly impact the severity of colitis or Th1 (IFN-γ) cytokine production. This differs from a previous report from our group that CEACAM1a ligation inhibits a delayed-type hypersensitivity response in the skin more so when performed during the phase of T cell priming and is consistent with our in vitro results with CEACAM1a ligation (18). The reason for these differences is unclear but may represent at its simplest variations between CEACAM1a function in the skin and gut. In contrast, given that administration of the CC1 mAb before the secondary exposure with antigen during the effector phase had major effects upon TNBS-associated immunopathology and Th1 (IFN-γ) cytokine production as reported here at a time when antigen-specific T cells have been activated and are presumably expressing significant levels of CEACAM1a (2, 19–21), an alternative explanation is possible. Specifically, our observation that a CEACAM1a-Fc fusion caused a reduction in IFN-γ production and associated immunopathology suggests that homophilic ligation of CEACAM1 on antigen-activated T cells leads to induction of an inhibitory pathway in T cells that is specifically linked to Th1 pathways. These studies thus suggest that CEACAM1a on activated T cells might be the target of the CC1 mAb. Although it is possible that the CC1 mAb mediates its effects in the TNBS model through interactions with other cell types that are known to express CEACAM1a such as macrophages, B cells, NK cells, and epithelial cells (3–7), as the TNBS colitis model is clearly regulated by Th1 pathways, the CC1 mAb induced down-regulation of IFN-γ production by T cells is likely to play an indispensable role in the suppression of the colitis observed. Moreover, the fact that production of Th2 cytokines was not affected by the CC1 mAb in the TNBS colitis model further suggests that CEACAM1a is directly and specifically linked to regulation of Th1 but not Th2 pathways in this model. Furthermore, the result that the MHV spike glycoprotein, which binds to the N-domain of CEACAM1a, specifically inhibited Th1 differentiation and activation by purified T cells directly supports a role for CEACAM1a regulating Th1 cytokine production by T cells. Taken together with the observation that a chimeric CEACAM1-Fc fusion, which is known to bind homophilically to CEACAM1a, could also reduce immunopathology and Th1 cytokine production in vivo, our results suggest that homophilic (CEACAM1a) or heterophilic (MHV or antibody) ligation of CEACAM1a can directly activate an inhibitory pathway on T cells that is linked to Th1 cytokine production. Preliminary observations that overexpression of a long cytoplasmic tail isoform of CEACAM1a in naive mouse T cells inhibits the development of colitis and IFN-γ production after transfer into RAG-deficient recipient mice directly supports this proposed mechanism of inhibition (unpublished data).
Because CEACAM1 ligation had no significant effect on IL-12 production that could account for suppression of Th1 T cell activity in colitis, we analyzed the expression and activation of Th1-associated transcription factors. Interestingly, a marked suppression of nuclear levels of T-bet, a master transcription factor for Th1 cells, was noted upon CEACAM1 ligation, whereas the levels of the IL-12–inducible transcription factor STAT-4 showed little or no change. These data suggest that CEACAM1 controls Th1 T cell activation and Th1-mediated immunopathology in vivo via its effects on T-bet. Blockade of CEACAM1 ligation in vivo may in turn suppress Th1 T cell activation via down-regulation of T-bet and its effects on chromatin remodeling and promoter activity in the IFN-γ gene locus (33, 43). The molecular basis for the effects of CEACAM1 on T-bet activation, however, remains to be defined. Furthermore, based upon the fact that human and mouse T cells express CEACAM1a isoforms containing a long cytoplasmic tail known to associate with SHP-1 when phosphorylated (18, 21, 22, 44), CEACAM1a ligation may induce negative signals provided by the ITIM-containing cytoplasmic tail of CEACAM1a. Although this remains to be proven, in preliminary support of this possibility, we have observed that transfection of a long but not short cytoplasmic tail containing isoform of CEACAM1 into human T cells results in inhibition of T cell receptor–CD3 complex signaling, which is dependent upon both ITIM domains and SHP-1 (unpublished data).
Ligation of a similar immune inhibitory receptor containing an ITIM, programmed death 1, down-regulates both Th1 and Th2 cytokines with the effect upon Th1 cytokines being larger than that on Th2 cytokines (45). Our data showing that ligation of CEACAM1 specifically inhibits IFN-γ but not IL-4 in these models are thus unique. However, it remains possible that like programmed death 1, CEACAM1 may inhibit both Th1 and Th2 pathways if the signal strength is either of high enough intensity and/or modified by other pathways.
Another interesting aspect of our study is that the CC1 mAb could also prevent oxazolone colitis via suppression of IFN-γ production by LPLs. This is interesting because oxazolone colitis was originally reported as a predominantly Th2-type colitis model (25). Our studies with IFN-γ−/− and IL-4−/− mice clearly indicate that oxazolone colitis is caused by both Th1 and Th2 cytokines in C57BL/6 and BALB/c mouse strains. The differences in the results described here relative to those previously reported (25) might be due to differences in the mouse strains used and/or in the bacterial flora of the animal facility in which the models were studied. It may also be possible that the contribution of cytokines for the induction of colitis varies during the course of oxazolone colitis similar to that which has been previously reported for the TNBS colitis and IL-10−/− colitis models in which Th1 and Th2 cytokines are operative during different phases of colitis (46, 47). As shown here, combination therapy with CC1 and anti–IL-4 mAbs to block both Th1 and Th2 pathways were additive in preventing oxazolone colitis in C57BL/6 mice. This is consistent with recent evidence that both Th1 and Th2 cells specific for the same bacterially associated antigen can drive the progression of colitis (48).
In summary, we have shown that CEACAM1a is associated with specific regulation of Th1 pathways in vivo as defined by an analysis of two T cell–dependent, hapten-mediated colitis models. Based upon an analysis of the effects of a CEACAM1a-Fc chimeric protein in vivo and a soluble natural ligand for the N-domain of CEACAM1a (the MHV spike glycoprotein) on T cells in vitro, the regulation of Th1 pathways by CEACAM1a is likely due to either homophilic or heterophilic ligation of the CEACAM1a N-domain on activated T cells. These results directly demonstrate an in vivo role for CEACAM1 as a functionally important activation-induced regulatory molecule on T cells as suggested by previous in vitro studies (3, 4, 18, 20, 21) and show that the major functional effect of CEACAM1 on T cells in vivo is inhibitory and not stimulatory as suggested by some prior in vitro studies (19, 22). Moreover, these studies point toward an important role for CEACAM1 in regulating Th1-mediated inflammation such as that associated with inflammatory bowel disease and, potentially, virus-mediated immunopathology.
Acknowledgments
H. Iijima was supported by the Naito Foundation. M.F. Neurath was supported by the Innovationsstiftung Rheinland-Pfalz, the SFB458, the Gerhard Hess program of the Deutsche Forschungsgemeinshaft, and a J. William Fulbright scholarship for senior scientists. E.E. Nieuwenhuis was supported by the Ter Meulen Fund, Royal Netherlands Academy of Arts and Sciences. R.S. Blumberg was supported by National Institutes of Health DK44319, DK51362, DK53056, and the Harvard Digestive Diseases Center.
The online version of this article contains supplemental material.
Abbreviations used in this paper: CEACAM1, carcinoembryonic antigen-related cellular adhesion molecule 1; ITIM, immunoreceptor tyrosine-based inhibitory motif; LPL, lamina propria lymphocyte; MHV, mouse hepatitis virus; STAT-4, signal transducer and activator of antigen 4; TNBS, trinitrobenzene sulfonic acid.
References
- 1.Beauchemin, N., P. Draber, G. Dveksler, P. Gold, S. Gray-Owen, F. Grunert, S. Hammarstrom, K.V. Holmes, A. Karlsson, M. Kuroki, et al. 1999. Redefined nomenclature for members of the carcinoembryonic antigen family. Exp. Cell Res. 252:243–249. [DOI] [PubMed] [Google Scholar]
- 2.Hammarstrom, S. 1999. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 9:67–81. [DOI] [PubMed] [Google Scholar]
- 3.Kammerer, R., D. Stober, B.B. Singer, B. Obrink, and J. Reimann. 2001. Carcinoembryonic antigen-related cell adhesion molecule 1 on murine dendritic cells is a potent regulator of T cell stimulation. J. Immunol. 166:6537–6544. [DOI] [PubMed] [Google Scholar]
- 4.Moller, M.J., R. Kammerer, F. Grunert, and S. von Kleist. 1996. Biliary glycoprotein (BGP) expression on T cells and on a natural-killer-cell sub-population. Int. J. Cancer. 65:740–745. [DOI] [PubMed] [Google Scholar]
- 5.Chen, T., W. Zimmermann, J. Parker, I. Chen, A. Maeda, and S. Bolland. 2001. Biliary glycoprotein (BGPa, CD66a, CEACAM1) mediates inhibitory signals. J. Leukoc. Biol. 70:335–340. [PubMed] [Google Scholar]
- 6.Skubitz, K.M., K.D. Campbell, and A.P. Skubitz. 2000. Synthetic peptides of CD66a stimulate neutrophil adhesion to endothelial cells. J. Immunol. 164:4257–4264. [DOI] [PubMed] [Google Scholar]
- 7.Coutelier, J.P., C. Godfraind, G.S. Dveksler, M. Wysocka, C.B. Cardellichio, H. Noel, and K.V. Holmes. 1994. B lymphocyte and macrophage expression of carcinoembryonic antigen-related adhesion molecules that serve as receptors for murine coronavirus. Eur. J. Immunol. 24:1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan, K., B.D. Zelus, R. Meijers, J.H. Liu, J.M. Bergelson, N. Duke, R. Zhang, A. Joachimiak, K.V. Holmes, and J.H. Wang. 2002. Crystal structure of murine sCEACAM1a[1,4]: a coronavirus receptor in the CEA family. EMBO J. 21:2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson, J.A., F. Grunert, and W. Zimmermann. 1991. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J. Clin. Lab. Anal. 5:344–366. [DOI] [PubMed] [Google Scholar]
- 10.Ravetch, J.V., and L.L. Lanier. 2000. Immune inhibitory receptors. Science. 290:84–89. [DOI] [PubMed] [Google Scholar]
- 11.Watt, S.M., A.M. Teixeira, G.Q. Zhou, R. Doyonnas, Y. Zhang, F. Grunert, R.S. Blumberg, M. Kuroki, K.M. Skubitz, and P.A. Bates. 2001. Homophilic adhesion of human CEACAM1 involves N-terminal domain interactions: structural analysis of the binding site. Blood. 98:1469–1479. [DOI] [PubMed] [Google Scholar]
- 12.Bos, M.P., D. Hogan, and R.J. Belland. 1999. Homologue scanning mutagenesis reveals CD66 receptor residues required for neisserial Opa protein binding. J. Exp. Med. 190:331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Virji, M., D. Evans, J. Griffith, D. Hill, L. Serino, A. Hadfield, and S.M. Watt. 2000. Carcinoembryonic antigens are targeted by diverse strains of typable and non-typable Haemophilus influenzae. Mol. Microbiol. 36:784–795. [DOI] [PubMed] [Google Scholar]
- 14.Virji, M., D. Evans, A. Hadfield, F. Grunert, A.M. Teixeira, and S.M. Watt. 1999. Critical determinants of host receptor targeting by Neisseria meningitidis and Neisseria gonorrhoeae: identification of Opa adhesiotopes on the N-domain of CD66 molecules. Mol. Microbiol. 34:538–551. [DOI] [PubMed] [Google Scholar]
- 15.Ergun, S., N. Kilik, G. Ziegeler, A. Hansen, P. Nollau, J. Gotze, J.H. Wurmbach, A. Horst, J. Weil, M. Fernando, et al. 2000. CEA-related cell adhesion molecule 1: a potent angiogenic factor and a major effector of vascular endothelial growth factor. Mol. Cell. 5:311–320. [DOI] [PubMed] [Google Scholar]
- 16.Izzi, L., C. Turbide, C. Houde, T. Kunath, and N. Beauchemin. 1999. cis-Determinants in the cytoplasmic domain of CEACAM1 responsible for its tumor inhibitory function. Oncogene. 18:5563–5572. [DOI] [PubMed] [Google Scholar]
- 17.Huang, J., J.D. Hardy, Y. Sun, and J.E. Shively. 1999. Essential role of biliary glycoprotein (CD66a) in morphogenesis of the human mammary epithelial cell line MCF10F. J. Cell Sci. 112:4193–4205. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima, A., H. Iijima, M.F. Neurath, T. Nagaishi, E.E. Nieuwenhuis, R. Raychowdhury, J. Glickman, D.M. Blau, S. Russell, K.V. Holmes, et al. 2002. Activation-induced expression of carcinoembryonic antigen-cell adhesion molecule 1 regulates mouse T lymphocyte function. J. Immunol. 168:1028–1035. [DOI] [PubMed] [Google Scholar]
- 19.Kammerer, R., S. Hahn, B.B. Singer, J.S. Luo, and S. von Kleist. 1998. Biliary glycoprotein (CD66a), a cell adhesion molecule of the immunoglobulin superfamily, on human lymphocytes: structure, expression and involvement in T cell activation. Eur. J. Immunol. 28:3664–3674. [DOI] [PubMed] [Google Scholar]
- 20.Morales, V.M., A. Christ, S.M. Watt, H.S. Kim, K.W. Johnson, N. Utku, A.M. Texieira, A. Mizoguchi, E. Mizoguchi, G.J. Russell, et al. 1999. Regulation of human intestinal intraepithelial lymphocyte cytolytic function by biliary glycoprotein (CD66a). J. Immunol. 163:1363–1370. [PubMed] [Google Scholar]
- 21.Boulton, I.C., and S.D. Gray-Owen. 2002. Neisserial binding to CEACAM1 arrests the activation and proliferation of CD4+ T lymphocytes. Nat. Immunol. 3:229–236. [DOI] [PubMed] [Google Scholar]
- 22.Donda, A., L. Mori, A. Shamshiev, I. Carena, C. Mottet, M.H. Heim, C. Beglinger, F. Grunert, C. Rochlitz, L. Terracciano, et al. 2000. Locally inducible CD66a (CEACAM1) as an amplifier of the human intestinal T cell response. Eur. J. Immunol. 30:2593–2603. [DOI] [PubMed] [Google Scholar]
- 23.Neurath, M.F., I. Fuss, B.L. Kelsall, E. Stuber, and W. Strober. 1995. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elson, C.O., K.W. Beagley, A.T. Sharmanov, K. Fujihashi, H. Kiyono, G.S. Tennyson, Y. Cong, C.A. Black, B.W. Ridwan, and J.R. McGhee. 1996. Hapten-induced model of murine inflammatory bowel disease: mucosa immune responses and protection by tolerance. J. Immunol. 157:2174–2185. [PubMed] [Google Scholar]
- 25.Boirivant, M., I.J. Fuss, A. Chu, and W. Strober. 1998. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J. Exp. Med. 188:1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nieuwenhuis, E.E., M.F. Neurath, N. Corazza, H. Iijima, J. Trgovcich, S. Wirtz, J. Glickman, D. Bailey, M. Yoshida, P.R. Galle, et al. 2002. Disruption of T helper 2-immune responses in Epstein-Barr virus-induced gene 3-deficient mice. Proc. Natl. Acad. Sci. USA. 99:16951–16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neurath, M.F., B. Weigmann, S. Finotto, J. Glickman, E. Nieuwenhuis, H. Iijima, A. Mizoguchi, E. Mizoguchi, J. Mudter, P.R. Galle, et al. 2002. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J. Exp. Med. 195:1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dveksler, G.S., M.N. Pensiero, C.B. Cardellichio, R.K. Williams, G.S. Jiang, K.V. Holmes, and C.W. Dieffenbach. 1991. Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 65:6881–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dveksler, G.S., C.W. Dieffenbach, C.B. Cardellichio, K. McCuaig, M.N. Pensiero, G.S. Jiang, N. Beauchemin, and K.V. Holmes. 1993. Several members of the mouse carcinoembryonic antigen-related glycoprotein family are functional receptors for the coronavirus mouse hepatitis virus-A59. J. Virol. 67:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith, A.L., C.B. Cardellichio, D.F. Winograd, M.S. de Souza, S.W. Barthold, and K.V. Holmes. 1991. Monoclonal antibody to the receptor for murine coronavirus MHV-A59 inhibits viral replication in vivo. J. Infect. Dis. 163:879–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wessner, D.R., P.C. Shick, J.H. Lu, C.B. Cardellichio, S.E. Gagneten, N. Beauchemin, K.V. Holmes, and G.S. Dveksler. 1998. Mutational analysis of the virus and monoclonal antibody binding sites in MHVR, the cellular receptor of the murine coronavirus mouse hepatitis virus strain A59. J. Virol. 72:1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallagher, T.M. 1997. A role for naturally occurring variation of the murine coronavirus spike protein in stabilizing association with the cellular receptor. J. Virol. 71:3129–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szabo, S.J., S.T. Kim, G.L. Costa, X. Zhang, C.G. Fathman, and L.H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 100:655–669. [DOI] [PubMed] [Google Scholar]
- 34.Mach, N., S. Gillessen, S.B. Wilson, C. Sheehan, M. Mihm, and G. Dranoff. 2000. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 60:3239–3246. [PubMed] [Google Scholar]
- 35.Neurath, M.F., I. Fuss, M. Pasparakis, L. Alexopoulou, S. Haralambous, K.H. Meyer zum Buschenfelde, W. Strober, and G. Kollias. 1997. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur. J. Immunol. 27:1743–1750. [DOI] [PubMed] [Google Scholar]
- 36.Glimcher, L.H., and H. Singh. 1999. Transcription factors in lymphocyte development–T and B cells get together. Cell. 96:13–23. [DOI] [PubMed] [Google Scholar]
- 37.Seder, R.A., R. Gazzinelli, A. Sher, and W.E. Paul. 1993. Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon gamma production and diminishes interleukin 4 inhibition of such priming. Proc. Natl. Acad. Sci. USA. 90:10188–10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maldonado-Lopez, R., T. De Smedt, P. Michel, J. Godfroid, B. Pajak, C. Heirman, K. Thielemans, O. Leo, J. Urbain, and M. Moser. 1999. CD8α1 and CD8α− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 189:587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gagneten, S., O. Gout, M. Dubois-Dalcq, P. Rottier, J. Rossen, and K.V. Holmes. 1995. Interaction of mouse hepatitis virus (MHV) spike glycoprotein with receptor glycoprotein MHVR is required for infection with an MHV strain that expresses the hemagglutinin-esterase glycoprotein. J. Virol. 69:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris, G.P., P.L. Beck, M.S. Herridge, W.T. Depew, M.R. Szewczuk, and J.L. Wallace. 1989. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 96:795–803. [PubMed] [Google Scholar]
- 41.Kanai, T., M. Watanabe, A. Okazawa, T. Sato, M. Yamazaki, S. Okamoto, H. Ishii, T. Totsuka, R. Iiyama, R. Okamoto, et al. 2001. Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn's disease. Gastroenterology. 121:875–888. [DOI] [PubMed] [Google Scholar]
- 42.Stuber, E., W. Strober, and M. Neurath. 1996. Blocking the CD40L–CD40 interaction in vivo specifically prevents the priming of T helper 1 cells through the inhibition of interleukin 12 secretion. J. Exp. Med. 183:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mullen, A.C., F.A. High, A.S. Hutchins, H.W. Lee, A.V. Villarino, D.M. Livingston, A.L. Kung, N. Cereb, T.P. Yao, S.Y. Yang, et al. 2001. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 292:1907–1910. [DOI] [PubMed] [Google Scholar]
- 44.Singer, B.B., I. Scheffrahn, R. Heymann, K. Sigmundsson, R. Kammerer, and B. Obrink. 2002. Carcinoembryonic antigen-related cell adhesion molecule 1 expression and signaling in human, mouse, and rat leukocytes: evidence for replacement of the short cytoplasmic domain isoform by glycosylphosphatidylinositol-linked proteins in human leukocytes. J. Immunol. 168:5139–5146. [DOI] [PubMed] [Google Scholar]
- 45.Latchman, Y., C.R. Wood, T. Chernova, D. Chaudhary, M. Borde, I. Chernova, Y. Iwai, A.J. Long, J.A. Brown, R. Nunes, et al. 2001. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2:261–268. [DOI] [PubMed] [Google Scholar]
- 46.Dohi, T., K. Fujihashi, H. Kiyono, C.O. Elson, and J.R. McGhee. 2000. Mice deficient in Th1- and Th2-type cytokines develop distinct forms of hapten-induced colitis. Gastroenterology. 119:724–733. [DOI] [PubMed] [Google Scholar]
- 47.Spencer, D.M., G.M. Veldman, S. Banerjee, J. Willis, and A.D. Levine. 2002. Distinct inflammatory mechanisms mediate early versus late colitis in mice. Gastroenterology. 122:94–105. [DOI] [PubMed] [Google Scholar]
- 48.Iqbal, N., J.R. Oliver, F.H. Wagner, A.S. Lazenby, C.O. Elson, and C.T. Weaver. 2002. T helper 1 and T helper 2 cells are pathogenic in an antigen-specific model of colitis. J. Exp. Med. 195:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]