

Abstract
In multiple sclerosis (MS), inflammation in the central nervous system (CNS) leads to damage of axons and myelin. Early during the clinical course, patients can compensate this damage, but little is known about the changes that underlie this improvement of neurological function. To study axonal changes that may contribute to recovery, we made use of an animal model of MS, which allows us to target inflammatory lesions to the corticospinal tract (CST), a major descending motor pathway. We demonstrate that axons remodel at multiple levels in response to a single neuroinflammatory lesion as follows: (a) surrounding the lesion, local interneurons show regenerative sprouting; (b) above the lesion, descending CST axons extend new collaterals that establish a “detour” circuit to the lumbar target area, whereas below the lesion, spared CST axons increase their terminal branching; and (c) in the motor cortex, the distribution of projection neurons is remodeled, and new neurons are recruited to the cortical motor pool. Behavioral tests directly show the importance of these changes for recovery. This paper provides evidence for a highly plastic response of the motor system to a single neuroinflammatory lesion. This framework will help to understand the endogenous repair capacity of the CNS and to develop therapeutic strategies to support it.
Keywords: demyelinating autoimmune disease, spinal cord, pyramidal tracts, axons, nerve regeneration
Introduction
Inflammatory disorders of the central nervous system (CNS) such as multiple sclerosis (MS) are often accompanied by substantial damage to axons and myelin (1). The extent of axonal damage is of particular importance for the clinical deficit that MS patients accumulate over time (2, 3). However, the clinical outcome will ultimately depend on the balance of damage inflicted on the CNS and the extent of endogenous repair that can counter it. Although axonal destruction in MS has been characterized extensively in recent years, very little is known about how it can be counterbalanced (4, 5). Substantial compensation is evident in the typical clinical course of MS. The relapsing–remitting form of the disease is defined by cycles of newly emerging clinical deficits that resolve within a few days or weeks. The regression of tissue edema and inflammation undoubtedly plays a major role early during remission, but these processes cannot mend structural damage caused by the inflammatory attack. Thus, additional compensatory mechanisms must be considered for complete recovery that often occurs despite substantial structural damage that persists at the site of inflammation.
The neuropathology of MS and its animal model, experimental autoimmune encephalomyelitis (EAE), suggests that limited repair of myelin damage can be achieved (“remyelination”), when oligodendrocytes form new myelin (5, 6) This gives rise to so-called shadow plaques, which are areas of myelin pallor in MS brain tissue. In addition, the redistribution of sodium-channels in demyelinated axons can help to compensate for myelin loss (7). However, given the close correlation between the extent of axon damage and clinical deficit in MS, one would expect compensation for axon loss to be an even more powerful means of recovery. Surprisingly, little is known about how lost axonal connections are repaired in MS. Early papers have documented changes suggestive of axonal outgrowth in lesions of MS, a finding subsequently confirmed for EAE (8–10). In addition, functional MRI studies provide evidence for cortical adaptation in MS patients (11, 12). The lack of understanding of how recovery from axonal damage occurs in MS is, in part, due to the disseminated nature of MS and EAE. Multiple neuroinflammatory lesions occur in unpredictable locations in these diseases and damage various axonal tracts with high interindividual variability. This makes the analysis of axonal compensation, which occurs equally in a disseminated and variable way, difficult if not impossible.
In the present paper, we have circumvented this problem by using a recently developed “targeted” EAE model in which we can reproducibly induce a single inflammatory, demyelinating lesion in the dorsal column area of the midthoracic spinal cord (13). This area comprises the corticospinal tract (CST), a major descending motor tract that connects cortical layer V neurons with spinal targets and is often affected in MS (14). Targeting lesions to this tract system combined with detection of growth-associated proteins, as well as a variety of anterograde, retrograde, and trans-synaptic tracing techniques allowed us to analyze precisely how axonal compensation is achieved. We demonstrate that a single EAE lesion in the spinal cord induces axonal remodeling on many levels: (a) on the spinal level, in the neuronal circuitry surrounding the lesion, where numerous axons sprout; (b) in descending motor tracts, which create “detour” circuits to circumnavigate the lesion; and (c) at the cortical level, where the map of motor neurons that project to spinal motor targets is remodeled. Finally, behavioral experiments show that at least some of these remodeled connections directly contribute to functional recovery.
In summary, our paper provides insight into the mechanisms of endogenous axonal remodeling in inflammatory CNS diseases. In addition, our results suggest a new strategy for intervention in MS, as they underscore the existence of an endogenous repair program that could be targeted therapeutically.
Materials and Methods
Animals
Adult female Lewis rats (160 and 220 g) were obtained from Harlan. All experiments were approved by the veterinary department of the Canton of Zurich.
Immunogen
An NH2-terminal fragment of rat myelin oligodendrocyte glycoprotein (rMOG; amino acids 1–125) was expressed in Escherichia coli, purified to homogeneity as described previously (15), dissolved in 6 M urea, and solubilized by dialysis against 20 mM sodium acetate buffer, pH 3.0.
Induction of a Targeted EAE Lesion
A focal EAE lesion was targeted to the CST area in the midthoracic spinal cord by minimal invasive injection of proinflammatory cytokines in MOG immunized animals as described previously (reference 13 and see supplemental information for detailed protocols).
Surgical Procedures
Thoracic Lesion of the CST.
A bilateral dorsolateral hemisection of the spinal cord at T8 was performed as described previously (16). The main dorsomedial as well as the minor dorsolateral components of the CST were transected.
Medullar Lesion of the CST.
A pyramidotomy was performed at the level of the medulla oblongata using a ventral approach as described previously (17). The entire CST was transected rostral to the decussation on both sides.
Histopathology
Histopathological evaluation was performed on 3-μm thick sections of spinal cords of animals with targeted EAE lesions at day 7 (n = 4), day 14 (n = 4), and day 28 (n = 4) after lesion induction (see online supplemental material for detailed protocols).
Immunohistochemistry
Cross sections of the spinal cord were cut on a vibratome or cryostat and analyzed using standard immunohistochemistry (see online supplemental material for detailed protocols). The following primary antibodies were used: rabbit anti–c-Jun (1:500; Santa Cruz Biotechnology, Inc.), mouse anti-GAP43 (1:1,000; clone 9-1E12; Chemicon), mouse anti-GFAP (1:200, clone G-A-5, Chemicon), mouse anti-MAP2 (1:100; clone AP20; Chemicon), rabbit antineurofilament (1:100, Chemicon), and mouse ED1 (1:1,000, clone ED1; Serotec).
Quantitative Analysis of Immunostainings
Expression of c-Jun and GAP43 was quantified on cryostat sections of EAE animals perfused at day 7, day 14, and day 28 (n = 4 for each time point), as well as from untreated control animals (n = 2) and animals receiving a cytokine injection but no immunization (n = 6). For comparison, c-Jun and GAP43 expression were analyzed in animals perfused at day 7, day 14, and day 28 after a traumatic spinal cord lesion (n = 5 for each time point). For analysis, we selected sections with comparable lesions size and counted the total number of c-Jun+, GAP43+ neurons as well as the number of c-Jun+/GAP43+ neurons (10–30 sections per animal). For four EAE animals at day 14 after lesion induction, we further reconstructed the localization of c-Jun+ and GAP43+ neurons on five consecutive sections.
Tracing of Axonal Tract Systems
The hindlimb CST was traced anterogradely and bilaterally with biotinylated dextran amine (BDA) as described previously (18). Contacts between hindlimb CST collaterals and propriospinal neurons (PSNs) were analyzed by anterograde tracing of the CST (BDA) and retrograde tracing of PSNs (fluoroemerald; for detailed tracing protocols, see online supplemental material).
Analysis of CST Damage
The hindlimb CST damage was quantified in targeted EAE animals as described previously (reference 13 and see online supplemental material for detailed protocols).
Analysis of CST Reorganization
We determined the sprouting of the hindlimb CST above and below the level of the EAE lesion on cross sections of the cervical (segments C3–C5, between 40 and 50 sections per animal were evaluated) and lumbar (segments L1–L3, between 20 and 25 sections per tract were evaluated) spinal cord at day 28 as follows. For each side of each section, we determined the number of CST collaterals (the number of fibers emerging from the CST tract and entering the gray matter) and the number of CST branches (the number of fibers crossing grid lines positioned in the gray matter [see Figs. 3 and 4]). To correct for variation in tracing efficiency, we normalized the number of CST collaterals and branches per section to the total number of labeled CST axons on each side (determined as the average of traced fibers on five cross sections between C3 and C5 or L1 and L3). As we detected a substantial contribution of collaterals derived from the minor ventral and lateral components of the CST in the lumbar spinal cord, we determined the number of labeled axons as well as the number of collaterals per lumbar section originating from these CST components and included them in the analysis. For the final analysis, we determined the following indices: the collateral index, as the average number of CST collaterals per section divided by the number of labeled CST fibers and multiplied by 1,000; the branch index, as the average number of CST branches per section divided by the number of labeled CST fibers and multiplied with 1,000; and the ratio of CST branches/collateral as the average number of CST branches per section divided by the average number of CST collaterals per section. We further reconstructed the cervical CST collaterals and branches on 10 consecutive sections derived from comparable levels of the cervical spinal cord in EAE (n = 10) and control animals (n = 4) using a camera lucida. In addition, hindlimb CST collaterals and PSNs were reconstructed on five consecutive sections of the cervical spinal cord in double-traced animals.
Figure 3.

Targeted EAE lesions induce new CST collaterals above the lesion. (a) Scheme of how the number of CST axons (dots in CST area), collaterals (open circles around fibers entering the gray matter), and branches (closed circles where fibers cross a line positioned at the ventral border of the central canal) were counted. (b) Cross section of the normal cervical spinal cord with very few collaterals (BDA-traced hindlimb CST outlined by gray line). (c) Numerous cervical collaterals (arrows) in a targeted EAE animal at day 28. (d–f) Cervical hindlimb CST reorganization in relation to extent of CST damage. Collaterals (d) and branches (e) increase equally. As their ratio remains constant (f), cervical reorganization is mainly due to formation of new collaterals. (b and c) Bars, 50 μm.
Figure 4.
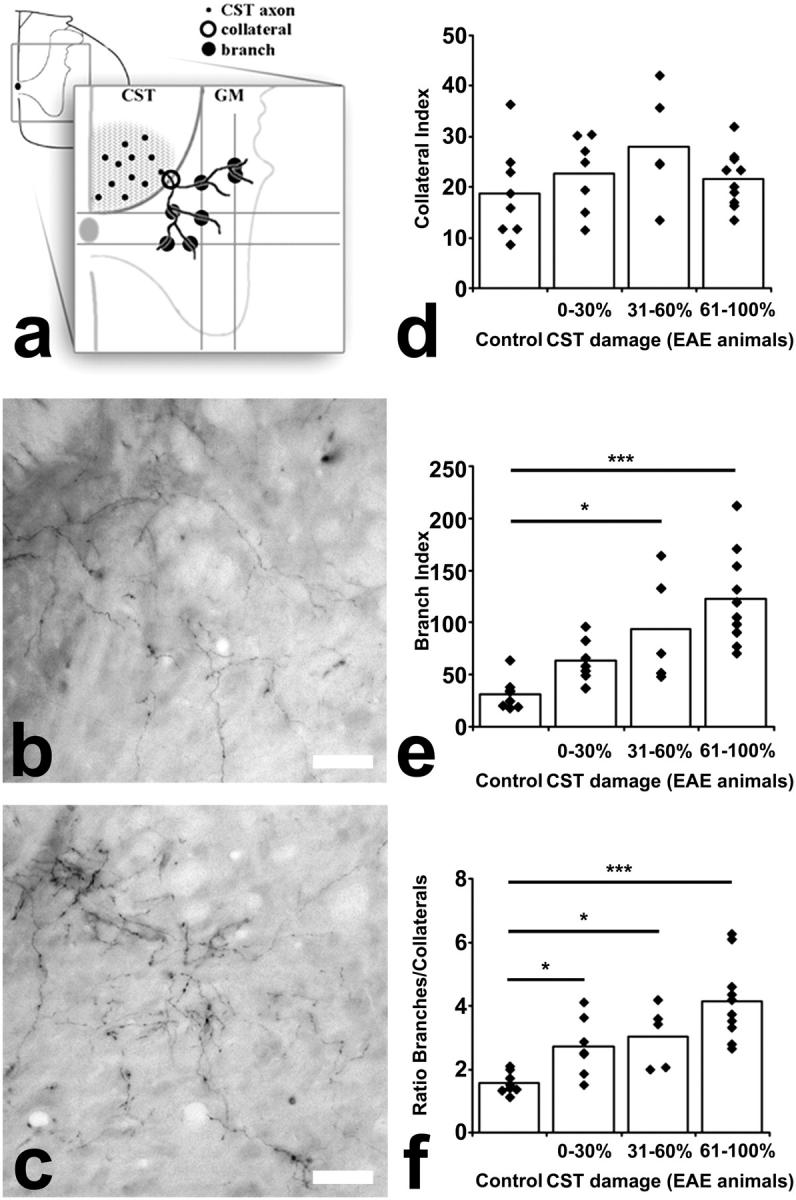
Targeted EAE lesions induce branching of CST collaterals below the lesion. (a) Scheme of how lumbar CST reorganization was evaluated (compare with Fig. 3). Because lumbar CST collaterals grow less directed than cervical ones, two horizontal and two vertical lines positioned in the gray matter were used to assess branching. (b) Cross section of the normal lumbar spinal cord with collaterals that branch in the gray matter. (c) In EAE animals, CST collateral branching is increased (D28). (d–f) Lumbar hindlimb CST reorganization in relation to extent of CST damage. The number of collaterals is similar between control and EAE animals (d), whereas the number of branches emerging from these collaterals is increased (e and f). These results indicate that lumbar remodeling of the CST is primarily due to an increased branching of existing collaterals. (b and c) Bars, 50 μm.
Transsynaptic Tracing of Axonal Connectivity
To assess changes induced by the EAE lesion in the connectivity between the lumbar spinal cord and the cortex, we used a retrograde transsynaptic tracer, GFP-labeled pseudo-rabies virus Bartha (PRV) as described previously (reference 18 and, for a detailed protocol, see online supplemental material). The number of neurons with PRV immunoreactivity in lamina V was counted throughout the entire cortex and normalized to 100 sections per animal. For all animals, we mapped the localization of PRV+ neurons in lamina V of the cortex in relation to existing maps of the hindlimb and forelimb motor area (19). The following groups of animals were analyzed: animals with a newly transected CST at the thoracic level (n = 8), animals with an unlesioned CST (n = 6), and animals 4 wk after a thoracic EAE lesion with (n = 8) or without (n = 8) an additional fresh thoracic transection of the CST.
Behavioral Testing
Three behavioral test paradigms were used to assess overall motor performance (standard EAE score; reference 13) as well as CST-related responses (hindlimb placing and proprioceptive adduction; references 18, 20). For a detailed description of the test paradigms, see online supplemental material.
Statistical Analysis
Statistical analysis was performed with Prism software (GraphPad). Differences in the anatomical data were analyzed using either an unpaired Student's t test or one-way analysis of variance followed by Newman-Keuls Multiple Comparisons tests. Nonparametric behavioral data were analyzed over time with a Friedman test and behavioral scores were compared with a Wilcoxon matched paired test. *, P < 0.05 was considered significant. **, P < 0.01. ***, P < 0.001. Data are presented as mean ± SEM.
Online Supplemental Material
The online supplemental material contains detailed protocols of the staining and tracing techniques used in this paper. It contains comparison of axonal remodeling after inflammatory and traumatic spinal cord lesions. Fig. S1 shows comparison of growth-associated gene expression in inflammatory and traumatic spinal cord lesions. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040452/DC1.
Results
This work explores how axonal damage is compensated for in an animal model of MS. We approached this problem in a reductionist way, by studying the compensatory response of the motor system to a single, localized neuroinflammatory lesion. A targeted EAE lesion was induced in the dorsal column of the midthoracic spinal cord by minimal invasive injection of proinflammatory cytokines in rats immunized with subthreshold levels of MOG(13). Previously, we had established that these targeted lesions reflect the major pathological hallmarks of MS, including focal inflammatory demyelination and extensive axonal damage (13). We have now characterized the evolution of inflammation, demyelination, and axonal damage in these lesions over time (Fig. 1). After the cytokine injection, a large inflammatory infiltration developed within a few days (Fig. 1 a, D7). However, both the area of inflammation (Fig. 1 d) as well as the density of cell infiltration (day 7: 4229 ± 379; day 14: 5503 ± 848; and day 28: 5428 ± 406 cells/mm2) remained largely unchanged for the remaining experimental period (days 7–28). Parameters of structural damage showed a similar time course. Extensive demyelination occurred up to day 7, but the area of demyelination did not change significantly between days 7 and 28 (Fig. 1, e–h). We did not detect remyelination at any time point. The axon density dropped substantially over the first days of lesion development and again remained unchanged from day 7 up to day 28 (Fig. 1, i–l). In accordance with this finding, acute axonal damage, as assessed by immunohistochemistry for APP, was prominent at an early disease stage (Fig. 1 m, D7) and substantially lower at later stages (Fig. 1, n and o, D14, D28). However, despite the persistent structural damage, the animals showed a nearly complete behavioral recovery over the same time period (Fig. 1 p). These findings point to the existence of compensatory mechanisms, which promote behavioral recovery in the presence of persisting structural deficits. The targeted EAE model now allows us to analyze these compensatory mechanisms on multiple anatomical levels.
Figure 1.

Histopathology of lesion development in the targeted EAE model. (a–c) Spinal cross sections of targeted EAE lesions stained with hematoxylin eosin at days 7 (a), 14 (b), and 28 (c) after lesions induction. (d) Quantification of the area of inflammation based on hematoxylin eosin–stained sections. (e–g) Spinal cross sections of targeted EAE lesions stained with Luxol fast blue at days 7 (e), 14 (f), and 28 (g) after lesion induction. (h) Quantification of the area of demyelination based in Luxol fast blue–stained sections. (i–k) Spinal cross sections of targeted EAE lesions stained by Bielschowsky silver impregnation at days 7 (i), 14 (j), and 28 (k) after lesions induction. (l) Quantification of axon density based on silver impregnated sections. (m–o) Spinal cross sections of targeted EAE lesions immunostained for APP (red, with nuclear counterstain in blue) at days 7 (m), 14 (n), and 28 (o) after lesion induction. (p) Quantification of the behavioral recovery over time in targeted EAE animals (mean ± SEM).
Growth-associated Proteins and Neurite Formation in Interneurons near Spinal EAE Lesions.
As a first step to understanding how neurons respond to inflammatory axonal damage, we focused our analysis on neurons in the immediate vicinity of the lesion. We asked whether proteins that are part of a growth-associated expression pattern, such as c-Jun and GAP43 (21–23), could be detected in neurons near the lesion and whether their expression correlated with axon outgrowth. We immunostained for c-Jun at various stages after induction of a targeted EAE lesion in the midthoracic spinal cord. Very little c-Jun immunoreactivity was detected in untreated control animals (1.2 ± 0.4 c-Jun+ neurons/section) and in animals receiving a cytokine injection but no immunization (day 7: 1.4 ± 0.2; day 14: 2.4 ± 0.3 c-Jun+ neurons/section). In contrast, in targeted EAE animals, many neurons were immunoreactive for c-Jun with a typical nuclear staining pattern (c-Jun+; Fig. 2 a and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20040452/DC1). The number of c-Jun+ neurons was strongly increased starting on day 7 (17.5 ± 2.9 c-Jun+ neurons/section), peaked at day 14 (23.9 ± 4.5 c-Jun+ neurons/section), and declined after. However, persistent expression of c-Jun was still detected up to day 28 (5.1 ± 1 c-Jun+ neurons/section). This is in contrast with the comparably short-lasting induction of c-Jun observed after traumatic spinal cord lesions (online supplemental material and Fig. S1).
Figure 2.
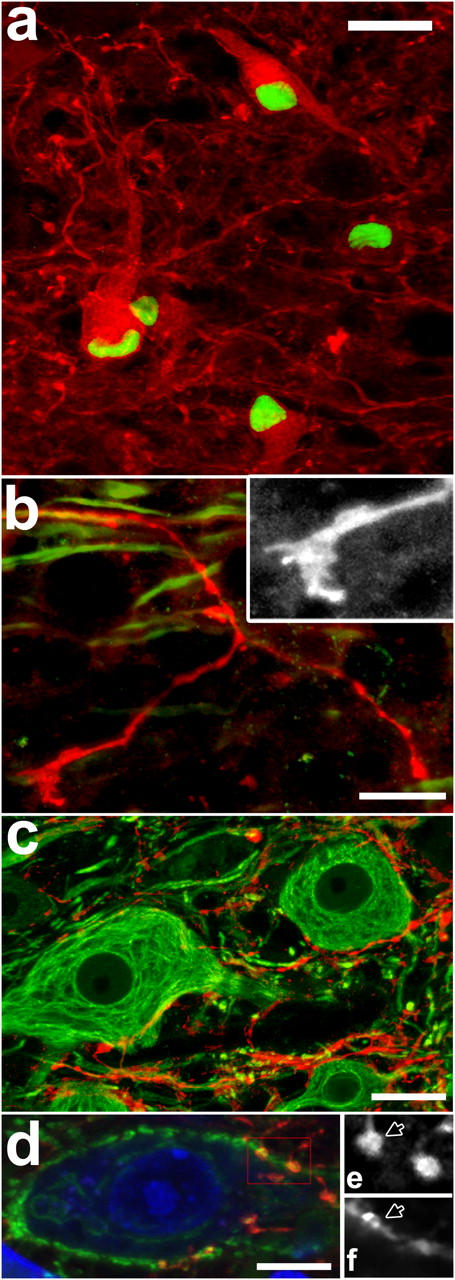
Targeted EAE lesions induce sprouting of spinal neurons. (a–d) Confocal images of GAP43+ neurons and axonal sprouts at day 28. (a) Coexpression of c-Jun (green) and GAP43 (red) in neurons (D28). (b) GAP43+ sprout (red) emerging from a neurofilament+ axon (NF, green) in the ventral white matter tipped by a complex growth cone (inset: red channel enlarged to 175%). (c) Dense network of GAP43+ sprouts (red), with close appositions to neuronal somata (NF, green). (d) Some appositions between GAP43+ sprouts (red) and neuronal somata (neurotrace, blue) are synaptophysin+ (green). (e and f) Single optical sections (at 175% magnification of red box in d) show colocalization of GAP43 (e) and synaptophysin (f) at two putative synaptic sites (one marked by an arrow). (a–d) Bars, 25 μm. γ ≠ 1 was used to improve visibility of synaptophysin staining.
Because the persistent expression of c-Jun is associated with axonal outgrowth, in particular if accompanied by other growth-associated proteins such as GAP43 (21, 23), we asked whether GAP43 was coexpressed with c-Jun in neurons near EAE lesions. In untreated control tissue, GAP43 staining displays a “punctuate” pattern surrounding unstained neuronal somata (0.0 GAP43+ neurons/section) as expected for a presynaptic protein such as GAP43. Similarly, only very few GAP43+ neurons were detected in animals receiving a cytokine injection but no immunization (day 7: 0.1 ± 0.1; day 14: 0.3 ± 0.2 GAP43+ neurons/section) or animals with a traumatic spinal cord injury (see online supplemental material and Fig. S1). In contrast, animals with a targeted EAE lesion showed GAP43 immunoreactivity in the somata of neurons (GAP43+ neurons; Fig. 2 a). The first GAP43+ neurons were detected at day 7. Their number peaked at day 14 and declined toward day 28 (day 7: 1.2 ± 0.5; day 14: 7.4 ± 0.8; day 28: 1.1 ± 0.1 GAP43+ neurons/section). Initially (day 7), the cells showed very little labeling along processes, but later a network of labeled processes became apparent (day 14 and most prominently at day 28). GAP43 immunoreactivity was closely associated with the prolonged presence of nuclear c-Jun, indicating that both genes are part of a common gene activation program (day 7: 87.8%, day 14: 84.4%, and day 28: 95.4% of GAP43+ neurons are c-Jun+, whereas only 10–30% of all MAP2+ neurons coexpress c-Jun over this time period). However, not all c-Jun+ neurons expressed GAP43, and are thus unlikely to initiate growth (at day 7: 5.8% of c-Jun+ neurons are also GAP43+, this number rises to 23.5% at day 14 and 20.4% at day 28). Although c-Jun+ neurons are evenly distributed throughout the gray matter, those coexpressing GAP43 are primarily confined to medium-sized neurons in the intermediate laminae of the spinal cord near the lesion border (Rexed laminae 5–7 and Fig. S1 d).
Next, we examined the formation of new processes using GAP43 as a marker for growing neurites (24, 25). In our model, GAP43+ neurites became apparent from day 14 on (see the previous paragraph) and often resembled sprouts, characterized by caliber variations, irregular growth path, and complex branching pattern (Fig. 2). Occasionally, GAP43+ neurites ended in growth conelike structures (Fig. 2 b). The vast majority of these GAP43+ neurites was observed in the gray matter adjacent to the lesion often originating from local interneurons (Fig. 2 c). Only a few sprouts appeared to grow into white matter, primarily extending into the unaffected ventral white matter and almost never into the lesion. At day 14, most GAP43+ neurites ended in small axonal bulbs. However, at day 28, some of the GAP43+ neurites showed varicosities adjacent to neuronal somata or proximal dendrites. Some of these varicosities were immunoreactive for synaptophysin (Fig. 2, d–f) and, thus, represent putative synaptic boutons.
The CST Forms New Collaterals Above the EAE Lesion.
Adaptive changes of descending motor tract systems were assessed at day 28 after the induction of a midthoracic EAE lesion. As the main part of the CST in rodents runs in the dorsal column, our targeted lesion transects a variable fraction of all hindlimb CST axons (ranging from 2 to 100% of hindlimb CST fibers [percentage of CST damage]). We wondered if this inflammatory damage would induce adaptive changes in descending axons before they entered the lesion site (i.e., in the cervical spinal cord). In principle, CST axons could give rise to more collaterals that enter the gray matter, and/or collaterals could show an increased formation of branches (for a definition of collaterals and branches, see Materials and Methods and Fig. 3 a).
In normal animals, very few collaterals and branches emerged from the hindlimb CST in the cervical spinal cord (mean collateral index: 0.38; mean branch index: 0.69; n = 7; Fig. 3 b). In contrast, a nearly 10-fold increased number of hindlimb CST collaterals and branches was detected in the cervical spinal cord of animals with a targeted EAE lesion (mean collateral index: 3.71, P < 0.001 vs. controls; mean branch index 4.1, P < 0.01 vs. controls; n = 29; Fig. 3, c, d, and e). However, the ratio of branches to collaterals was comparable between EAE and control animals (mean ratio of 1.14 CST branches/collateral in EAE animals vs. 1.8 branches/collaterals in control animals; Fig. 3 f). This de novo formation of cervical collaterals is similar to the induction of CST collaterals observed after traumatic spinal cord lesions (see online supplemental material).
To understand whether this reorganization depends on the severity of the lesion, we quantified the extent of CST collateral formation in spinal cords in response to EAE lesions of varying severity. We found that the more severe the lesion is, the more pronounced is the increase in collateral formation (Fig. 3 d).
CST Branches Form below the EAE Lesion.
To determine the contribution of spared hindlimb CST fibers to axonal remodeling, we quantified the number of hindlimb collaterals and branches and their ratio in the lumbar spinal cord of animals at day 28 after the induction of a targeted EAE lesion (Fig. 4).
Compared with the CST of control animals (n = 8), the CST of animals with a targeted EAE lesion (n = 22) showed a threefold increase in the number of branches (mean branch index: 97.1 in EAE animals vs. mean branch index: 31.3 in control animals, P < 0.001; Fig. 4, b, c, and e). However, in contrast with the cervical spinal cord, the number of collaterals remained similar between both groups (mean collateral index: 23.4 in EAE animals vs. mean collateral index: 18.8 in control animals, P > 0.05; Fig. 4 d). Accordingly, the branch-to-collateral ratio is increased in EAE animals (mean ratio of 3.43 branches/collateral in EAE animals vs. mean ratio of 1.56 branches/collateral in control animals, P < 0.001; Fig. 4 f). Again, lumbar reorganization was dependent on the number of damaged CST fibers with terminal branching increasing with the amount of CST damage (Fig. 4 e).
Cervical CST Collaterals Contact PSNs That Relay to the Lumbar Spinal Cord.
To investigate the functional role of cervical CST collaterals formed in response to thoracic EAE lesions, we reconstructed the projection pattern of these collaterals in animals with a targeted EAE lesion (n = 10) and control animals (n = 4; Fig. 5). The newly formed collaterals showed a very uniform projection pattern toward the intermediate layers of the spinal cord gray matter (Fig. 5, c and d), suggesting that they may contact a specific target neurons. The most attractive candidate targets in this location are long PSNs, which stand out for several reasons. First, whereas the cell bodies of PSNs are located in laminae V–VIII, their axons travel through a ventral pathway to the lumbar spinal cord (26, 27). This leaves them unaffected by a dorsolateral EAE lesion used in this work. Second, some PSN axons are known to directly contact lumbar motor neurons and, thus, offer an alternate detour circuit to these CST target neurons (26). Third, we have previously found that long PSNs are part of such a CST detour circuit in a dorsal hemisection model of spinal cord injury and that the number of contacts between PSNs and lumbar motor neurons increases in response to complete CST transection (18). To assess whether long PSNs are targeted by CST collaterals, we traced hindlimb CST fibers anterogradely and long PSNs retrogradely in animals with a targeted EAE lesion. At 28 d after lesion induction, newly formed CST collaterals form a dense network around the somata of PSNs, and individual CST branches form close appositions with PSNs (Fig. 5, e–n). This suggests that newly formed CST collaterals contact propriospinal interneurons as part of the remodeling process induced by an EAE lesion.
Figure 5.
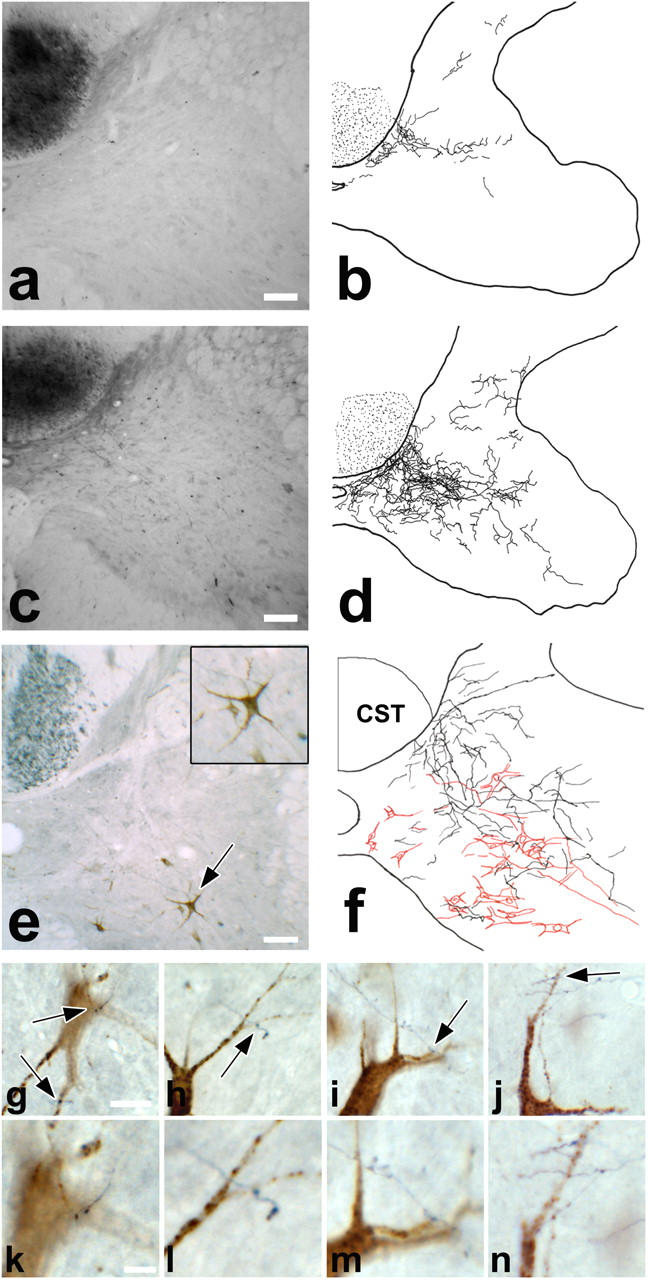
New cervical CST collaterals contact PSNs to form a detour circuit. (a) Cross section of the normal cervical spinal cord with a traced hindlimb CST, and (b) camera lucida reconstruction of CST collaterals on 10 consecutive cervical sections. Very few collaterals are seen in control animals. (c) Comparable cross section, and (d) camera lucida reconstruction at day 28 in an EAE animal. Numerous collaterals project to the intermediate laminae. (e) Cervical cross section of an EAE animal at day 28, with anterograde labeling of the hindlimb CST (BDA, black) and retrograde tracing of long PSNs (fluoroemerald, brown). Inset shows 175% magnification of PSN indicated by arrow. (f) Camera lucida drawings of CST collaterals (black) and PSNs (red) on five consecutive sections. CST collaterals form a dense network around long PSNs in the intermediate layers. (g–j) Examples of close appositions (arrows) between CST collaterals and PSNs in double-traced targeted EAE animals. (k–n) Higher magnifications of these contact sites. (a, c, and e) Bars, 100 μm. (g–j) Bars, 25 μm. (k–n) Bars, 10 μm.
Reorganization of CST Connections Contributes to Functional Recovery.
We further analyzed whether these newly formed collaterals can contribute to functional recovery by following the recovery of overall motor function and CST-specific motor responses using specific behavioral tests in animals with targeted EAE lesions (Fig. 6). To show directly that newly formed CST collaterals contribute to functional recovery, the CST was cut above the level where collaterals formed in targeted EAE animals; i.e., at the level of the pyramids in the medulla oblongata, where the CST can be selectively transected without destroying any other spinal motor tracts (“pyramidotomy”; reference 17). If done in animals that previously showed recovery, the drop in motor test performance after pyramidotomy establishes the contribution of the CST to recovery of function (Fig. 6 a). To differentiate recovery induced by the formation of new collaterals from functions dependent on unlesioned CST fibers, we performed the behavioral analysis in animals with both anatomically and functionally nearly complete CST lesions. (n = 8; mean CST damage 99.56%, a range of 98.56–99.97%; complete loss of CST-dependent responses at day 3 after lesion in seven out of eight animals).
Figure 6.

Rewiring of corticospinal connections contributes to functional recovery. (a) Scheme of the pyramidotomy (PTX) experiment. Transection of the CST above the cervical level removes new CST collaterals and permits assessment of their contribution to the recovery of CST function. (b and c) Functional recovery of targeted EAE animals with a complete inflammatory CST lesion from baseline (BL) to day 28 in behavioral test paradigms. CST-related responses such as hindlimb placing (b) and proprioceptive adduction (c) are nearly completely lost at day 3 but afterwards slowly recover up to day 28. (d and e) Comparison of behavioral tests before (corresponds to D28) and 2 d after PTX. Hindlimb placing (d) and proprioceptive adduction (e) lose a significant part of their previous recovery, suggesting the involvement of cervical reorganization in recovery.
At day 3, all targeted EAE animals showed high initial deficits in the overall motor performance (median EAE score: 2.5) as well as a complete loss of CST-related responses (Fig. 6, b and c). The CST-related responses as well as the overall motor function (at day 28: median EAE score: 1.5) slowly recovered over the course of the next 4 wk (Fig. 6, b and c). However, 2 d after pyramidotomy, the animals had lost a substantial part of the previously recovered CST function (Fig. 6, d and e). In contrast, the overall motor performance of these animals, which is insensitive to CST damage, did not change significantly in response to pyramidotomy (1.5 vs. 1.5 median EAE score before/after pyramidotomy [P > 0.05]). This strongly suggests that recovered CST function is lost as a specific consequence of CST transection by pyramidotomy and not as an unspecific consequence of the surgical procedure. As virtually no CST fibers crossed the EAE lesion in these animals, these results indicate that recovery of CST function in these experiments is at least partially due to reorganization of CST connections between the midthoracic EAE lesion and the pyramidotomy in the medulla oblongata.
Intraspinal Rewiring Is Accompanied by Reorganization of Cortical Motor Representation.
Finally, we investigated to what extent the changes of axonal connectivity in the spinal cord are complemented by changes in the cortical motor representation (Fig. 7). For this purpose, we retrogradely traced cortical neurons, which are directly or indirectly connected to motor pools of hindlimb muscles with a transsynaptic tracer, PRV. As we have established previously, PRV reaches the lumbar spinal cord in 2–3 d, the cervical spinal cord in 4–5 d, and the cortex in 6–7 d after PRV injection into hindlimb muscles (18). In normal animals, an average of 304 cortical projection neurons were labeled by PRV per brain, located almost exclusively in or directly adjacent to the hindlimb motor cortex (79% in hindlimb motor cortex; n = 6; Fig. 7, a and e). Control animals with a fresh mechanical transection of the CST showed virtually no labeled cortical projection neurons (average 31 PRV+ neurons per brain; n = 8; Fig. 7, d and e), indicating that the CST is the major descending connection between the lumbar spinal cord and the cortex that can be traced with PRV. However, targeted EAE animals traced 4 wk after lesion induction showed an average of 174 labeled cortical projection neurons per brain (n = 8; Fig. 7, b and e). This number is similar to the increased number of PRV+ neurons observed 3 wk after a traumatic spinal cord lesion (see online supplemental material). In EAE animals, these labeled neurons could have been traced both via remaining unlesioned CST fibers and via de novo formed cortico-spinal detour circuits. Two arguments support the conclusion that the labeled cortical projection neurons in EAE animals are at least partly traced through newly formed detour circuits. First, in targeted EAE animals, in which the remaining unlesioned CST fibers were mechanically transected just before PRV injection by a dorsal thoracic hemisection, an average of 105 PRV+ projection neurons were found per brain (n = 8; Fig. 7, c and e). This represents a significant increase both in the total number of PRV+ cortical neurons as well as in the number of PRV+ neurons in the original hindlimb motor area compared with freshly lesioned control animals (105 vs. 31; P < 0.01 for the total PRV+ neurons and 69 vs. 18; P < 0.01 for the PRV+ neurons in the hindlimb motor area; Fig. 7, e and g). Second, in contrast with unlesioned control animals, the PRV+ projection neurons in targeted EAE animals are only partially located in the hindlimb motor cortex (44% of PRV+ neurons in hindlimb motor cortex in targeted EAE animals vs. 79% in unlesioned control animals) and partially in other cortical areas, in particular the forelimb motor cortex (37% of PRV+ neurons in forelimb motor cortex in targeted EAE animals vs. 7% in unlesioned control animals; Fig. 7 f).
Figure 7.

Targeted EAE lesions induce a reorganization of the cortical motor representation. (a–d) Position of PRV+ neurons in hindlimb (blue) and forelimb (red) motor cortex. Neurons outside both areas are depicted in yellow. (a) In controls, numerous cortical projection neurons are almost exclusively found in the hindlimb motor area, which lose their connection to lumbar targets after fresh mechanical transection of the thoracic CST (d). In contrast, PRV+ neurons in EAE animals after remodeling (b) are located within and outside of the hindlimb motor area (particularly in the forelimb motor cortex). Additional mechanical transection of the thoracic CST does not disconnect all of these neurons (c), suggesting that many use a detour pathway to contact their lumbar targets. (e) Total numbers of PRV+ cortical neurons (CST-Tr.+/−, presence or absence of mechanical CST lesion; EAE+/−, presence or absence of an EAE lesion). (f) Percentage of PRV+ cortical neurons located in the forelimb motor area, which is significantly higher in EAE animals (CST Tr.−/EAE+) compared with control animals (CST Tr.−/EAE−). This indicates that in animals with a targeted EAE lesion, additional projection neurons have gained access to the lumbar spinal cord. (g) The number of PRV+ neurons in the hindlimb motor cortex, which retain access to the lumbar spinal cord after mechanical CST transection is significantly higher in EAE animals (CST Tr.+/EAE+) than in freshly transected control animals (CST Tr.+/EAE−). This suggests that some of the original hindlimb CST neurons have regained access to their targets.
Discussion
In this work, we investigate how axons remodel in diseases like MS. We show that a single inflammatory lesion in the spinal cord is sufficient to induce changes in axonal connectivity at multiple anatomical levels in the motor system (Fig. 8).
Figure 8.

A single MS-like lesion induces axonal remodeling on multiple anatomical levels. In the intact spinal cord, the hindlimb CST (green) forms a direct connection to lumbar motor circuits (1) and in some cases, contacts lumbar motor neurons (blue). A targeted neuroinflammatory lesion in the dorsal column (2, orange) interrupts among others the hindlimb CST, as well as local spinal circuits (red). Extensive remodeling leads to the restoration of damaged connections and contributes to functional recovery. Near the lesion local interneurons sprout (5, red), whereas the descending CST is remodeled in two ways. Below the lesion, spared hindlimb CST fibers increase their branching (6) to contact more target cells. (4) Above the lesion, damaged CST fibers extend new collaterals to contact preserved spinal interneurons, e.g., long PSNs (red) that relay to lumbar motor circuits. Remodeling of intraspinal connections is complemented by reorganization of the cortical motor representation (3).
This work also provides a unique opportunity to compare changes induced by neuroinflammation to those found after other forms of lesion (e.g., traumatic spinal cord injury). To investigate the effect of the neuroinflammatory environment on these remodeling processes we have studied axonal sprouting as well as the expression of two proteins, c-Jun and GAP43, that are paradigmatic for factors associated with axonal outgrowth (21–23, 28), in the vicinity of inflammatory EAE lesions as well as traumatic spinal cord lesions.
In the targeted EAE model, local interneurons sprout extensively close to the EAE lesion. Sprouting is paralleled by increased expression of c-Jun and GAP43 in these interneurons. This is in line with previous studies suggesting that successful neuronal outgrowth in the periphery and in the CNS requires activation of many genes, including c-Jun and GAP43 (21–23). Of these, c-Jun is expressed early and appears to indicate a cellular stress response. Current models suggest that c-Jun activation is double edged; it can lead to neuronal apoptosis, if transient and followed by the expression of genes such as JunD, or to axonal growth and regeneration, if sustained and accompanied by the expression of genes such as GAP43 (23). Our results suggest that a large number of spinal interneurons express c-Jun and, thus, mount an initial stress response. We found no evidence of neuronal apoptosis in EAE animals (unpublished data), but a subfraction of c-Jun+ interneurons also expressed GAP43 and started sprouting, primarily close to the lesion in the intermediate layers of the gray matter. This is in line with earlier studies that show that the ability to enter a regeneratory gene expression pattern is limited to subpopulations of neurons in the CNS (29). The extensive local growth response induced in the vicinity of an EAE lesion is in contrast with the comparably minor and short-lasting response induced by traumatic lesions (see online supplemental material). Several properties of the EAE lesion can contribute to this enhanced local growth response. First, it is well established that the infiltrating immune cells themselves can support neuronal outgrowth most likely due to the secretion of soluble growth promoting factors such as neurotrophins (30, 31). The finding that neuronal outgrowth is primarily induced in spinal neurons located in the gray matter immediately adjacent to the inflammatory infiltrate would support this assumption. Second, the extensive demyelination caused by the targeted EAE lesions is expected to lead to a reduced effect of myelin inhibition on neurite outgrowth in and around the lesion area (32, 33). Third, the temporal characteristics of the inflammatory damage are likely to trigger a more prolonged neuronal stress response, which in turn would favor the induction of a neuronal growth program. The distinct temporal profile of prolonged c-Jun and GAP43 expression, which is observed after inflammatory but not after traumatic damage, would support this notion.
On the level of the descending motor tract systems, some of the changes induced by an EAE lesion resemble those found after traumatic injury to these axons (i.e., the formation of new cervical CST collaterals (18). However, in contrast with commonly used spinal cord injury models, neuroinflammatory lesions lead only to a partial transection of the axonal tract system and, thus, allow for an additional level of reorganization based on the unlesioned fibers of the tract. We found that remodeling of CST after neuroinflammatory lesions follows two basic principles. First, below the lesion, untransected fibers spared by the EAE lesion increase the number of branches from their collaterals in the lumbar spinal cord. Second, above the lesion, in the cervical cord, CST fibers form new collaterals (that show a relatively normal degree of branching). The finding that the number of these collaterals increases with the extent of CST damage, and that new cervical collaterals are found in animals with complete CST transections, suggests that new collaterals are primarily formed by transected CST axons. These newly formed collaterals contact PSNs in the cervical cord. The formation of collaterals above the lesion provides new access to the cervical spinal cord, but this would be of no functional benefit unless a detour connection to the original target is established. The formation of such a detour connection in targeted EAE animals is suggested by the three following observations. First, cervical CST collaterals form new contacts with PSNs, which provide an intact connection to the lumbar spinal cord. Second, CST neurons can be transsynaptically traced from hindlimb muscles with PRV. Third, recovery is partially lost after pyramidotomy in animals with near complete thoracic CST lesion, showing that transected CST fiber input had previously reestablished a functional connection to the lumbar hindlimb motor pools.
The potential of these detour pathways is underlined by transsynaptic tracing experiments. We show that a substantial fraction of cortical neurons in hindlimb motor cortex have regained access to lumbar motor pools after only 4 wk. This timeframe coincides with the formation of CST collaterals and the recovery of CST function (18). Many neurons that can be traced from the hindlimb muscles are found in the original hindlimb motor cortex and must have been connected after the lesion. The mechanical transection of preserved CST fibers demonstrates that a substantial part of these hindlimb motor neurons were reconnected to their target area using remodeled intraspinal pathways. Furthermore, the distribution of PRV+ neurons in cortex after a targeted EAE lesion indicates that the additional recruitment of cortical neurons contributes to cortical reorganization. Cortical neurons located outside the primary hindlimb motor cortex (e.g., in the forelimb motor cortex) can be traced from the hindlimb after remodeling. It is plausible that anatomical changes in cortical projection observed by PRV tracing could account for some adaptive changes in cortical activation patterns as occur in response to various types of CNS lesions in animals and humans (34, 35). For example, it has been shown previously that hand movements activate a larger area in functional magnetic resonance images in MS patients, likely representing an attempt to functionally compensate the impaired hand function (11, 12). This provides a remarkable parallel to our work, where animals with impaired hindlimb function recruit new cortical projection neurons outside the hindlimb motor area, which in turn should result in a larger cortical area activated during hindlimb movements. Interestingly, the adaptive changes of the cortical motor representation in response to targeted EAE lesions are highly reminiscent of those induced by traumatic spinal cord lesions (18). This suggests that on the cortical level a default program of reorganization is implemented after injury, which depends on the extent of injury, but not on the type of injury. This is in contrast with the local reorganization on the spinal level, which is influenced not only by the extent of injury but also by its type.
From this work and other studies (18, 36), a common theme arises: structural reconnection and functional recovery can be achieved as long as sufficient “reserve capacity” for remodeling is present in the CNS, where true long-distance regeneration does not occur. Our work provides insight into the anatomical basis of this reserve capacity. Although spinal interneurons can sprout neurites, descending motor fibers can grow collaterals, and cortical projections can be reorganized, the success of all this critically depends on the presence of preserved detour pathways. Only as long as sufficient numbers of intact axonal pathways exists that allow damaged pathways to reestablish connections with reasonable probability and efficiency, recovery will be possible. The concept of a limited reserve capacity of axonal pathways in the CNS is particularly informative when explored in the context of MS (37). Early in the disease, deficits can often be readily compensated despite the fact that they are accompanied by substantial axonal damage. We speculate that during this stage the reserve capacity of the CNS is still largely intact. Thus, damage to axonal tract systems can be compensated for by both the remaining unlesioned tract fibers as well as by the formation of detour circuits. Although this leads to complete or partial recovery, it also increases the vulnerability of axonal systems as they depend on the patency of increasingly widespread detour pathways that are at high risk of being destroyed during subsequent inflammatory episodes. In parallel, each cycle of transection and detour formation decreases the remaining reserve of parallel pathways that can substitute for lost axonal connections. Therefore, compensation of clinical deficits is limited as this reserve becomes depleted. As the disease progresses, clinical deficits begin to remain, as new lesions become more and more likely to “hit” an axonal pathway that has exhausted its reserve, and at some point starts to progress steadily (as almost all axonal pathways have reached this point of no return). Once the reserve capacity is exhausted, new axonal damage is directly translated into clinical deficit giving rise to a progressive disease course.
This concept has far-reaching implications for the therapy of MS. First, our results clearly emphasize the importance of preventing axonal damage throughout the disease course of MS. This becomes even more pertinent in later stages of the disease: first, to preserve the reserve capacity for remodeling and, second, because the CNS will become more vulnerable to subsequent axonal damage after it underwent several cycles of damage and remodeling. As long as a sufficient number of connections to the target area exist, endogenous processes of axonal rewiring can be implemented and compensate arising functional deficits. Aggressive attempts to limit axonal damage even late during the relapsing-remitting stage, may prevent or at least delay the transition of the disease to the progressive stage. Second, therapeutic strategies that foster endogenous reorganization (e.g., by including unlesioned tract systems in the rewiring process) may provide additional benefits by enlarging the endogenous reserve capacity. This could be achieved in several complementary ways. Physical therapy can support remodeling processes, as has been demonstrated in numerous studies in humans and animals (34, 38). Growth-promoting therapies might help reestablish axonal connections, either by promoting sprouting or by disinhibiting endogenous growth restriction (33, 39, 40).
In summary, our work underscores the compensatory potential of the adult CNS, and raises the hope that the presence of powerful rewiring machinery in the CNS can ultimately be therapeutically harnessed to benefit patients with MS.
Acknowledgments
The authors wish to thank J. Scholl and O. Weinmann for excellent technical assistance, R. Schoeb for expert help with the figures, and Drs. R. Hohlfeld and L. Godinho for valuable suggestions and critical reading of the paper.
M. Kerschensteiner and T. Misgeld are supported by postdoctoral Emmy-Noether fellowships of the Deutsche Forschungsgemeinschaft (DFG grant nos. Ke774/2-1 and Mi694/1-1) and a grant from the Dana Foundation. This work was further supported by the medical faculty of the University of Goettingen (D. Merkler and C. Stadelmann, junior research group), the Swiss National Science Foundation (grant no. 31-63633 to M.E. Schwab), and the NCCR “Neural Plasticity and Repair” (to M.E. Schwab).
The authors have no conflicting financial interests.
Abbreviations used in this paper: BDA, biotinylated dextran amine; CNS, central nervous system; CST, corticospinal tract; EAE, experimental autoimmune encephalomyelitis; MS, multiple sclerosis; PRV, pseudo-rabies virus Bartha; PSN, propriospinal neuron.
References
- 1.Steinman, L. 2001. Multiple sclerosis: a two-stage disease. Nat. Immunol. 2:762–764. [DOI] [PubMed] [Google Scholar]
- 2.De Stefano, N., P.M. Matthews, L. Fu, S. Narayanan, J. Stanley, G.S. Francis, J.P. Antel, and D.L. Arnold. 1998. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain. 121:1469–1477. [DOI] [PubMed] [Google Scholar]
- 3.Bjartmar, C., G. Kidd, S. Mork, R. Rudick, and B.D. Trapp. 2000. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann. Neurol. 48:893–901. [PubMed] [Google Scholar]
- 4.Trapp, B.D., J. Peterson, R.M. Ransohoff, R. Rudick, S. Mork, and L. Bo. 1998. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 338:278–285. [DOI] [PubMed] [Google Scholar]
- 5.Kornek, B., M.K. Storch, R. Weissert, E. Wallstroem, A. Stefferl, T. Olsson, C. Linington, M. Schmidbauer, and H. Lassmann. 2000. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 157:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raine, C.S., and E. Wu. 1993. Multiple sclerosis: remyelination in acute lesions. J. Neuropathol. Exp. Neurol. 52:199–204. [PubMed] [Google Scholar]
- 7.Waxman, S.G. 1998. Demyelinating diseases–new pathological insights, new therapeutic targets. N. Engl. J. Med. 338:323–325. [DOI] [PubMed] [Google Scholar]
- 8.Doinikow, B. 1915. Ueber De- und Regenerationserscheinungen an Achsenzylindern bei der multiplen Sklerose. Z. ges Neurol. Psych. 27:151–178. [Google Scholar]
- 9.Raine, C.S., and A.H. Cross. 1989. Axonal dystrophy as a consequence of long-term demyelination. Lab. Invest. 60:714–725. [PubMed] [Google Scholar]
- 10.Dahl, D., G. Perides, and A. Bignami. 1989. Axonal regeneration in old multiple sclerosis plaques. Immunohistochemical study with monoclonal antibodies to phosphorylated and non-phosphorylated neurofilament proteins. Acta. Neuropathol. 79:154–159. [DOI] [PubMed] [Google Scholar]
- 11.Reddy, H., S. Narayanan, R. Arnoutelis, M. Jenkinson, J. Antel, P.M. Matthews, and D.L. Arnold. 2000. Evidence for adaptive functional changes in the cerebral cortex with axonal injury from multiple sclerosis. Brain. 123:2314–2320. [DOI] [PubMed] [Google Scholar]
- 12.Pantano, P., G.D. Iannetti, F. Caramia, C. Mainero, S. Di Legge, L. Bozzao, C. Pozzilli, and G.L. Lenzi. 2002. Cortical motor reorganization after a single clinical attack of multiple sclerosis. Brain. 125:1607–1615. [DOI] [PubMed] [Google Scholar]
- 13.Kerschensteiner, M., C. Stadelmann, B.S. Buddeberg, D. Merkler, F.M. Bareyre, D.C. Anthony, C. Linington, W. Bruck, and M.E. Schwab. 2004. Targeting EAE lesions to a predetermined axonal tract system allows for refined behavioral testing in an animal model of multiple sclerosis. Am. J. Pathol. 164:1455–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Luca, G.C., G.C. Ebers, and M.M. Esiri. 2004. Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain. 127:1009–1018. [DOI] [PubMed] [Google Scholar]
- 15.Adelmann, M., J. Wood, I. Benzel, P. Fiori, H. Lassmann, J.M. Matthieu, M.V. Gardinier, K. Dornmair, and C. Linington. 1995. The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J. Neuroimmunol. 63:17–27. [DOI] [PubMed] [Google Scholar]
- 16.Schnell, L., and M.E. Schwab. 1990. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 343:269–272. [DOI] [PubMed] [Google Scholar]
- 17.Thallmair, M., G.A. Metz, W.J. Z'Graggen, O. Raineteau, G.L. Kartje, and M.E. Schwab. 1998. Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nat. Neurosci. 1:124–131. [DOI] [PubMed] [Google Scholar]
- 18.Bareyre, F.M., M. Kerschensteiner, O. Raineteau, T.C. Mettenleiter, O. Weinmann, and M.E. Schwab. 2004. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat. Neurosci. 7:269–277. [DOI] [PubMed] [Google Scholar]
- 19.Neafsey, E.J., E.L. Bold, G. Haas, K.M. Hurley-Gius, G. Quirk, C.F. Sievert, and R.R. Terreberry. 1986. The organization of the rat motor cortex: a microstimulation mapping study. Brain Res. 396:77–96. [DOI] [PubMed] [Google Scholar]
- 20.Deryck, M., J. Vanreempts, H. Duytschaever, B. Vandeuren, and G. Clincke. 1992. Neocortical localization of tactile proprioceptive limb placing reactions in the rat. Brain Res. 573:44–60. [DOI] [PubMed] [Google Scholar]
- 21.Chaisuksunt, V., Y. Zhang, P.N. Anderson, G. Campbell, E. Vaudano, M. Schachner, and A.R. Lieberman. 2000. Axonal regeneration from CNS neurons in the cerebellum and brainstem of adult rats: correlation with the patterns of expression and distribution of messenger RNAs for L1, CHL1, c-jun and growth-associated protein-43. Neuroscience. 100:87–108. [DOI] [PubMed] [Google Scholar]
- 22.Raivich, G., M. Bohatschek, C. Da Costa, O. Iwata, M. Galiano, M. Hristova, A.S. Nateri, M. Makwana, L. Riera-Sans, D.P. Wolfer, et al. 2004. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 43:57–67. [DOI] [PubMed] [Google Scholar]
- 23.Herdegen, T., P. Skene, and M. Bahr. 1997. The c-Jun transcription factor—bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 20:227–231. [DOI] [PubMed] [Google Scholar]
- 24.Chen, P., D.E. Goldberg, B. Kolb, M. Lanser, and L.I. Benowitz. 2002. Inosine induces axonal rewiring and improves behavioral outcome after stroke. Proc. Natl. Acad. Sci. USA. 99:9031–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curtis, R., D. Green, R.M. Lindsay, and G.P. Wilkin. 1993. Up-regulation of GAP-43 and growth of axons in rat spinal cord after compression injury. J. Neurocytol. 22:51–64. [DOI] [PubMed] [Google Scholar]
- 26.Giovanelli, B.M., and H.G. Kuypers. 1969. Propriospinal fibers interconnecting the spinal enlargements in the cat. Brain Res. 14:321–330. [DOI] [PubMed] [Google Scholar]
- 27.Alstermark, B., A. Lundberg, M. Pinter, and S. Sasaki. 1987. Subpopulations and functions of long C3-C5 propriospinal neurones. Brain Res. 404:395–400. [DOI] [PubMed] [Google Scholar]
- 28.Bomze, H.M., K.R. Bulsara, B.J. Iskandar, P. Caroni, and J.H. Skene. 2001. Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat. Neurosci. 4:38–43. [DOI] [PubMed] [Google Scholar]
- 29.Fawcett, J.W. 1992. Intrinsic neuronal determinants of regeneration. Trends Neurosci. 15:5–8. [DOI] [PubMed] [Google Scholar]
- 30.Kerschensteiner, M., E. Gallmeier, L. Behrens, V. Vargas Leal, T. Misgeld, W.E. Klinkert, R. Kolbeck, E. Hoppe, R.L. Oropeza-Wekerle, I. Bartke, et al. 1999. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J. Exp. Med. 189:865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hauben, E., O. Butovsky, U. Nevo, E. Yoles, G. Moalem, E. Agranov, F. Mor, R. Leibowitz-Amit, E. Pevsner, S. Akselrod, et al. 2000. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J. Neurosci. 20:6421–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang, D.W., L. McKerracher, P.E. Braun, and S. David. 1999. A therapeutic vacine approacch to stimulate axon regeneration in the adult mammalien spinal cord. Neuron. 24:639–647. [DOI] [PubMed] [Google Scholar]
- 33.Schwab, M.E. 2004. Nogo and axon regeneration. Curr. Opin. Neurobiol. 14:118–124. [DOI] [PubMed] [Google Scholar]
- 34.Nudo, R.J., B.M. Wise, F. SiFuentes, and G.W. Milliken. 1996. Neural substrates for the effects of rehabilitative training on motor recovery after ischemic infarct. Science. 272:1791–1794. [DOI] [PubMed] [Google Scholar]
- 35.Payne, B.R., and S.G. Lomber. 2001. Reconstructing functional systems after lesions of cerebral cortex. Nat. Rev. Neurosci. 2:911–919. [DOI] [PubMed] [Google Scholar]
- 36.Blight, A.R. 2004. Just one word: plasticity. Nat. Neurosci. 7:206–208. [DOI] [PubMed] [Google Scholar]
- 37.Bjartmar, C., J.R. Wujek, and B.D. Trapp. 2003. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J. Neurol. Sci. 206:165–171. [DOI] [PubMed] [Google Scholar]
- 38.Taub, E., G. Uswatte, and T. Elbert. 2002. New treatments in neurorehabilitation founded on basic research. Nat. Rev. Neurosci. 3:228–236. [DOI] [PubMed] [Google Scholar]
- 39.Thoenen, H., and M. Sendtner. 2002. Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat. Neurosci. 5:1046–1050. [DOI] [PubMed] [Google Scholar]
- 40.Silver, J., and J.H. Miller. 2004. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 5:146–156. [DOI] [PubMed] [Google Scholar]