

Abstract
Prediabetes and diabetes in nonobese diabetic (NOD) mice have been targeted by a variety of immunotherapies, including the use of a soluble form of cytotoxic T lymphocyte antigen 4 (CTLA-4) and interferon (IFN)-γ. The cytokine, however, fails to activate tolerogenic properties in dendritic cells (DCs) from highly susceptible female mice early in prediabetes. The defect is characterized by impaired induction of immunosuppressive tryptophan catabolism, is related to transient blockade of the signal transducer and activator of transcription (STAT)1 pathway of intracellular signaling by IFN-γ, and is caused by peroxynitrite production. Here, we show that soluble CTLA-4 imparts suppressive properties to DCs from early prediabetic NOD female mice through mechanisms that rely on autocrine signaling by IFN-γ. Although phosphorylation of STAT1 in response to IFN-γ is compromised in those mice, CTLA-4 obviates the defect. IFN-γ–driven expression of tryptophan catabolism by CTLA-4–immunoglobulin is made possible through the concomitant activation of the Forkhead Box class O (FOXO) transcription factor FOXO3a, induction of the superoxide dismutase gene, and prevention of peroxynitrite formation.
Keywords: NOD mice, dendritic cells, CTLA-4, superoxide dismutase, signal transduction
Introduction
The nonobese diabetic (NOD) strain of mice has become a prototypic model of autoimmune disease (1). Most female mice die of type 1 diabetes, reflecting the onset of insulitis at ∼4 wk of age and the T cell–mediated destruction of pancreatic β cells. Several defects have been described in NOD mice, including impaired expression of cytotoxic T lymphocyte antigen 4 (CTLA-4; reference 2), aberrant APC function (3), and peroxynitrite formation (4). Prediabetes and diabetes have been treated with age-related outcomes by numerous, apparently disparate approaches, including administration of soluble CTLA-4 (CTLA-4–Ig; reference 5) and the use of IFN-γ (6, 7).
We have recently proposed a model of tolerance induction that might trace the activity of the synthetic immunomodulator CTLA-4–Ig (8), CTLA-4–expressing regulatory T cells (9), and recombinant IFN-γ (10) to a unitary mechanism. In this model, CTLA-4–Ig acts through B7-mediated signaling in tolerogenic DCs to promote release of IFN-γ, whose autocrine effects result in activation of the immunosuppressive pathway of tryptophan catabolism (11). However, IFN-γ is unable to induce tolerizing properties in DCs from NOD female mice at 4 wk of age, and this is apparently due to peroxynitrite nitration of STAT1 and blockade of IFN-γ signaling (12). The enzyme indoleamine 2,3-dioxygenase (IDO) is in fact responsible for tryptophan degradation by DCs (11), and IDO induction by IFN-γ requires STAT1 signaling (8).
Transcription factors of the Forkhead Box class O (FOXO) family are known to act as signal transducers at the confluence of multiple signaling pathways (13). For example, they represent important downstream targets of the phosphatidylinositol 3 kinase (PI3K) pathway, which has been shown to play a critical role in cell proliferation and survival (14). FOXO factors function under the control of insulin/insulin-like signaling (15). Through the involvement of FOXO family members, insulin regulates hepatic gluconeogenesis (16, 17) and reduces expression of pyruvate dehydrogenase kinase 4 activity, which is increased by glucocorticoids and diabetes (18). A member of the FOXO family protects quiescent cells from oxidative stress (19).
In this study, we provide evidence that CTLA-4–Ig can bypass the DC defect in tolerogenesis of prediabetic mice, enabling IFN-γ responsiveness and tryptophan catabolism to occur. The activity of CTLA-4–Ig relies on activation of members of the FOXO family of transcription factors, induction of superoxide dismutase (SOD), and blockade of peroxynitrite formation. Thus, composite mechanisms contribute to the therapeutic efficacy of CTLA-4–Ig in autoimmune diabetes in NOD mice. Although damage from oxidative stress can compromise early tolerogenesis in these animals, the immunosuppressive activity of DCs can be fully rescued by coordinate actions of CTLA-4–Ig on cytokine production and the oxidant/antioxidant balance.
Materials and Methods
Mice and Reagents.
Female and male NOD/LtJ mice, 4 and 8 wk of age, were purchased from The Jackson Laboratory. NOD female mice genetically deficient in IFN-γ (NOD.IFN-γnull; reference 20) or the β chain subunit of IFN-γ receptor (NOD.IFN-γRB null; reference 21) were as described previously. The enzyme inhibitors 1-methyl-dl-tryptophan (1-MT; Sigma-Aldrich), guanidinoethyl disulphide (GED; Sigma-Aldrich), 2-methoxyestradiol (2-MeOE2; Calbiochem), and LY294002 (Cell Signaling Technology) were commercially available products. The SOD mimetic agent Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP) was from Calbiochem. LY294002 is a specific inhibitor for PI3Ks, and at a concentration of 50 μM, it has no inhibitory effect on a range of protein kinases, including the c-AMP–dependent protein kinase and c-Src (22, 23). For 2-MeOE2, a SOD inhibitor, previous studies have demonstrated that the agent, when used at concentrations up to 100 μM, has limited or no effects on a variety of enzymatic functions, indicating that inhibition of SOD by 2-MeOE2 is probably specific (24). The pharmacological characterization and selectivity of GED as an inhibitor of nitric oxide (NO) synthase have been described previously (25). The issue of selectivity of 1-MT in inhibiting IDO has thoroughly been discussed elsewhere (26). All in vivo studies were performed in compliance with National and Perugia University Animal Care and Use Committee guidelines.
DC Preparations and Treatments and Immunization.
Splenic DCs were prepared from pools of at least five mice and fractionated according to CD11c/CD8α expression using positive selection columns in combination with CD11c and CD8α microbeads (10, 12, 27). For cytokine activation (27), DCs were subjected to overnight exposure to 100 ng/ml rIL-12 (CD8− DCs) or 200 U/ml IFN-γ (CD8+ DCs) in the presence or absence of 2 μM 1-MT, 250 μM GED, or 25 μM LY294002. Cells were instead exposed to 100 μM MnTBAP for 5 h before the addition of IFN-γ. CD8+ DC activation with CTLA-4–Ig involved exposure to 40 μg/ml of the synthetic immunomodulator with or without 2 μM 1-MT or 10 μg/ml XMG1.2-neutralizing anti–IFN-γ antibody (9). Cells were exposed to 2 μM 2-MeOE2 for 5 h before the addition of CTLA-4–Ig. 40 μg/ml of native IgG3 was always used as a control in all experiments involving CTLA-4–Ig (8). In all treatments involving CTLA-4–Ig, IFN-γ, MnTBAP, or enzyme inhibitors, either singly or in combination, cell viability remained >70% at the end of the incubation period. For immunization, cells were washed before peptide loading (5 μM for 2 h at 37°C), irradiation, and i.v. injection into recipient hosts. 3 × 105 IL-12–treated CD8− DCs were injected in combination with 9 × 103 CD8+ DCs either as such or treated with different agents as indicated above.
Skin Test Assay.
A skin test assay was used for measuring class I–restricted delayed-type hypersensitivity responses to the synthetic NRP-A7 peptide, as described previously (9, 10), using 4-wk-old NOD female mice as recipients. Results were expressed as the increase in footpad weight of peptide-injected footpads over that of vehicle-injected counterparts, which served as an internal control for each individual animal. Data are the mean ± SD for at least six mice per group. The statistical analysis was performed using Student's paired t test by comparing the mean weight of experimental footpads with that of control counterparts. The mean weight of control (vehicle-injected) footpads was in the 130–150 mg range, depending on individual experiments. In a given experiment, the different treatments of mice with DCs would not change per se the actual weights of control footpads.
Kynurenine Assay.
IDO functional activity was measured in vitro in terms of ability of DCs to metabolize tryptophan to kynurenine, whose concentrations were measured by HPLC as described previously (28).
Western Blot Analyses.
IDO expression was investigated as described previously (9) using a specific antibody in CD8+ DCs either untreated or exposed overnight to 200 U/ml IFN-γ, with or without MnTBAP, or to 40 μg/ml CTLA-4–Ig. On studying STAT1 phosphorylation, CD8+ DCs were exposed for 5 h to MnTBAP, and then treated with 200 U/ml IFN-γ for 10 min. After SDS-PAGE resolution, immunoblotting was performed by sequential exposure to anti-phosphoSTAT1 and anti-STAT1 antibodies (both from Cell Signaling Technology). For analysis of SOD expression, cells either untreated or treated overnight with CTLA-4–Ig were subjected to Western blot by means of an anti-MnSOD (SOD-111) or an anti-Cu/ZnSOD (SOD-101) rabbit polyclonal antibody specific for SOD2 and SOD1, respectively (Stressgen Biotechnology). Analysis of FOXO protein expressions in cytosolic (29) and nuclear (30) extracts of CD8+ DCs treated overnight with CTLA-4–Ig involved the use of rabbit polyclonal anti-FKHRL1 (FOXO3a) antibody (Upstate Biotechnology) according to manufacturer's instructions. For immunoprecipitation of NIH/3T3 cells, an anti-FKHRL1 antibody (Santa Cruz Biotechnology, Inc.) and a control rabbit polyclonal antibody were used. Anti-aldolase and anti-lamin A/C antibody reagents were from Santa Cruz Biotechnology, Inc. Anti–phospho-FKHRL1 (Thr32) was also from Upstate Biotechnology. Anti–human PTEN (clone 6H2.1), cross-reactive with its murine counterpart, was from Cascade Bioscience. Rabbit polyclonal anti-AKT and anti–phospho-AKT reagents were from Cell Signaling Technology.
Nitrotyrosine ELISA and Intracellular Superoxide Assessment.
The former assay was performed using a Nitrotyrosine ELISA kit (HyCult Biotechnology b.v.) according to the manufacturer's instructions (12). The latter is based on the chemical properties of hydroethidine, a weak blue fluorescent dye that is selectively converted by superoxide anion to ethidium with a bright red fluorescence (31). The control and drug-treated cells (1.5 × 106/sample) were incubated with 20 ng/ml hydroethidine for 30 min, washed, and analyzed by flow cytometry using the red laser channel.
Luciferase Assay.
6 × 106 DCs were electroporated (230 V, 75 ohms, 1,500 microfarads) with 40 μg FHRE-Luc plasmid. The construct contains three FKHRL1 elements from the human Fas ligand promoter (32) and was provided by M.E. Greenberg (Harvard Medical School, Boston, MA). 1 μg of another reporter plasmid, pRL-TK (Promega) encoding renilla luciferase, was coelectroporated as an internal control of the transfection process. Cells were seeded in 48-well plates at 106/ml. The next day, cells were stimulated for 6 or 24 h with CTLA-4–Ig or LY294002 before lysis. Luciferase assays were performed using a dual luciferase reporter assay kit (Promega). Relative light units (RLUs) from the firefly luciferase were normalized for transfection efficiency to the renilla luciferase RLU in each lysate. The renilla luciferase RLU values in 40-μl lysates were in the same range (0.224–0.256, typically in one experiment at 24 h) for control, CTLA-4–Ig, or LY294002 treatments of DCs either as such or subjected to gene silencing.
Small Interfering RNA (siRNA) Synthesis and Transfection.
The siRNA sequences specific for murine FOXO3 (sense, 5′-GCUCCUCACUGUAUUCAGtt-3′; antisense, 5′-CUGAAUACA-GUGAGGAGCCtg-3′) were selected, synthesized, and annealed by the manufacturer (Ambion). For transfection, 6.7 μg siRNA in 30 μl of transfection buffer (20 mM Hepes, 150 mM NaCl, pH 7.4) were pipetted into a sterile eppendorf tube. In a separate polystyrene tube, 6.7 μg DOTAP (1,2 dioleoyl-3-trimethylammonium-propane) was mixed with 30 μl of transfection buffer, and then both solutions were mixed gently by pipetting several times. After incubation at room temperature for 20 min, the mixture was added to 1 ml of complete medium containing 106 DCs and incubated for 20 h at 37°C. Cells were then recovered, washed, and immediately used for in vitro or in vivo experiments.
Results
A SOD Mimetic Restores IFN-γ Responsiveness in DCs from 4-wk-old NOD Female Mice.
The tolerogenic potential of IFN-γ is specifically lost in CD8+ DCs from early prediabetic NOD female mice, in which peroxynitrite-mediated nitration of STAT1 is observed (12). Peroxynitrite is a highly reactive oxidant resulting from the interaction of NO with superoxide. SOD can scavenge superoxide and thus limit peroxynitrite formation. MnTBAP is a synthetic mimetic of SOD that does not scavenge NO. Because of the ability of the inhibitor of NO synthase and direct scavenger of peroxynitrite, GED, to restore IFN-γ responsiveness in vitro in CD8+ DCs from 4-wk-old NOD female mice (12), we comparatively analyzed the effect of GED and MnTBAP on the in vivo ability of those cells to modulate CD8− DC priming to NRP-A7, a peptide mimotope for autoimmune diabetes in mice (33, 34). A combination of immunogenic CD8− and tolerogenic CD8+ DCs were injected into NOD mice that were assayed at 2 wk for skin test reactivity to the eliciting peptide. DCs from 4-wk-old NOD female mice were thus fractionated, treated with cytokines, loaded with the peptide, and injected into recipient hosts. IL-12 was used to enhance the priming ability of CD8− DCs (35) and IFN-γ was used to induce tolerizing properties in CD8+ DCs (10). Groups of CD8+ DCs were treated with IFN-γ in the presence of GED or MnTBAP, with or without 1-MT, which is a competitive inhibitor of IDO (Fig. 1). As expected, IFN-γ treatment alone of CD8+ DCs did not enable these cells to prevent host priming to NRP-A7. However, the presence of GED or MnTBAP during IFN-γ activation rescued the ability of CD8+ DCs to suppress immunity. The effect was dependent on tryptophan catabolism, as it was negated by the addition of 1-MT to the combination of IFN-γ and GED or MnTBAP. No effect was afforded by the use of GED or MnTBAP in the absence of IFN-γ, further indicating that the latter is required for the induction of tolerance in this experimental setting.
Figure 1.

Ability of the SOD mimetic MnTBAP to activate IDO-dependent tolerizing properties in CD8+ DCs from early prediabetic NOD female mice. Combinations of IL-12–treated CD8− and CD8+ DCs subjected to various treatments (indicated) were loaded with NRP-A7 and injected into recipient mice to be assayed at 2 wk for skin test reactivity to the eliciting peptide. CD8− DCs were used after overnight activation with 100 ng/ml IL-12, whereas CD8+ DCs were used either as such or after activation with 200 U/ml IFN-γ in the presence or absence of 250 μM GED or 100 μM MnTBAP. Groups of CD8+ DCs were also treated with 2 μM 1-MT. *, P < 0.001, experimental versus control footpads. One experiment is reported representative of three.
To confirm that the mechanism of action of MnTBAP involved the rescue of IFN-γ signaling through STAT1 and enhanced transcription of the IDO gene, we analyzed STAT1 phosphorylation and IDO protein expression by Western blot in CD8+ DCs (Fig. 2 A). At the same time, we measured IDO functional activity in terms of ability to metabolize tryptophan to kynurenine in vitro (Fig. 2 B). CD8+ DCs from 8-wk-old NOD female mice were used as a control in both assays. Similar to GED (12), MnTBAP restored, at least in part, STAT1 phosphorylation and IDO protein expression as well as kynurenine production in DCs from 4-wk-old NOD female mice treated with IFN-γ.
Figure 2.
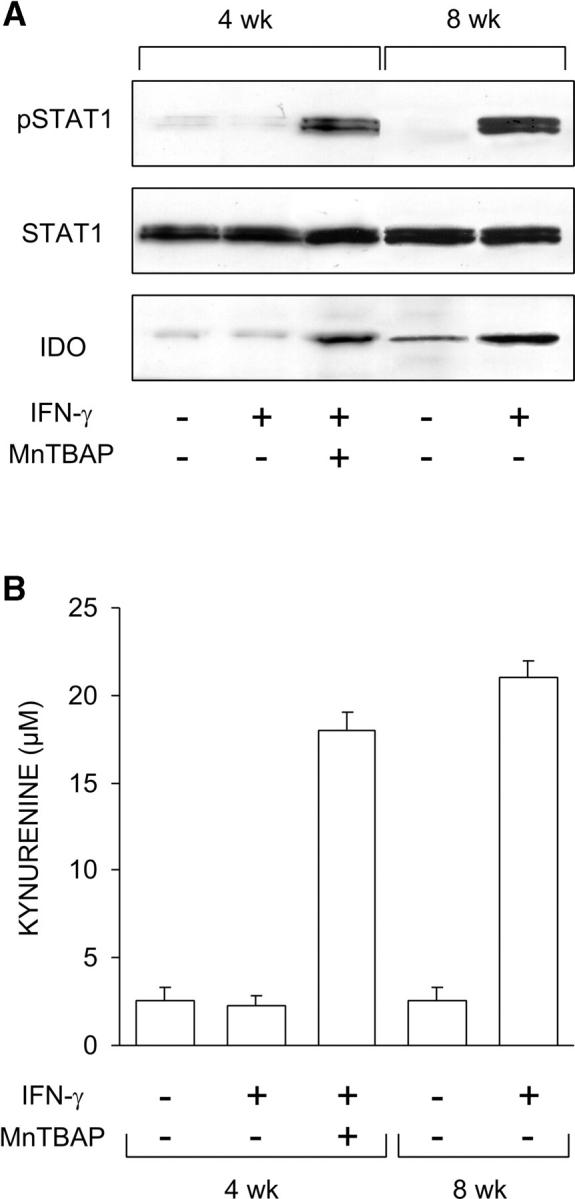
Ability of MnTBAP to restore IFN-γ responsiveness in CD8+ DCs from early prediabetic NOD female mice. (A) MnTBAP was examined for ability to restore STAT1 phosphorylation and IDO expression in response to IFN-γ. CD8+ DCs from 4-wk-old or control 8-wk-old NOD female mice were treated with IFN-γ as specified in Materials and Methods and assayed by Western blot. IDO expression was investigated with an IDO-specific polyclonal antibody, whereas STAT1 phosphorylation was analyzed with an anti-phosphoSTAT1 reagent. One experiment representative of three is shown. (B) MnTBAP was also studied for its ability to restore tryptophan conversion to kynurenine in response to IFN-γ, using CD8+ DCs from the same donors. Kynurenine levels in supernatants were measured by HPLC, and results are the mean ± SD of triplicate samples in one of three experiments.
CTLA-4–Ig Acts through Autocrine IFN-γ to Obviate the Tolerogenic Defect in 4-wk-old NOD Female Mice.
The ability of CTLA-4–Ig to prime DCs from conventional strains of mice for tolerogenic, IDO-dependent effects requires autocrine IFN-γ and STAT1-mediated signaling (8, 9, 36). Therefore, we investigated whether CTLA-4–Ig would be able to rescue the tolerizing ability of CD8+ DCs from 4-wk-old NOD female mice. We adopted an experimental model analogous to that shown in Fig. 1, in which a combination of CD8− and CD8+ DCs was injected into NOD mice to be assayed at 2 wk for skin test reactivity to NRP-A7. DCs from 4-wk-old NOD female mice were fractionated, treated with IL-12 (CD8−) or IFN-γ (CD8+), loaded with the peptide, and injected into recipient hosts. In an alternative to IFN-γ, CD8+ DCs were treated with CTLA-4–Ig. Groups of CD8+ DCs were coexposed to 1-MT or neutralizing antibody to IFN-γ during activation with CTLA-4–Ig. In addition, prediabetic NOD female mice genetically deficient in IFN-γ or IFN-γ receptor β chain expression were used as a source of CD8+ DCs to be used in the skin test as well as kynurenine production assays. As expected, IFN-γ treatment of CD8+ DCs from NOD mice did not enable these cells to prevent priming to NRP-A7 (Fig. 3 A). In contrast, CTLA-4–Ig was fully capable of blocking induction of immunity. However, the copresence of 1-MT or anti–IFN-γ during CD8+ cell exposure to CTLA-4–Ig ablated the ability of the latter to prevent priming. Similar to the effect of 1-MT or anti–IFN-γ, the genetic deficiency of IFN-γ or IFN-γ receptor expression prevented CTLA-4–Ig effects on CD8+ DCs. In addition, CTLA-4–Ig initiated IFN-γ–dependent tryptophan catabolism in vitro in CD8+ DCs from 4-wk-old NOD female mice (Fig. 3 B) and induced STAT1 phosphorylation in the same cells (Fig. 3 C). However, no STAT1 phosphorylation was observed in response to IFN-γ when mice genetically deficient in IFN-γ receptor expression were used as a source of DCs (Fig. 3 C). The levels of IFN-γ induced by CTLA-4–Ig in vitro in CD8+ DCs from NOD mice, whether or not deficient in IFN-γ receptor expression, were similar to those in conventional strains of mice (unpublished data). These data might imply that although CTLA-4–Ig induces IFN-γ and this, in turn, will cause peroxynitrite formation (12), the fusion protein also activates mechanisms capable of opposing peroxynitrite toxicity.
Figure 3.
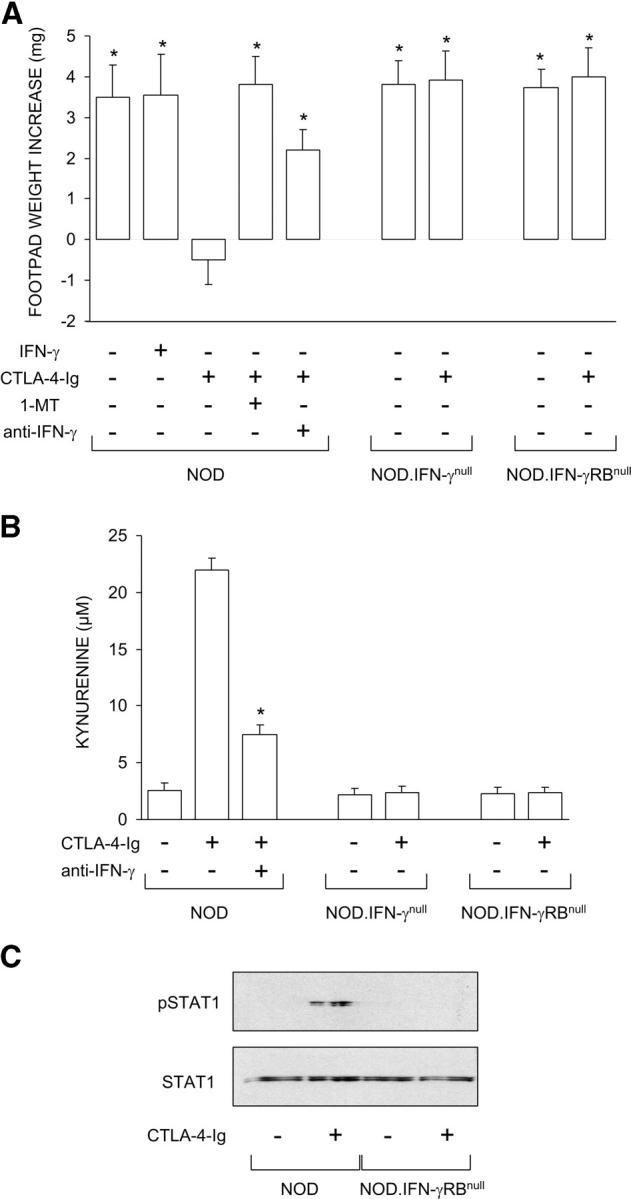
Ability of CTLA-4–Ig to rescue the tolerizing properties of CD8+ DCs from early prediabetic NOD female mice through mechanisms dependent on IFN-γ and IDO. (A) Mixtures of CD8− and CD8+ DCs were loaded with NRP-A7 and injected into recipient mice that were later assayed for skin test reactivity to the peptide. CD8− DCs were used after activation with IL-12, whereas CD8+ DCs were exposed overnight to IFN-γ or CTLA-4–Ig. Groups of CD8+ DCs were also treated with 1-MT or anti–IFN-γ (indicated). Mice deficient in IFN-γ or IFN-γ receptor β chain were also used as a source of CD8+ DCs. *, P < 0.001, experimental versus control footpads. One experiment is reported representative of three. (B) CTLA-4–Ig was examined for its ability to initiate tryptophan catabolism in CD8+ DCs from the same donors as in A. Cells were exposed overnight to CTLA-4–Ig in the presence or absence of anti–IFN-γ, and kynurenine levels were measured in supernatants. *, P < 0.001, presence versus absence of anti–IFN-γ during CTLA-4–Ig exposure. One experiment representative of three is shown. (C) CTLA-4–Ig fails to induce STAT1 phosphorylation in NOD mice deficient in IFN-γ receptor β chain. CD8+ DCs, treated overnight with CTLA-4–Ig, were assayed for STAT1 phosphorylation by Western blot analysis. One of two experiments with similar results is shown.
Both MnTBAP and CTLA-4–Ig Oppose Induction of Peroxynitrite by IFN-γ in Early Prediabetic NOD Female Mice.
Peroxynitrite is produced in acutely diabetic NOD mice (4) and may impair cell signaling via nitration of tyrosine residues (37). We have previously shown that IFN-γ will induce high levels of peroxynitrite in CD8+ DCs from NOD females selectively at ∼4 wk of age (12). To investigate the effects of MnTBAP and CTLA-4–Ig on peroxynitrite production, we analyzed IFN-γ–induced production of nitrotyrosine in CD8+ DCs from 4-wk-old NOD female mice. Cells were either treated with MnTBAP during IFN-γ exposure or pretreated overnight with CTLA-4–Ig before IFN-γ treatment (Fig. 4). In line with previous results (12), no nitrotyrosine formation was observed in cell supernatants of DCs unexposed to IFN-γ. The cytokine induced high levels of peroxynitrite, but this effect was greatly impaired by the use of MnTBAP or CTLA-4–Ig.
Figure 4.

Antagonistic effect of MnTBAP and CTLA-4–Ig on induction of peroxynitrite by IFN-γ in CD8+ DCs from early prediabetic NOD female mice. CD8+ DCs were recovered from these animals to be incubated overnight with IFN-γ. Groups of cells were cotreated with MnTBAP or pretreated with CTLA-4–Ig before exposure to IFN-γ. Nitrotyrosine in cell supernatants was assayed by sandwich ELISA, the lower detection limit of the assay being 2 nM. Data are means ± SD of replicate samples in one of three experiments. *, P < 0.001, combined treatment with MnTBAP or CTLA-4–Ig versus IFN-γ alone.
A Specific SOD Inhibitor Blocks CTLA-4–Ig Effects on CD8+ DCs.
2-MeOE2 is an estrogen metabolite that has been shown to inhibit SOD activity and is capable of in vivo effects that can be traced to this property (24, 31). We became interested in ascertaining the effect of 2-MeOE2 on the ability of CTLA-4–Ig to rescue IDO-dependent functions in CD8+ DCs from 4-wk-old NOD female mice. Cells were exposed sequentially to 2-MeOE2 and CTLA-4–Ig to be assayed for nitrotyrosine formation in supernatants, functional activity in a skin test assay with the NRP-A7 peptide, and tryptophan conversion to kynurenine (Fig. 5). In all three assays, the use of 2-MeOE2 was found to affect the activity of CTLA-4–Ig, resulting in enhanced production of nitrotyrosine in vitro, ablation of the tolerogenic potential of the CD8+ DC subset in vivo, and reduced tryptophan conversion to kynurenine.
Figure 5.

Antagonistic effect of 2-MeOE2 on CTLA-4–Ig activity. Early prediabetic NOD female mice were used as a source of CD8+ DCs that were exposed to 2-MeOE2 for 5 h before overnight incubation with CTLA-4–Ig. Nitrotyrosine in cell supernatants was assayed by ELISA or cells were tested in a kynurenine production assay of IDO functional activity, according to conditions specified above. Alternatively, the CD8+ DCs were loaded with NRP-A7, admixed with IL-12–treated and peptide-loaded CD8− DCs, and used in a skin test assay. *, P < 0.001, presence versus absence of 2MeOE2 during CTLA-4–Ig treatment; **, P < 0.001, experimental versus control footpads. One experiment representative of three is shown.
CTLA-4–Ig Induces SOD2 Expression and Activity in CD8+ DCs.
In general, cells scavenge superoxide with the help of an inducible MnSOD (SOD2) as well as a constitutive Cu/ZnSOD (SOD1). To gain insight into the mechanistic basis for the SOD-dependent effects of CTLA-4–Ig on CD8+ DCs from prediabetic mice, we analyzed IDO, SOD1, and SOD2 expression by Western blot using specific antibodies (Fig. 6 A). On comparing female and male mice of 4 or 8 wk of age, we found a selective defect in basal SOD2 protein expression in female mice at 4 wk. However, overnight exposure to CTLA-4–Ig increased protein expression to an extent comparable to the basal levels in age-matched male and 8-wk-old female mice, in which no major changes were induced by CTLA-4–Ig. At the same time, CTLA-4–Ig led to comparable IDO protein expression in the three groups of animals. In contrast, CTLA-4–Ig had no major effect on the expression of SOD1, which did not vary in NOD mice as a function of age or gender.
Figure 6.
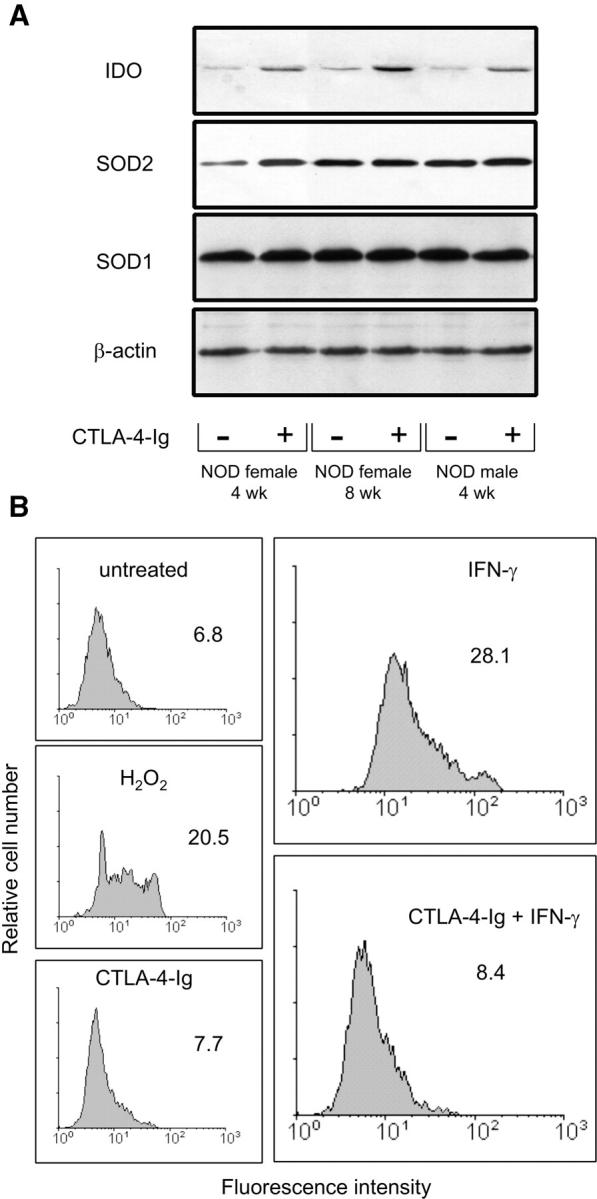
Ability of CTLA-4–Ig to restore SOD2 expression and function in CD8+ DCs from 4-wk-old NOD female mice. (A) CTLA-4–Ig induces SOD2 and IDO expression. Cells from different types of donor (indicated) were treated overnight with CTLA-4–Ig to be assayed by Western blot. IDO expression was investigated with an IDO-specific polyclonal antibody, whereas SOD2 and SOD1 were analyzed with specific antibody reagents. Loading controls consisted of samples reprobed with β actin–specific antibody. One experiment representative of three is shown. (B) Superoxide accumulation in CD8+ DCs treated with IFN-γ and reversal of the effect by CTLA-4–Ig. Cells were assayed for intracellular superoxide content by flow cytometry after treatment with IFN-γ or sequential exposure to CTLA-4–Ig and IFN-γ (indicated). Cells treated with CTLA-4–Ig alone were also assayed along with cells exposed to 200 μM of a nonspecific inducer of superoxide, H2O2. Control cells (untreated) were freshly harvested DCs. Mean channel fluorescence intensity values are also indicated. One of two experiments is shown.
SOD enzymes are critical in eliminating the superoxide radical. To confirm that CTLA-4–Ig induced increased SOD2 function, we used hydroethidine, a compound specifically converted by superoxide to highly fluorescent ethidium, allowing measurement of cellular superoxide contents by flow cytometry. Using CD8+ DCs from early prediabetic female mice, we found that cell treatment with IFN-γ greatly increased superoxide levels, an effect that was reversed by cell exposure to CTLA-4–Ig before IFN-γ treatment (Fig. 6 B).
CTLA-4–Ig Modulates FOXO Expression and Activates FOXO-dependent Transcription and Suppressive Activity in DCs.
Recent evidence indicates that FOXO-regulated genes include the SOD2 gene, thus revealing a role for FOXO transcription factors, in particular FOXO3a, in the control of oxidative stress (19). We examined whether CTLA-4–Ig treatment of CD8+ DCs from prediabetic NOD mice affects FOXO3a expression. Using Western blot analysis, we analyzed the pattern of expression of FOXO3a in the cytoplasm and the nucleus (Fig. 7 A). The experimental design included the use of controls for quality of fractionation as well as the analysis of an immunoprecipitate from a reference cell line as obtained with a FOXO3a-specific antibody. CTLA-4–Ig caused an increased level of FOXO3a in the cytoplasm at 2 h, followed by a substantial decline at 6–24 h. This was associated with the appearance at 6 h of FOXO3a in the nucleus, which was followed by an increase at 24 h.
Figure 7.
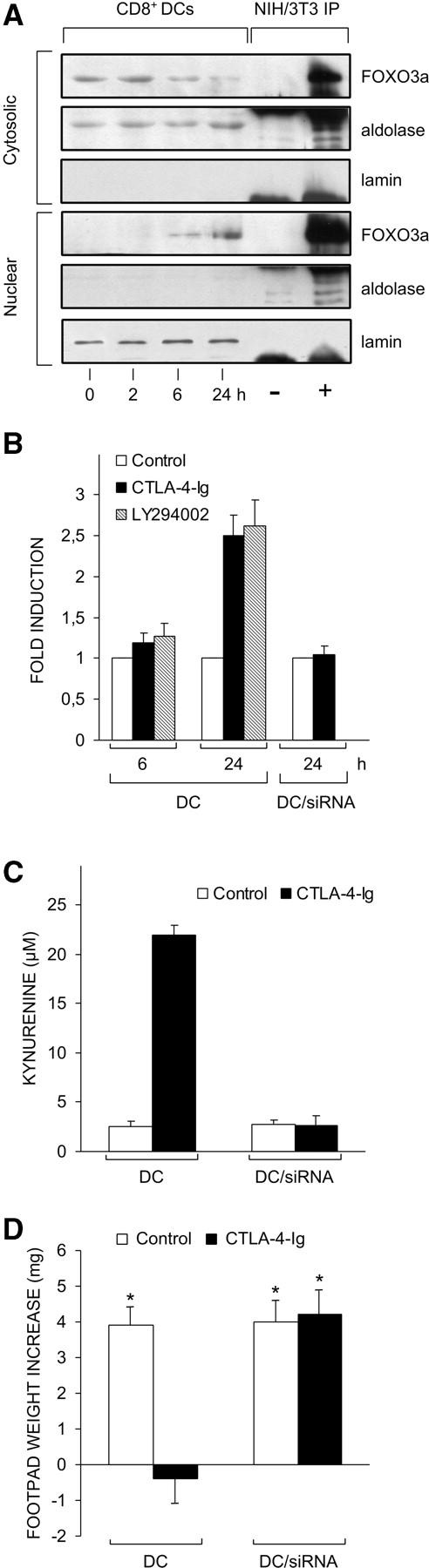
Ability of CTLA-4–Ig or a PI3K inhibitor to activate FOXO as revealed by different assays. (A) CTLA-4–Ig regulates FOXO expression. Cytoplasmic and nuclear extracts from CD8+ DCs of prediabetic mice were subjected to immunoblot analysis for FOXO3a protein expression. Cells were either untreated (time 0) or treated with CTLA-4–Ig for different times (indicated). The analysis was also conducted on NIH/3T3 cell immunoprecipitates (IP) obtained with control (−) or FOXO3a-specific antibody (+). Controls for fractionation included the use of antibodies specific for the cytosolic marker aldolase and the nuclear marker lamin. These antibodies nonspecifically reacted with the heavy chain of the Igs used for immunoprecipitation of NIH/3T3 cells. One experiment representative of three is shown. (B) CTLA-4–Ig and the PI3K inhibitor activate FOXO-dependent transcription in DCs. CD8+ DCs purified from prediabetic NOD female mice were transfected with pRL-TK in combination with FHRE-Luc. After 24 h, cells were incubated with CTLA-4–Ig or the PI3K inhibitor LY294002. Luciferase activity was monitored at 6 and 24 h. Results are expressed as fold induction (mean ± SD of three independent experiments) of the sample incubated with CTLA-4–Ig or LY294002 versus the corresponding untreated sample, the control value being 1. Also reported in B are values of luciferase activity in siRNA-treated DCs in which the FOXO3a gene had been silenced. In mock-transfected DCs (i.e., cells treated with DOTAP alone), the fold induction at 24 h of CTLA-4–Ig treatment was 2.4 ± 0.3. (C) siRNA-FOXO3a inhibits tryptophan catabolism in DCs treated with CTLA-4–Ig. CD8+ DCs from early prediabetic female mice, either mock-transfected (DC) or treated with siRNA-FOXO3a (DC/siRNA), were exposed overnight to CTLA-4–Ig, and kynurenine levels were measured in supernatants. One experiment representative of two is shown. (D) siRNA-FOXO3a inhibits the tolerogenic potential conferred by CTLA-4–Ig on DCs. Combinations of CD8+ DCs (either mock or siRNA-FOXO3a transfected) and IL-12–treated CD8− DCs were loaded with NRP-A7 and injected into recipient mice to be assayed at 2 wk for skin test reactivity. Either type of CD8+ DC was used after control or CTLA-4–Ig treatment. *, P < 0.001, experimental versus control footpads. One experiment is reported representative of two.
PI3K-regulated phosphorylation of FOXO transcription factors is pivotal in modulating their functional activity. In the absence of phosphorylation, these factors localize in the nucleus where they act as potent activators of transcription (13–15). To confirm that FOXO factors were among the transcriptional mediators of CTLA-4–Ig effects in CD8+ DCs from prediabetic NOD mice, we used promoter-driven expression of luciferase activity in DCs transfected with a reporter plasmid containing the luciferase gene in combination with a FOXO3a-regulated promoter. We electroporated CD8+ DCs from 4-wk-old NOD female mice with the FHRE-Luc reporter plasmid regulated by three FKHRL1 elements from the human Fas ligand promoter. Cells were treated with CTLA-4–Ig or the PI3K inhibitor, LY294002 (Fig. 7 B). The results showed that both CTLA-4–Ig and the PI3K inhibitor induced an ∼2.5-fold increase in the expression of luciferase activity controlled by the FOXO-regulated promoter at ∼24 h of treatment.
RNA interference is a mechanism of posttranscriptional gene silencing that can be used for immune modulation in DC gene expression (38). RNA interference has experimentally been used for silencing genes coding for transcription factors in human DCs (39). Therefore, we became interested in ascertaining the effects of CTLA-4–Ig in siRNA-FOXO3a–treated DCs. We used promoter-driven expression of luciferase activity under the conditions described above. Fig. 7 B shows that siRNA-FOXO3a completely abolished the expression of FOXO-regulated luciferase activity induced by CTLA-4–Ig at 24 h of treatment.
To directly investigate the effects of FOXO3a gene silencing in the modulation of DC function by CTLA-4–Ig, we measured IDO functional activity in vitro and tolerogenic potential in vivo in CD8+ DCs from 4-wk-old NOD female mice treated or not with siRNA-FOXO3a before CTLA-4–Ig activation. Fig. 7 C shows that FOXO3a gene silencing abolished the ability of CD8+ DCs to metabolize tryptophan to kynurenine in response to CTLA-4–Ig. At the same time, the siRNA-treated DCs could not be primed by CTLA-4–Ig for suppression of footpad reactivity to NRP when a combination of CD8− and CD8+ DCs was injected into NOD recipient mice according to the experimental design shown in Fig. 1 (Fig. 7 D).
Combined Effects of IFN-γ and LY294002 on Functional Activity of CD8+ DCs.
The ability of the PI3K inhibitor to activate FOXO-dependent gene transcription prompted us to evaluate the possibility that a mixture of IFN-γ and LY294002 could affect the functional activity of CD8+ DCs from prediabetic NOD females in a manner comparable to CTLA-4–Ig, which apparently combines the effects of both agents. CD8+ DCs from 4-wk-old animals were exposed overnight to LY294002 and IFN-γ to be assayed for interference with the induction of immunity by cotransferred CD8− DCs in a skin test assay with the NRP-A7 peptide. A group of CD8+ DCs was instead treated with CTLA-4–Ig (Fig. 8). Contrary to the effects of IFN-γ alone, cotreatment with the PI3K inhibitor fully rescued the tolerogenic potential of CD8+ DCs, thus mimicking the activity of CTLA-4–Ig.
Figure 8.

IFN-γ in combination with a PI3K inhibitor can induce tolerizing properties in CD8+ DCs from early prediabetic NOD female mice. Combinations of CD8+ and IL-12–treated CD8− DCs were loaded with NRP-A7 and injected into recipient mice to be assayed at 2 wk for skin test reactivity. CD8+ DCs were used after exposure to IFN-γ in the presence or absence of LY294002. In an alternative, CD8+ DCs were treated with CTLA-4–Ig. *, P < 0.001, experimental versus control footpads. One experiment is reported representative of three.
Effect of CTLA-4–Ig on Regulatory Mechanisms of FOXO3a Expression.
Although the molecular targets of PI3K appear to be multiple, many functional effects of PI3K activation are thought to be mediated by the serin/threonin kinase AKT/PKB. Critical in attenuation of PI3K signaling is dephosphorylation of the lipid products generated by PI3K by the phosphatase and tensin homologue, PTEN, a phosphoinositide phosphatase (13–15, 40). Therefore, we became interested in ascertaining whether CTLA-4–Ig effects on FOXO might involve interference with positive (e.g., PTEN-mediated) or negative (e.g., AKT-mediated) regulation of FOXO activity. We performed immunoblot analysis of PTEN expression in DCs from NOD female mice of 4 versus 8 wk of age, either as such or after stimulation with CTLA-4–Ig in vitro (Fig. 9 A). The results showed that early prediabetic female mice displayed impaired expression of PTEN, a defect that was nevertheless rectified by CTLA-4–Ig. This suggested that unrestrained PI3K signaling might occur in prediabetic mice. We examined AKT phosphorylation in those animals under different experimental conditions, namely after treatment with IFN-γ or combinations of IFN-γ with CTLA-4–Ig or LY294002. We also assessed FOXO3a phosphorylation in the same samples. For comparison, we analyzed phosphorylation of AKT and FOXO3a in 8-wk-old female mice. Fig. 9 B shows that marked phosphorylation of AKT and FOXO3a was observed in early prediabetic mice in response to IFN-γ. However, cotreatment with CTLA-4–Ig or LY294002 negated the effect of IFN-γ.
Figure 9.

Effect of CTLA-4–Ig alone or in combination with IFN-γ on the expression of PTEN, AKT/phospho-AKT, and FOXO3a/phospho-FOXO3a in early prediabetic NOD female mice. (A) Defective PTEN expression and reversal by CTLA-4–Ig in 4-wk-old mice. CD8+ DCs from female donors of different ages were treated overnight with CTLA-4–Ig before Western blot analysis. PTEN expression was investigated with a specific monoclonal antibody. Loading controls consisted of samples reprobed with β actin–specific antibody. One experiment representative of three is shown. (B) Enhanced phosphorylation of AKT and FOXO3a in response to a 30-min activation with IFN-γ in the same cells as in A and reversal by pretreatment with CTLA-4–Ig (18 h) or LY294002 (5 h). CD8+ DCs from 8-wk-old NOD females were used as control.
Discussion
In addition to DCs (41), CD4+ CD25+ regulatory T cells are essential in the protection from organ-specific autoimmune diseases. In the pancreas, they inhibit actions of autoreactive T cells and thereby prevent diabetes progression. Environmental stimuli can regulate the size of the intraislet CD4+ CD25+ T cell pool (42). Regulatory T cells express surface CTLA-4, and both cell-bound (9) and soluble CTLA-4 (8) can signal DCs through B7 molecules to initiate the immunosuppressive pathway of tryptophan catabolism. Therefore, it is of interest that reduced expression of CTLA-4 has been observed in NOD mice (2), that a defect in tryptophan catabolism impairs tolerance in those animals (12), and that CTLA-4–Ig (5) as well as regulatory T cells (43–45) will protect susceptible hosts from diabetes development. Recombinant IFN-γ is also protective, either when administered to diabetic mice (7) or when used to condition DCs in vitro before transfer into prediabetic recipients (6). However, cell signaling by IFN-γ is compromised in prediabetic NOD female mice, owing to peroxynitrite-induced nitration of STAT1 (12). Because the immunosuppressive effects of CTLA-4–Ig in conventional strains of mice require autocrine signaling by IFN-γ (8, 9, 36), these data raise the question of whether modulation of tryptophan catabolism contributes to the therapeutic efficacy of CTLA-4–Ig in prediabetes.
In this study, we found that CTLA-4–Ig did induce IFN-γ–dependent activation of IDO, thus implying effective autocrine signaling by the cytokine in DCs from early prediabetic NOD female mice, in which IFN-γ would be ineffective per se. Because the signaling defect of recombinant IFN-γ was rectified by coexposure to an inhibitor of peroxynitrite as well as a synthetic mimetic of SOD, MnTBAP, we investigated whether CTLA-4–Ig might affect the oxidant/antioxidant balance in target DCs. The redox status of NOD mice is abnormal (46). SOD levels in NOD female mice are extremely low, whereas in males, values are considerably higher than in females but still lower than in control mice (47). A metalloporphyrin-based SOD mimetic inhibits adoptive transfer of autoimmune diabetes by a diabetogenic T cell clone (48). We found that both MnTBAP and CTLA-4–Ig opposed the induction of peroxynitrite by IFN-γ in prediabetic mice. Moreover, a SOD inhibitor was capable of blocking CTLA-4–Ig activity in CD8+ DCs, leading to enhanced production of nitrotyrosine, reversal of the tolerogenic potential of those cells, and impaired tryptophan catabolism. Thus, the ability of CTLA-4–Ig to rescue CD8+ DC function in NOD mice was contingent on both the induction of autocrine IFN-γ and the oxygen-scavenging activity of SOD.
Transcriptional regulation of the murine inducible SOD2 gene is complex, and multiple potential regulatory elements are present for binding distinct classes of transcription factors (49). The FOXO family of transcription factors regulates expression of genes that have a prominent role in the control of cell cycle, death, and metabolism (13, 14). They represent important downstream targets of the PI3K pathway, such that phosphorylation prevents their access to the nucleus and inhibits expression of their function (15). Much evidence is accumulating to indicate that unrestrained PI3K signaling contributes to autoimmunity (40). Among the genes regulated by FOXO factors is the SOD2 gene (19). Therefore, we wanted to investigate the effect of CTLA-4–Ig or a PI3K inhibitor on SOD-mediated functions in CD8+ DCs from prediabetic mice. We found that both agents were capable of activating FOXO-dependent gene transcription in this DC subtype, and CTLA-4–Ig modulated the cytoplasmic versus nuclear expression of FOXO and induced SOD2 protein expression. At the same time, the PI3K inhibitor allowed externally added IFN-γ to prime CD8+ DCs for induction of tolerogenic effects. On the other hand, silencing of the FOXO3a gene blocked CTLA-4–Ig effects on CD8+ DCs from prediabetic mice, as demonstrated by assessment of tryptophan catabolism in vitro and tolerogenic function in vivo. Thus, FOXO modulation by either CTLA-4–Ig or a PI3K inhibitor appears to be a prerequisite for the occurrence of SOD-dependent effects that will allow endogenous or external IFN-γ to initiate tolerogenesis in CD8+ DCs from prediabetic mice.
The mechanisms whereby CTLA-4–Ig, different from IFN-γ, can activate FOXO are presently unclear. Recent evidence indicates that CTLA-4 determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3K function (50). Activation of PI3K by receptor stimulation initiates events leading to phosphorylation of AKT. Phosphorylated AKT migrates into the nucleus and phosphorylates FOXO transcription factors, resulting in nuclear exclusion and inhibition of transcription. The PI3K/AKT/FOXO signaling module appears to be remarkably conserved during evolution. Although the regulation and function of FOXO activity has been resolved in detail through genetic analysis in the nematode worm Caenorhabditis elegans, in mammalian cells, FOXO regulation and function have only recently started to be explored. An important concept that has emerged from several studies is that PTEN-mediated dephosphorylation of the lipid products generated by PI3K is crucial in modulating PI3K signaling (13–15, 40). On examining phosphorylation of AKT and FOXO3a in NOD mice, we observed marked phosphorylation of both in response to IFN-γ in early prediabetic mice, an effect that was ablated by cotreatment with CTLA-4–Ig. Interestingly, defective expression of PTEN was demonstrated in those mice, and again the defect was rectified by CTLA-4–Ig. Taken together, these data suggest that the effects of CTLA-4–Ig on FOXO3a expression might involve interference with physiological regulators of FOXO factors. A hypothetical model for the role of CTLA-4–Ig in correcting the defect in tolerogenesis of NOD mice is depicted in Fig. 10. Although the mechanisms of CTLA-4–Ig interference with PTEN expression are presently unknown, it is interesting to note that the genetic deficiency of either CTLA-4 (51) or PTEN (40) results in a fatal lymphoproliferative disease and/or development of autoimmunity.
Figure 10.

Hypothetical model for the role of CTLA-4–Ig in correcting the defective tolerogenesis of early prediabetic NOD female mice. CTLA-4–Ig may induce IFN-γ production in CD8+ DCs and, at the same time, enable cell responsiveness to the autocrine STAT1-dependent effects of the cytokine, leading to activation of the tolerogenic pathway of tryptophan catabolism. Although aberrant peroxynitrite formation would normally impede STAT1 phosphorylation and IFN-γ responsiveness in diabetes-prone animals (dotted line), CTLA-4–Ig may reduce peroxynitrite formation via enhanced SOD2 expression. This, in turn, could result from increased activity of the transcription factor FOXO3a, whose nuclear persistence is conditioned by positive and negative regulators such as PTEN and phospho-AKT, both of which appear to be affected by cell treatment with CTLA-4–Ig.
In general, activation of FOXOs causes growth suppression in a variety of cell types, whereas in cells of the immune system, most notably T cells, programmed cell death is often induced (13). Therefore, the observation of FOXO-dependent gene transcription by CTLA-4–Ig in DCs is of interest, further suggesting that FOXO activation results in cell type–specific gene regulation. In certain cells of the immune system or under conditions in which a rapid turnover of cells is required for homeostasis, the stress-induced activation of FOXO factors may result in cell death. In contrast, in cells that are not continuously produced, or when a critical function would be compromised by oxidative stress, FOXO activity will result in protection, for example by up-regulation of oxidant-scavenging proteins (19).
The antecedent of diabetes in NOD mice is insulitis, which begins in lymph nodes when β cell–specific T lymphocytes encounter APCs presenting β cell–derived peptides. The stimulus initiating insulitis appears to be a ripple of β cell death that occurs physiologically in juvenile mice of all strains. Although the mechanisms whereby this event results in induction of immunity rather than tolerance in diabetes-prone NOD mice is unclear, an important element could be represented by the recruitment and function of CD8− DCs (52). Our current data are consistent with the notion that changes in APCs underlie the initiation of autoantigen exposure, and may provide some insight into the nature of the imbalance between immunity and tolerance in the priming process. Aberrant IL-12 production (53), potentiating immunity by CD8− DCs, and oxidative stress (54), impairing tolerogenesis by CD8+ DCs, may combine with defective expression of regulatory T cell activity (2) to swing the balance in favor of the immunogenic presentation of autoantigens. Soluble CTLA-4 will oppose oxidative stress not only by virtue of SOD2 induction, but also by rescuing the activity of IDO, which uses superoxide anion as a substrate (11). Improved tolerogenesis resulting from the antioxidant and IFN-γ–inducing abilities of CTLA-4–Ig may thus skew DCs toward unresponsiveness to autoantigens.
In conclusion, the current data expand on our previous hypothesis that a defect in tryptophan catabolism impairs tolerance in NOD mice, provide new interpretive clues to mechanisms of the beneficial action of CTLA-4–Ig in prediabetic mice, and indicate that B7-mediated signaling in DCs may lead to a multiplicity of downstream effects. Furthermore, the new data reveal a role for FOXO factors in regulating the redox status of DCs, and emphasize the importance of oxidative stress in the pathogenesis of autoimmunity in NOD mice. It is conceivable that experimental studies targeting prediabetes by concordant actions on the redox status of APCs and T cell regulation of APC function may provide a framework for the design of innovative forms of immunomodulation in humans.
Acknowledgments
The authors have no conflicting financial interests.
U. Grohmann and P. Puccetti are senior authors on this paper.
Abbreviations used in this paper: 1-MT, 1-methyl-dl-tryptophan; 2-MeOE2, 2-methoxyestradiol; CTLA-4, cytotoxic T lymphocyte antigen 4; FOXO, Forkhead Box class O; GED, guanidinoethyl disulphide; IDO, indoleamine 2,3-dioxygenase; MnTBAP, Mn(III)tetrakis(4-benzoic acid)porphyrin chloride; NO, nitric oxide; NOD, nonobese diabetic; PI3K, phosphatidylinositol 3 kinase; RLU, relative light unit; siRNA, small interfering RNA; SOD, superoxide dismutase.
References
- 1.Atkinson, M.A., and E.H. Leiter. 1999. The NOD mouse model of type 1 diabetes: as good as it gets? Nat. Med. 5:601–604. [DOI] [PubMed] [Google Scholar]
- 2.Colucci, F., M.L. Bergman, C. Penha-Goncalves, C.M. Cilio, and D. Holmberg. 1997. Apoptosis resistance of nonobese diabetic peripheral lymphocytes linked to the Idd5 diabetes susceptibility region. Proc. Natl. Acad. Sci. USA. 94:8670–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serreze, D.V., H.R. Gaskins, and E.H. Leiter. 1993. Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J. Immunol. 150:2534–2543. [PubMed] [Google Scholar]
- 4.Suarez-Pinzon, W.L., J.G. Mabley, K. Strynadka, R.F. Power, C. Szabo, and A. Rabinovitch. 2001. An inhibitor of inducible nitric oxide synthase and scavenger of peroxynitrite prevents diabetes development in NOD mice. J. Autoimmun. 16:449–455. [DOI] [PubMed] [Google Scholar]
- 5.Lenschow, D.J., S.C. Ho, H. Sattar, L. Rhee, G. Gray, N. Nabavi, K.C. Herold, and J.A. Bluestone. 1995. Differential effects of anti–B7-1 and anti–B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J. Exp. Med. 181:1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shinomiya, M., S.M. Fazle Akbar, H. Shinomiya, and M. Onji. 1999. Transfer of dendritic cells (DC) ex vivo stimulated with interferon-gamma (IFN-γ) down-modulates autoimmune diabetes in non-obese diabetic (NOD) mice. Clin. Exp. Immunol. 117:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobel, D.O., J. Han, J. Williams, J.W. Yoon, H.S. Jun, and B. Ahvazi. 2002. Gamma interferon paradoxically inhibits the development of diabetes in the NOD mouse. J. Autoimmun. 19:129–137. [DOI] [PubMed] [Google Scholar]
- 8.Grohmann, U., C. Orabona, F. Fallarino, C. Vacca, F. Calcinaro, A. Falorni, P. Candeloro, M.L. Belladonna, R. Bianchi, M.C. Fioretti, and P. Puccetti. 2002. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3:1097–1101. [DOI] [PubMed] [Google Scholar]
- 9.Fallarino, F., U. Grohmann, K.W. Hwang, C. Orabona, C. Vacca, R. Bianchi, M.L. Belladonna, M.C. Fioretti, M.L. Alegre, and P. Puccetti. 2003. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 4:1206–1212. [DOI] [PubMed] [Google Scholar]
- 10.Grohmann, U., R. Bianchi, M.L. Belladonna, S. Silla, F. Fallarino, M.C. Fioretti, and P. Puccetti. 2000. IFN-γ inhibits presentation of a tumor/self peptide by CD8α− dendritic cells via potentiation of the CD8α+ subset. J. Immunol. 165:1357–1363. [DOI] [PubMed] [Google Scholar]
- 11.Grohmann, U., F. Fallarino, and P. Puccetti. 2003. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol. 24:242–248. [DOI] [PubMed] [Google Scholar]
- 12.Grohmann, U., F. Fallarino, R. Bianchi, C. Orabona, C. Vacca, M.C. Fioretti, and P. Puccetti. 2003. A defect in tryptophan catabolism impairs tolerance in nonobese diabetic mice. J. Exp. Med. 198:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birkenkamp, K.U., and P.J. Coffer. 2003. FOXO transcription factors as regulators of immune homeostasis: molecules to die for? J. Immunol. 171:1623–1629. [DOI] [PubMed] [Google Scholar]
- 14.Burgering, B.M., and G.J. Kops. 2002. Cell cycle and death control: long live Forkheads. Trends Biochem. Sci. 27:352–360. [DOI] [PubMed] [Google Scholar]
- 15.Van Der Heide, L.P., M.F. Hoekman, and M.P. Smidt. 2004. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 380:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puigserver, P., J. Rhee, J. Donovan, C.J. Walkey, J.C. Yoon, F. Oriente, Y. Kitamura, J. Altomonte, H. Dong, D. Accili, and B.M. Spiegelman. 2003. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature. 423:550–555. [DOI] [PubMed] [Google Scholar]
- 17.Altomonte, J., A. Richter, S. Harbaran, J. Suriawinata, J. Nakae, S.N. Thung, M. Meseck, D. Accili, and H. Dong. 2003. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am. J. Physiol. Endocrinol. Metab. 285:E718–E728. [DOI] [PubMed] [Google Scholar]
- 18.Kwon, H.S., B. Huang, T.G. Unterman, and R.A. Harris. 2004. Protein kinase B-α inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes. 53:899–910. [DOI] [PubMed] [Google Scholar]
- 19.Kops, G.J., T.B. Dansen, P.E. Polderman, I. Saarloos, K.W. Wirtz, P.J. Coffer, T.T. Huang, J.L. Bos, R.H. Medema, and B.M. Burgering. 2002. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 419:316–321. [DOI] [PubMed] [Google Scholar]
- 20.Serreze, D.V., H.D. Chapman, C.M. Post, E.A. Johnson, W.L. Suarez-Pinzon, and A. Rabinovitch. 2001. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J. Immunol. 166:1352–1359. [DOI] [PubMed] [Google Scholar]
- 21.Serreze, D.V., C.M. Post, H.D. Chapman, E.A. Johnson, B. Lu, and P.B. Rothman. 2000. Interferon-γ receptor signaling is dispensable in the development of autoimmune type 1 diabetes in NOD mice. Diabetes. 49:2007–2011. [DOI] [PubMed] [Google Scholar]
- 22.Walker, E.H., M.E. Pacold, O. Perisic, L. Stephens, P.T. Hawkins, M.P. Wymann, and R.L. Williams. 2000. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell. 6:909–919. [DOI] [PubMed] [Google Scholar]
- 23.Vlahos, C.J., W.F. Matter, K.Y. Hui, and R.F. Brown. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269:5241–5248. [PubMed] [Google Scholar]
- 24.Golab, J., D. Nowis, M. Skrzycki, H. Czeczot, A. Baranczyk-Kuzma, G.M. Wilczynski, M. Makowski, P. Mroz, K. Kozar, R. Kaminski, et al. 2003. Antitumor effects of photodynamic therapy are potentiated by 2-methoxyestradiol. A superoxide dismutase inhibitor. J. Biol. Chem. 278:407–414. [DOI] [PubMed] [Google Scholar]
- 25.Szabo, C., R. Bryk, B. Zingarelli, G.J. Southan, T.C. Gahman, V. Bhat, A.L. Salzman, and D.J. Wolff. 1996. Pharmacological characterization of guanidinoethyldisulphide (GED), a novel inhibitor of nitric oxide synthase with selectivity towards the inducible isoform. Br. J. Pharmacol. 118:1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munn, D.H., M. Zhou, J.T. Attwood, I. Bondarev, S.J. Conway, B. Marshall, C. Brown, and A.L. Mellor. 1998. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 281:1191–1193. [DOI] [PubMed] [Google Scholar]
- 27.Grohmann, U., F. Fallarino, R. Bianchi, M.L. Belladonna, C. Vacca, C. Orabona, C. Uyttenhove, M.C. Fioretti, and P. Puccetti. 2001. IL-6 inhibits the tolerogenic function of CD8α+ dendritic cells expressing indoleamine 2,3-dioxygenase. J. Immunol. 167:708–714. [DOI] [PubMed] [Google Scholar]
- 28.Fallarino, F., C. Vacca, C. Orabona, M.L. Belladonna, R. Bianchi, B. Marshall, D.B. Keskin, A.L. Mellor, M.C. Fioretti, U. Grohmann, and P. Puccetti. 2002. Functional expression of indoleamine 2,3-dioxygenase by murine CD8α+ dendritic cells. Int. Immunol. 14:65–68. [DOI] [PubMed] [Google Scholar]
- 29.Fallarino, F., U. Grohmann, C. Vacca, R. Bianchi, C. Orabona, A. Spreca, M.C. Fioretti, and P. Puccetti. 2002. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 9:1069–1077. [DOI] [PubMed] [Google Scholar]
- 30.Grohmann, U., M.L. Belladonna, R. Bianchi, C. Orabona, E. Ayroldi, M.C. Fioretti, and P. Puccetti. 1998. IL-12 acts directly on DC to promote nuclear localization of NF-κB and primes DC for IL-12 production. Immunity. 9:315–323. [DOI] [PubMed] [Google Scholar]
- 31.Huang, P., L. Feng, E.A. Oldham, M.J. Keating, and W. Plunkett. 2000. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 407:390–395. [DOI] [PubMed] [Google Scholar]
- 32.Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- 33.Anderson, B., B.J. Park, J. Verdaguer, A. Amrani, and P. Santamaria. 1999. Prevalent CD8+ T cell response against one peptide/MHC complex in autoimmune diabetes. Proc. Natl. Acad. Sci. USA. 96:9311–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amrani, A., J. Verdaguer, P. Serra, S. Tafuro, R. Tan, and P. Santamaria. 2000. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature. 406:739–742. [DOI] [PubMed] [Google Scholar]
- 35.Grohmann, U., R. Bianchi, M.L. Belladonna, C. Vacca, S. Silla, E. Ayroldi, M.C. Fioretti, and P. Puccetti. 1999. IL-12 acts selectively on CD8α− dendritic cells to enhance presentation of a tumor peptide in vivo. J. Immunol. 163:3100–3105. [PubMed] [Google Scholar]
- 36.Grohmann, U., R. Bianchi, C. Orabona, F. Fallarino, C. Vacca, A. Micheletti, M.C. Fioretti, and P. Puccetti. 2003. Functional plasticity of dendritic cell subsets as mediated by CD40 versus B7 activation. J. Immunol. 171:2581–2587. [DOI] [PubMed] [Google Scholar]
- 37.Matata, B.M., and M. Galinanes. 2002. Peroxynitrite is an essential component of cytokines production mechanism in human monocytes through modulation of nuclear factor-κB DNA binding activity. J. Biol. Chem. 277:2330–2335. [DOI] [PubMed] [Google Scholar]
- 38.Hill, J.A., T.E. Ichim, K.P. Kusznieruk, M. Li, X. Huang, X. Yan, R. Zhong, E. Cairns, D.A. Bell, and W.P. Min. 2003. Immune modulation by silencing IL-12 production in dendritic cells using small interfering RNA. J. Immunol. 171:691–696. [DOI] [PubMed] [Google Scholar]
- 39.Laderach, D., D. Compagno, O. Danos, W. Vainchenker, and A. Galy. 2003. RNA interference shows critical requirement for NF-κB p50 in the production of IL-12 by human dendritic cells. J. Immunol. 171:1750–1757. [DOI] [PubMed] [Google Scholar]
- 40.Okkenhaug, K., A. Bilancio, J.L. Emery, and B. Vanhaesebroeck. 2004. Phosphoinositide 3-kinase in T cell activation and survival. Biochem. Soc. Trans. 32:332–335. [DOI] [PubMed] [Google Scholar]
- 41.Steinman, R.M., and M.C. Nussenzweig. 2002. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA. 99:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng, Y., Y. Laouar, M.O. Li, E.A. Green, and R.A. Flavell. 2004. TGF-β regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc. Natl. Acad. Sci. USA. 101:4572–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang, Q., K.J. Henriksen, M. Bi, E.B. Finger, G. Szot, J. Ye, E.L. Masteller, H. McDevitt, M. Bonyhadi, and J.A. Bluestone. 2004. In vitro–expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 199:1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tarbell, K.V., S. Yamazaki, K. Olson, P. Toy, and R.M. Steinman. 2004. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J. Exp. Med. 199:1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herman, A.E., G.J. Freeman, D. Mathis, and C. Benoist. 2004. CD4+ CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J. Exp. Med. 199:1479–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murata, Y., M. Amao, and J. Hamuro. 2003. Sequential conversion of the redox status of macrophages dictates the pathological progression of autoimmune diabetes. Eur. J. Immunol. 33:1001–1011. [DOI] [PubMed] [Google Scholar]
- 47.Papaccio, G., S. Frascatore, F.A. Pisanti, M.V. Latronico, and T. Linn. 1995. Superoxide dismutase in the nonobese diabetic (NOD) mouse: a dynamic time-course study. Life Sci. 56:2223–2228. [DOI] [PubMed] [Google Scholar]
- 48.Piganelli, J.D., S.C. Flores, C. Cruz, J. Koepp, I. Batinic-Haberle, J. Crapo, B. Day, R. Kachadourian, R. Young, B. Bradley, and K. Haskins. 2002. A metalloporphyrin-based superoxide dismutase mimic inhibits adoptive transfer of autoimmune diabetes by a diabetogenic T-cell clone. Diabetes. 51:347–355. [DOI] [PubMed] [Google Scholar]
- 49.Jones, P.L., G. Kucera, H. Gordon, and J.M. Boss. 1995. Cloning and characterization of the murine manganous superoxide dismutase-encoding gene. Gene. 153:155–161. [DOI] [PubMed] [Google Scholar]
- 50.Pandiyan, P., D. Gartner, O. Soezeri, A. Radbruch, K. Schulze-Osthoff, and M.C. Brunner-Weinzierl. 2004. CD152 (CTLA-4) determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3 kinase function. J. Exp. Med. 199:831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salomon, B., and J.A. Bluestone. 2001. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu. Rev. Immunol. 19:225–252. [DOI] [PubMed] [Google Scholar]
- 52.Turley, S., L. Poirot, M. Hattori, C. Benoist, and D. Mathis. 2003. Physiological β cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J. Exp. Med. 198:1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu, J., and D. Beller. 2002. Aberrant production of IL-12 by macrophages from several autoimmune-prone mouse strains is characterized by intrinsic and unique patterns of NF-κB expression and binding to the IL-12 p40 promoter. J. Immunol. 169:581–586. [DOI] [PubMed] [Google Scholar]
- 54.Gurlo, T., K. Kawamura, and H. von Grafenstein. 1999. Role of inflammatory infiltrate in activation and effector function of cloned islet reactive nonobese diabetic CD8+ T cells: involvement of a nitric oxide-dependent pathway. J. Immunol. 163:5770–5780. [PubMed] [Google Scholar]