

Abstract
T cell–APC conjugation as mediated by leukocyte function-associated antigen-1 (LFA-1)–intercellular adhesion molecule (ICAM)-1 binding is followed by formation of the supramolecular activation cluster (SMAC) at the immunological synapse. The intracellular processes that regulate SMAC formation and its influence on T cell function are important questions to be addressed. Here, using a mutational approach, we demonstrate that binding of adaptor adhesion and degranulation promoting adaptor protein (ADAP) to SLP-76 differentially regulates peripheral SMAC (pSMAC) formation relative to conjugation. Although mutation of the YDDV sites (termed M12) disrupted SLP-76 SH2 domain binding and prevented the ability of ADAP to increase conjugation and LFA-1 clustering, M12 acted selectively as a dominant negative (DN) inhibitor of pSMAC formation, an effect that was paralleled by a DN effect on interleukin-2 production. ADAP also colocalized with LFA-1 at the immunological synapse. Our findings identify ADAP–SLP-76 binding as a signaling event that differentially regulates SMAC formation, and support a role for SMAC formation in T cell cytokine production.
Keywords: integrins, adaptor proteins, immunological synapse
Introduction
The interaction of T cells with MHC antigen–bearing APCs initiates signaling events that are needed for the proliferation and effector function of T cells. The process of conjugation is mediated in part by leukocyte function-associated antigen-1 (LFA-1 or CD11a/CD18) binding to the intercellular adhesion molecules (ICAMs) 1 and 2 on APCs. Conjugation is accompanied by a rearrangement of receptors that form a supramolecular activation cluster (SMAC) at the immunological synapse (1–4). The SMAC is comprised of a central SMAC enriched with TCR-CD3, CD4, CD28, and other receptors that are surrounded by a peripheral SMAC (pSMAC) enriched with LFA-1 and talin (1, 2). Despite the marked nature of these events, the identity of the intracellular proteins that regulate SMAC formation has been unclear, as has the role of the SMAC in influencing T cell activation. This uncertainty may be related to the opposing effects of receptor clustering and degradation in the SMAC (5, 6). Tyrosine phosphorylation and phosphatidylinositol 3-kinase (PI-3K) signaling can occur outside the SMAC (4, 7, 8), and the CD2-associated protein–deficient T cells show the combined features of unstable SMAC formation and hyperproliferation (9). Alternately, the mere clustering of receptors and kinases in the central SMAC would be predicted to facilitate serial ligation of receptors, repeated kinase activation, and sustained signaling in T cells. SMAC formation is also needed for the directed release in viral transmission (10), but is not required for the killing by cytolytic T cells (11).
The outer ring of the SMAC (i.e., pSMAC) is comprised of LFA-1 linked to intracellular proteins talin and RapL (regulator for cell adhesion and polarization enriched in lymphoid tissues; references 12, 13). The conversion of LFA-1 from low to higher affinity forms is a complex process that involves conformation changes combined with an increase in avidity due to clustering (13–15). TCR-CD3 ligation and chemokines promote this conversion by means of inside-out signaling. The process is influenced by various intracellular signaling proteins such as p56lck, Ras family member Rap-1, and the exchange complex Cbl–CrkL–C3G (16, 17). Rap1 binds to its effector molecule RapL (regulator of adhesion and polarization enriched in lymphoid tissues), whereas dominant negative (DN) forms of RapL can inhibit LFA-1 clustering, ICAM-1 binding, and T cell–APC conjugation (18). In addition, the head region of talin can affect conformational changes in LFA-1 (19), although it may negatively regulate LFA-1 in neutrophils (20). RhoA has also been linked to the chemokine induction of high affinity LFA-1 on lymphocytes (21, 22).
Aside from catalytic proteins, adaptor proteins or molecular scaffolds play central roles in the signaling events needed for T cell function (23, 24). These proteins lack enzymatic domains and instead carry binding domains and sites that are needed for the assembly of complexes. LAT and SLP-76 (Src homology 2 domain–containing leukocyte protein of 76 kD) couple the TCR complex to the activation of phospholipase Cγ1 (PLCγ1), Ca2+ mobilization, and NFAT activity (25–29). SLP-76 carries two NH2-terminal YESP sites that are phosphorylated by ZAP-70 (Zeta-associated protein-70) and ITK (inducible T cell kinase)/RLK (resting lymphocyte kinase; references 30–33), and that bind to the SH2 domain of the hematopoietic guanine nucleotide exchange factor VAV-1 and the adaptor NCK (34–36). The COOH-terminal Src homology 3 domain of Nck can recruit Wiskot Aldrich Syndrome protein (WASP), whereas the localization and activation of Cdc42 and WASP requires Vav1 (37). GTP-bound Cdc42 binds the WASP, which in turn regulates the Arp2/3 complex, an initiator of actin filament formation (38).
Although the NH2-terminal region of SLP-76 is needed for the activation of PLCγ1 and Ca2+ mobilization, the SH2 domain is not needed for these events, but is nevertheless needed, in unexplained ways, for T cell proliferation (39, 40). In this context, we and others cloned an immune cell–specific adaptor adhesion and degranulation promoting adaptor protein (ADAP; previously termed Fyn-T–binding protein [FYB] or SLP-76–associated protein [SLAP]) that binds to the SH2 domain of SLP-76 (41, 42). It is also preferentially phosphorylated by p59fyn and binds to the SH2 domain of the kinase (41, 43). Two isoforms (ADAP-120/130) exist with the 120-kD isoform preferentially expressed in the thymus and the 130-kD isoform in the peripheral T cell compartment (44). Each isoform has a proline-rich region, two putative nuclear localization sites, multiple tyrosine sites, and two SH3 domains (41–43). Each isoform also has two identical ExYDDV motifs (EVY595/Y651 DDV motifs in ADAP-120) that bind to the SH2 domain of SLP-76, and a YDGI motif that binds to same domain in p59fyn (42, 43, 45, 46). A similar DxYDDV site in hematopoetic protein kinase 1 (HPK-1) binds to the SH2 domain of BLNK/SLP-65 (47). An alternate YGYI site for SLP-76 SH2 domain binding has been proposed (48), but is the subject of some uncertainty (46, 47). Further downstream, ADAP binds to SKAP-55 (49–51), and has an EVH1 (Ena [enabled]/VASP [vasodilator-stimulated phospho protein] homology 1 domain) binding site for VASP, a regulator of actin filament elongation (52).
Although the subject of some initial debate (42), ADAP positively regulates T cell function as shown in transfection studies (44, 46) and by the phenotype of the ADAP−/− mice (53, 54). This was clearly observed in ADAP-deficient mice that show a defect in LFA-1 adhesion and clustering on T cells (53, 54). No defects in other aspects of T cell signaling were apparent. The adaptor also modulates β1 integrin-dependent cell migration (55) as well as the clustering and adhesion of β1 integrins on basophils (56–58). ADAP can bind to MIST (mast cell immunoreceptor signal transducer)/Clnk (for cytokine-dependent hemopoietic cell linker), leading to enhanced β-hexosaminidase release in mast cells (56–58). A functional connection to SKAP-55 has been implied by the ability of SKAP-55 to regulate LFA-1 adhesion/clustering and T cell–APC conjugation (59). Loss of the SKAP-55 SH3 domain that binds ADAP abrogates the ability of SKAP-55 to influence these events (59).
The combined observation that the SLP-76 SH2 domain is needed for T cell function and binds to ADAP, an adaptor that in turn is needed for LFA-1 adhesion suggested that this intermolecular interaction might constitute a link in TCR-CD3–mediated activation of LFA-1 (i.e., inside-out signaling). In this work, using a mutational approach, we confirm that ADAP binding to the SLP-76 SH2 domain is needed for TCR-induced LFA-1 adhesion and T cell–APC conjugation. In addition, we further show that the ADAP–SLP-76 interaction differentially regulates pSMAC formation versus general LFA-1 clustering and conjugation. Although the loss of the SLP-76 SH2 domain binding sites on ADAP (i.e., M12) interfered with the ability of ADAP to promote conjugation and general LFA-1 clustering, it did not operate as a DN. In contrast, the same mutations converted ADAP into a potent DN in the blockade of SMAC formation, and concurrently, IL-2 production. ADAP also colocalized with LFA-1 at the immunological synapse. Our findings identify a specific intermolecular event that couples the TCR with SMAC formation in T cells, and support a role for SMAC formation in T cell function.
Materials and Methods
Cell Culture and Antibodies.
Reagents were obtained from the following sources: anti–mouse ADAP (Transduction Laboratories); TRITC-conjugated phalloidin (Sigma-Aldrich); antihemaglutinin (HA11; Babco); antiphosphotyrosine (4G10; obtained from T. Roberts, Dana-Farber Cancer Institute, Boston, MA); anti–SLP-76 (obtained from P.R. Findell, Syntex, Palo Alto, CA); anti-CD3 and anti-CD11a (BD Biosciences); murine ICAM-1 human Fc (R&D Systems); fibronectin (Sigma-Aldrich); hybridoma T8.1/L625 cells (a gift from O. Acuto, Institute Pasteur, Paris, France); and tetanus toxoid (Ttox) 830–843 peptide (Research Genetics). Cells were cultured in DMEM supplemented with 200 nM methotrexate, 1 mg/ml G418, 10% FCS, 10 mM Hepes, 2 mM l-glutamine, 100 U/ml pen/strep, and 5 × 10−5 M 2-mercaptoethanol. Freshly isolated CD4+ T cells from DO11.10 mice (R. Lechler, Imperial College London, London, England) were cultured in RPMI 1640 medium supplemented with 2 mM l-glutamine, 100 U/ml pen/strep, 10% heat-inactivated FCS, and 5 × 10−5 M 2-mercaptoethanol. OVA 323–339 peptide was purchased from AnaSpec Inc.
Transfection/Infection Protocols.
ADAP, GFP, and M12 were expressed in a pSRα mammalian expression vector containing an influenza hemaglutinin (HA) epitope tag at the NH2 terminus (45, 59). The SH2 domain of SLP-76 was amplified by PCR and cloned into a pEBG mammalian expression vector containing a GST epitope tag. Transfection was conducted as described previously (45). For retroviral infection, ADAP or M12 was cloned into IRES-GFP–based pMXF5 retroviral expression vector using BamHI and NotI sites or using the Pinco-GFP–based retroviral expression vector (C. Casimir, Imperial College London, London, England) using BamHI and EcoRI sites, and T cells were infected as described using the ecotropic Phoenix retroviral producer cell line (59, 60). Primary mouse CD4+ cells were isolated using Dynabeads with mouse anti-CD4 (Dynal Biotech) and stimulated with Con A (Sigma-Aldrich) for 48 h before infection. After two washes, CD4+ T cells were cultured and washed in 10−5 M methyl-α-d-manno-pyranoside (Sigma-Aldrich) to remove the lectin. After three infections, T cells were collected and used in the conjugation experiments.
T Cell–APC Conjugate Assay.
Antigen-induced T cell adhesion was quantified using a colorimetric assay for measuring intracellular succinate dehydronase content with MTT (3(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide) as described previously (59). Cleaved MTT was dissolved in DMSO, transferred into a 96-well plate, and measured for optical density at 595 nm using an ELISA reader (Bio-Rad Laboratories). The level of T8.1 cell adhesion to L625.7-presenting cells was calculated by subtracting the background signal obtained with L625.7 presenting cells alone. Alternatively, T8.1 cells were stained with PKH67 green or PKH26 red dyes (Sigma-Aldrich) and seeded on adherent L625.7 cells. After a period of culture, the supernatant containing nonadherent T8.1 cells was aspirated, and the remaining cells were observed by fluorescent microscope. Conjugates between DO11.10 Tg cells and A20 cells were measured as described previously (59). Fluorescence was analyzed using a FACSCalibur flow cytometer (Becton Dickinson) equipped with CELLQuest software. Immunoprecipitation and immunoblotting were conducted as described previously (46, 59). Levels of bound Ab were using horseradish peroxidase–conjugated rabbit anti–mouse followed by detection with enhanced chemiluminescence (Amersham Biosciences). Measurements of IL-2 were performed using GFP, ADAP-GFP, or M12-GFP–infected T8.1 cells cocultured with L625.7 cells pulsed with different concentrations of Ttox peptide for 48 h as described previously (44, 46). The supernatants were collected, and IL-2 was measured using ELISA with rat anti–mouse IL-2 monoclonal antibody (ELISA Capture) and biotinylated rat anti–mouse IL-2 monoclonal antibody (ELISA Detection).
Integrin Adhesion and Clustering Assays.
Binding to ICAM-1 or fibronectin was measured using 96-well flat-bottom plates coated with 3.4 μg of murine ICAM-1–Fc, 30 μg/ml fibronectin (Sigma-Aldrich), or 10 μg/ml BSA (Sigma-Aldrich) as described previously (59). LFA-1 capping on retroviral infected T8.1 cells was conducted as described previously (59).
Immunofluorescent and Confocal Microscopy.
Immunofluorescence microscopy was conducted as described previously (59). Cells were incubated with anti-HA antibody, followed by FITC-labeled secondary antibody and TRITC-labeled phalloidin (Sigma-Aldrich). Image acquisition was performed with an Eclipse E800 microscope (Nikon) and RT Slider digital camera (Diagnostics, Inc.). For LFA-1 confocal imaging at the T cell–APC interface, L625.7 cells were seeded on the 12-mm coverslips in 24-well plates overnight and pulsed with 2.5 μg/ml Ttox cells for 2 h. Infected T8.1 cells were added for 30 min followed by fixation with 2% paraformaldehyde for 20 min at 4°C. Cells were exposed to in a blocking solution (5% vol/vol FCS and 3% wt/vol BSA in Perm/Wash buffer; BD Biosciences) for 1 h at 4°C followed by an incubation with anti-CD11a (1 μg/250 ml) in blocking solution for 1 h at 4°C. After three washes with 0.1% Tween 20/PBS, Alexa Fluor 568–conjugated goat anti–rat antibody (1 μg/250 ml) was incubated with cells for 1 h at 4°C and washed three times in 0.1% Tween 20/PBS. Cells were viewed under a 63× oil immersion objective using a confocal laser scanning microscope (TCS SP2; Leica) equipped with argon/krypton and helium/neon lasers using excitation wavelengths of 488, 568, and 633 nm as described previously (61). Conjugates were scanned in the xy-direction every 0.3 μm throughout the z-plane. The face of the immunological synapse was reconstructed using a maximum intensity projection (Confocal Software; Leica). The percentage of fluorescence intensity at immunological synapse relative to the whole cell membranes was calculated.
Online Supplemental Material.
Conjugates and LFA-1 staining were conducted as before and scanned in the xy-direction every 0.3 μm throughout the z-plane. The face of the immunological synapse was reconstructed using a maximum intensity projection (Confocal Software; Leica). Three-dimensional movies were reconstructed using Volocity software. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040780/DC1.
Results
ADAP Regulates T Cell–APC Conjugate Formation and Colocalizes with F-actin at the Immunological Synapse.
To assess whether ADAP can regulate conjugate formation, a conjugation assay was initially used using a T cell hybridoma T8.1 expressing an antigen receptor for Ttox peptide in the context of HLA-DR*1102 (62). Adhesion was measured by use of a 3(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Materials and Methods). Ttox peptide induced a three- to fourfold increase in T cell adhesion, with maximal binding at 15–30 min, followed by a reduction in binding from 30–60 min (Fig. 1 a, top). Adhesion required the presence of adherent cells because the binding of T cells to tissue culture plates without adherent cells was negligible (unpublished data). Transfection with ADAP resulted in a further two- to threefold increase in T cell binding to APCs in the presence of Ttox. This resulted in an increase from 35–40 to 80–90% in the percentage of available T cells to adhere to APCs. ADAP also increased the binding of T cells in the absence of peptide (i.e., antigen-independent adhesion). Similar results were obtained using an alternate procedure where PKH-67 was used to stain T8.1 cells followed by incubation with APCs and visualization by incandescent and immunofluorescent microscopy (Fig. 1 b). As was observed in the MTT assay, ADAP overexpression increased the binding of T cells to Ttox/APCs by two- to threefold relative to Ttox-exposed vector control cells. Furthermore, conjugation was partially dependent on LFA-1 binding to ICAM-1 as shown by its partial inhibition by anti–LFA-1 (Fig. 1 c). These effects were observed with a low to moderate three- to fourfold overexpression of ADAP as detected by blotting (Fig. 1 a, bottom left). Expression of the transfected protein was confirmed by anti-HA blotting (Fig. 1 a, bottom right). These observations show that ADAP is capable of increasing conjugate formation between T cells and APCs.
Figure 1.
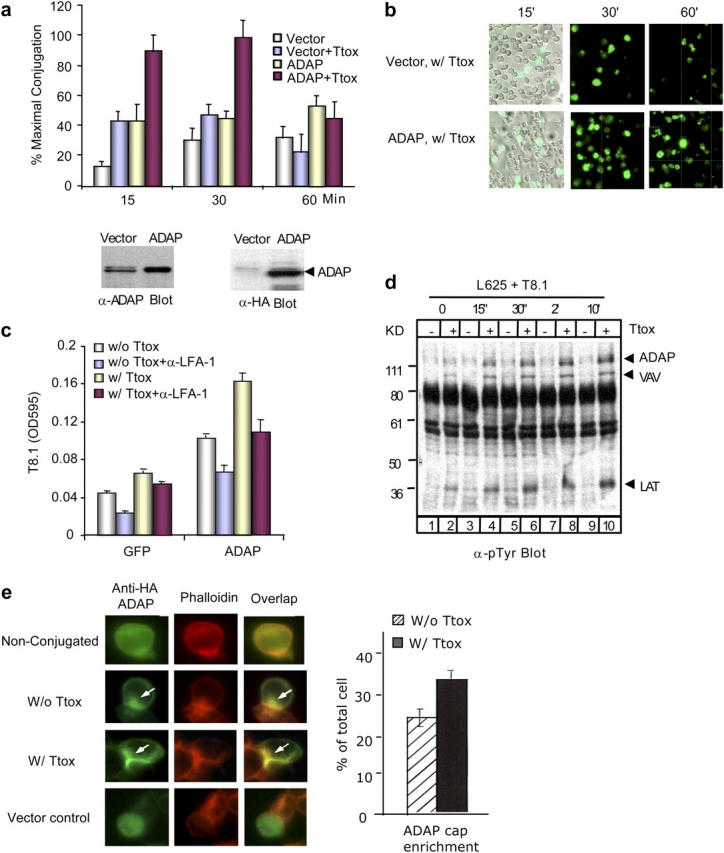
ADAP increases T cell–APC conjugation and colocalizes with F-actin at the immunological synapse. (a, top) pSRα vector or ADAP-transfected T8.1 cells were cocultured with L625.7 cells (APCs) in the absence or presence of Ttox peptide for indicated time. Conjugation was measured by MTT assay. (bottom) Lysates of cells transfected with pSRα vector and ADAP were blotted for ADAP expression using anti-ADAP (left) or anti-HA (right). (b) pSRα or ADAP-transfected T8.1 cells were stained with PKH-67 fluorescent dye and cocultured with APCs for the indicated time. Bright field images are shown for one of each transfection to confirm equivalent L625.7 cells. Immunofluorescence microscopy images show the T8.1 cells that bound to APCs. (c) ADAP enhancement of conjugation is partially dependent on LFA-1. GFP vector or ADAP-transfected T8.1 cells were pretreated with anti–LFA-1 (CD11a and CD18 antibodies) or untreated, and conjugates were measured by an MTT assay. (d) T8.1 and L625.7 cells were cocultured in the absence or presence of Ttox for indicated time, and lystates were subjected to antiphosphotyrosine blotting. Bands corresponding to ADAP, VAV, or LAT were identified by precipitation and immunoblotting assays (not depicted). (e) ADAP transfected T8.1 alone and conjugates in the absence or presence of Ttox, and GFP vector–transfected T8.1 cells were stained with anti-HA and TRITC-labeled phalloidin.
ADAP also underwent a change in tyrosine phosphorylation as a result of incubation between T cells/APCs, as detected by antiphosphotyrosine blotting (Fig. 1 d). This was time dependent and required the presence of peptide agonist (lanes 2, 4, 6, 8, 10). Phosphorylation was initially detected as early as 15 s of culture, peaked from 30 s to 2 min, and reached a plateau after 10 min. We showed previously that phosphorylation is stable for hours after anti-CD3 ligation (43). Little phosphorylation was observed in the absence of peptide (lanes 1, 3, 5, 7, 9). Other adaptors such as VAV and LAT also underwent peptide-dependent phosphorylation, the identity being confirmed by precipitation and blotting studies (unpublished data). These findings indicate that ADAP becomes phosphorylated on tyrosine residues during antigen presentation in an agonist-dependent manner.
ADAP also localized at the interface between T cells and APCs (Fig. 1 e). Cells were transfected with HA-tagged ADAP, followed by staining with anti-HA plus FITC-conjugated goat anti–mouse. TRITC-labeled phalloidin was used to visualize F-actin. Although unconjugated cells showed ADAP throughout the cytoplasm (Fig. 1, top), the adaptor localized at the T cell–APC contact area of conjugates formed in the presence and absence of peptide (Fig. 1, middle, arrows). Often this localization was accompanied by a concurrent loss of the adaptor in other regions of the cytoplasm. Ttox-stimulated conjugates had a more compressed localization of ADAP at the interface than observed in unstimulated conjugates (Fig. 1, bottom middle vs. top middle). Furthermore, the phalloidin subcap closely overlapped with ADAP in stimulated conjugates as shown by yellow fluorescence (Fig. 1, arrows, overlay of red and green fluorescence). The overlap was less coincident in unstimulated conjugates. As a negative control, vector-transfected cells exhibited no localization (Fig. 1, bottom). Overall, these observations demonstrate that ADAP can up-regulate T cell–APC conjugate formation in a process that is accompanied by changes in the phosphorylation and localization of ADAP.
T Cell–APC Conjugation Depends on SLP-76 Binding Sites on ADAP.
We previously identified two tyrosine-based motifs (i.e., EVY595/651DDV sites) in ADAP that bind to the SH2 domains of SLP-76 (45, 46). Therefore, next we investigated whether SLP-76 binding to ADAP was needed to couple the TCR with the downstream regulation of conjugation. ADAP mutated at the tyrosine residues (i.e., Y-F) in these motifs (termed M12) was incorporated into a pMXF5-GFP–based bicistronic vector for retroviral-mediated gene transfer (60). FACS profiles showed that cells expressed moderate levels (i.e., 48–62%) of GFP, ADAP-GFP, and M12-GFP (Fig. 2). Infected T cells were generally sorted before their use in conjugation assays. Significantly, although ADAP increased conjugate formation, the M12 mutant had little if any effect (Fig. 2 b). This defect in the function of M12 was also observed in the PKH26-labeling assay (Fig. 2 c). Although ADAP expression increased T cell adhesion (middle vs. left), the M12 mutant failed to increase conjugation in the presence of Ttox. These observations clearly indicate that the two single mutations needed for SLP-76 binding interfere with ADAP enhancement of conjugation.
Figure 2.

T cell–APC conjugation depends on SLP-76 binding sites on ADAP. (a, top) FACS profiles show the expression level of GFP, ADAP, or M12 in retroviral infected T8.1 cells. (bottom) GFP, ADAP-GFP, and M12-GFP–infected T8.1 cells were cocultured with L625.7 cells, and conjugates were assayed by MTT. (b) GFP, ADAP-GFP, and M12-GFP–infected T8.1 cells were stained with PKH26 and cocultured with L625.7 cells in the absence or presence of Ttox for 30 min. Immunofluorescence microscopy images show the T8.1 cells that bound to APCs. (c, top) FACS profiles show the expression level of GFP, ADAP, or M12 in retroviral infected DO11.10 mouse Tg cells. (bottom) GFP, ADAP-GFP, and M12-GFP–infected DO11.10 Tg cells were cocultured with PKH26 fluorescent dye-labeled A20 cells (APCs) in the presence or absence of OVA peptide for 30 min, and conjugates were enumerated by a FACS analysis. Histograms show the percentage of Tg cells forming conjugates.
M12 Is Defective in Primary T Cell–APC Conjugation.
To confirm this observation in primary T cells, retroviral gene transfer was used to infect activated CD4 positive lymph node/spleen cells from transgenic DO11.10 mice (60, 63). T cells from this mouse expresses a TCR for chicken OVA peptide (residues 323–339), which can be presented by A20 cells (59). Retroviral gene transfer resulted in expression of GFP, ADAP-GFP, or M12-GFP in 18–27% of cells (Fig. 2 e). Conjugation was assessed by FACS analysis for the binding of GFP-expressing cells with PKH26 labeled A20 cells. Exposure to OVA peptide increased conjugation by two- to threefold relative to vector GFP control cells (i.e., 2–3 to 7–8%; Fig. 2 c). Infection with ADAP-GFP further increased conjugation with a two- to threefold shift in binding to APCs (i.e., 7–8 to 20%). In contrast, as noted with the T8.1 cells, the expression of GFP-M12 failed to increase conjugation. These findings indicate that ADAP can enhance conjugate formation of primary murine T cells in a manner analogous to the T8.1 system, and that the loss of the SLP-76 binding sites abrogates this effect.
SLP-76 SH2 Domain, But Not VASP Binding Mutant, Blocks Conjugation.
Although the loss of the SLP-76 binding sites abrogated conjugation, it was formally possible that another as yet identified protein binds the ADAP EVYDDV sites and acts to couple the TCR with conjugation. To address this, the SH2 domain of SLP-76 was expressed as a GST fusion protein in T cells and assessed for an effect on conjugation (Fig. 3 a). Although the expression of GST alone had no effect, expression of GST-SLP-76-SH2 reduced conjugation to nonpeptide-treated levels. GST and GST-SLP-76-SH2 were expressed at similar levels as shown in anti-GST blotting (Fig. 3 a, bottom). SLP-76 SH2 domain precipitated a major band corresponding to ADAP in addition to two weak bands (unpublished data). Although tempered by the fact that SH2 domains bind to more than one protein, the predominate binding to ADAP and its inhibition in conjugation provides further evidence in support of the importance of ADAP–SLP-76 interaction in conjugation.
Figure 3.

SLP-76 SH2 domain expression blocks conjugation, whereas VASP-ADAP binding does not influence conjugate formation. (a, bottom) GST or GST-SLP-76-SH2–transfected T8.1 cells were cocultured with APCs for 30 min, and conjugates were measured by an MTT assay. (bottom) Cell lysates from GST or GST-SLP-76-SH2–transfected T8.1 cells were blotted with anti-GST. (b) pSRα vector, ADAP, M617, and M617/131/132-transfected T8.1 cells were cocultured with L625.7 cells for 30 min, and conjugates were measured by an MTT assay.
Interestingly, in contrast, we also included a variant of ADAP with a mutation in the FPPPPDDDI motif (residues 616–624) that had been reported to bind to the EVH1 domain of VASP (Fig. 3 b and reference 52). A crucial proline at residue 617 was substituted with an alanine that disrupts EVH1 domain binding (termed M617; reference 64). Surprisingly, M617 enhanced conjugation to the same extent as WT ADAP. Similarly, M617/131/132 with additional mutations in another possible EVH1 binding motif (i.e., FPWPP) at residues 131/132 enhanced conjugation. This indicates that VASP binding to ADAP at these sites is not apparently needed for conjugation.
ADAP–SLP-76 Binding Regulates Integrin Adhesion and Clustering.
Given the importance of conjugation, it was of interest to determine whether this interaction was needed for integrin-mediated adhesion and aggregation (Fig. 4). Previous studies by ourselves and others have shown a requirement for ADAP expression in integrin binding (53, 54). For this, infected T cells were incubated on plates with immobilized ICAM-1 and assessed for binding as described previously (59). Anti-CD3 increased the binding of GFP-infected control cells to ICAM-1 (Fig. 4 a). ADAP infection enhanced binding by twofold relative to this control. In contrast, M12 failed to enhance adhesion. A similar analysis was conducted using purified fibronectin immobilized on plates (Fig. 4 b). In this case, the combination of immobilized fibronectin plus anti-CD3 led to a fivefold increase in binding as compared with BSA-coated plates. Similarly, ADAP-infected cells showed twofold binding increased relative to control cells. M12 infection failed to augment binding, exhibiting binding comparable to the GFP vector control. Treatment with phorbol ester served as a positive control for inside-out signaling. These data indicate that the previously documented importance of ADAP in integrin binding depends on SLP-76 SH2 binding to ADAP.
Figure 4.

ADAP–SLP-76 binding regulates integrin adhesion and clustering. GFP, ADAP-GFP, and M12-GFP–infected T8.1 cells were unstimulated or stimulated with 2C11 or PMA for 30 min and incubated on BSA, ICAM-1Fc–coated plates (a) or fibronectin (FN)-coated plates (b). (c) GFP, ADAP-GFP, and M12-GFP–infected T8.1 cells were stimulated with 2C11 for 30 min, fixed, and stained with anti-CD11a and Alexa Fluor 546–conjugated anti–rat antibody. Anti–LFA-1 capping was defined by the presence of a discrete polarized cap at one end of the cell. (bottom) Histogram showing the percentage of T8.1 cells with LFA-1 clustering. (d) GFP, ADAP-GFP, and M12-GFP–infected T8.1 cells were cocultured with Ttox pulsed L625.7 cells for 30 min, fixed, and stained for LFA-1 as before. Imaging and measurements of staining at the immunological synapse amongst individual conjugates (>45) were performed by laser-scanning microscopy. (top) DIC image (left) showed the conjugate and fluorescence image (right) showed LFA-1 clustering at the interface between T8.1 and L625.7 cells. (bottom) Histogram showed the percentage of LFA-1 at the interface relative to the total surface LFA-1.
Because integrin clustering increases the avidity of integrin binding to substrate (65), M12 was next assessed for an effect on these events (Fig. 4 c). LFA-1 capping was defined by the presence of a discrete polarized cap (59). Anti-CD3 increased the percentage of vector-transfected control cells to form LFA-1 caps from 6 to 12%. Infection of cells with ADAP increased this further by twofold where 22–25% of T cells showed clustering after 30 min of anti-CD3 stimulation. However, in contrast, expression of M12 failed to increase levels (Fig. 4 c, bottom right). It also appeared to have a slight inhibitory effect, reducing clustering below that observed for vector-infected cells, although this was not statistically significant as judged by standard deviations.
Clustering of LFA-1 was imaged at the T cell–APC interface in conjugates formed over 30 min in the presence of Ttox. Anti-CD11a was used followed by an Alexa Fluor 568–conjugated anti–rat staining. Imaging of the immunological synapse amongst individual conjugates (>40) was conducted by laser-scanning confocal microscopy. As shown in Fig. 4 d, LFA-1 could readily be visualized at the interface of T cell–APC conjugates that had been stimulated by Ttox peptide (Fig. 4 d, top). LFA-1 at the interface was quantified as the percentage of LFA-1 located in the immunological synapse relative to the total surface of the T cell. There was considerable variation in the level of LFA-1 clustering amongst individual conjugates. Retroviral expression of ADAP-GFP increased the mean slightly, whereas M12 failed to support an increase, consistent with the observation in the capping of surface LFA-1 (Fig. 4 c). M12 also appeared to decrease the mean slightly, although this was a marginal effect relative to the overall heterogeneity amongst conjugates. Overall, these observations indicate that M12 failed to support LFA-1 clustering as determined by two independent assays.
M12 Acts as a DN in Blocking pSMAC Formation and IL-2 Production.
Importantly, so far, the importance of ADAP–SLP-76 binding was evident by the fact that M12 did not support an increased conjugation and general LFA-1 clustering observed with wild-type ADAP. However, at the same time, M12 did not act as a potent DN in interfering with the function of endogenous ADAP. In other words, M12 did not reduce in a statistically significant manner the level of conjugation or general LFA-1 clustering below the vector control. In this regard, SMAC formation is thought to occur subsequent to general LFA-1 clustering in a process that involves the segregation of LFA-1 from the TCR in the formation of the p- and c-SMAC (1, 2). Given this, next we assessed whether the characteristic ring-shaped clustering of LFA-1 at the immunological synapse (i.e., the formation of the pSMAC) was influenced by ADAP–SLP-76 binding. Imaging of the immunological synapse amongst individual conjugates (>38) was conducted by laser-scanning microscopy as described in Materials and Methods. Conjugates formed over 30 min in the presence of Ttox were stained for LFA-1 using CD11a antibody and Alexa Fluor 568–conjugated anti–rat antibody. ADAP was stained with anti-ADAP mAb and Cy5-conjugated anti–mouse. As seen in Fig. 5 a, ADAP staining generally colocalized with LFA-1 at the interface with the segregation of LFA-1 receptors that characterize a pSMAC (Fig. 5, b vs. c). Anti–LFA-1 staining was visualized perpendicular to the interfacing APCs in the context of ADAP versus M12 expression. Samples of pSMAC at the interface between T cells and APCs are shown in Fig. 5 b (top). Although 53% of conjugates of GFP-expressing cells showed the presence of a pSMAC at the immunological synapse (i.e., 21/39 conjugates), there was an increase in pSMAC formation in cells expressing ADAP-GFP, up to 71% of conjugates (i.e., 27/38 conjugates). In contrast, M12 had a marked effect as a DN, whereas only 20% of conjugates possessed a pSMAC (i.e., 8/39 conjugates; Fig. 5 a, bottom). This represents a 62% reduction in pSMAC formation relative to the GFP control cells. Three-dimensional views of the pSMAC formation are shown in Fig. S1 (available at http://www.jem.org/cgi/content/full/jem.20040780/DC1). A gallery of the patterns for the different conjugates is shown in Fig. S2 (available at http://www.jem.org/cgi/content/full/jem.20040780/DC1). It is noteworthy that, although there were significant effects of M12 on pSMAC formation, the overall intensity of LFA-1 at the immunological synapse was similar for the vector, ADAP, and M12-infected cells, a result consistent with the notion that general LFA-1 clustering was only marginally affected by M12 (i.e., not a DN). Therefore, unlike in the case of conjugation and LFA-1 clustering, M12 acted as a potent DN in abrogating the formation of pSMACs, indicating that SLP-76 binding to ADAP is needed for pSMAC formation. Thus, although other processes such as actin polymerization are known to affect the extent of accumulation of proteins at the immunological synapse, here we have identified an intracellular event that regulates assembly of the LFA-1–rich pSMAC while minimally effecting accumulation of LFA-1 at the immunological synapse itself.
Figure 5.
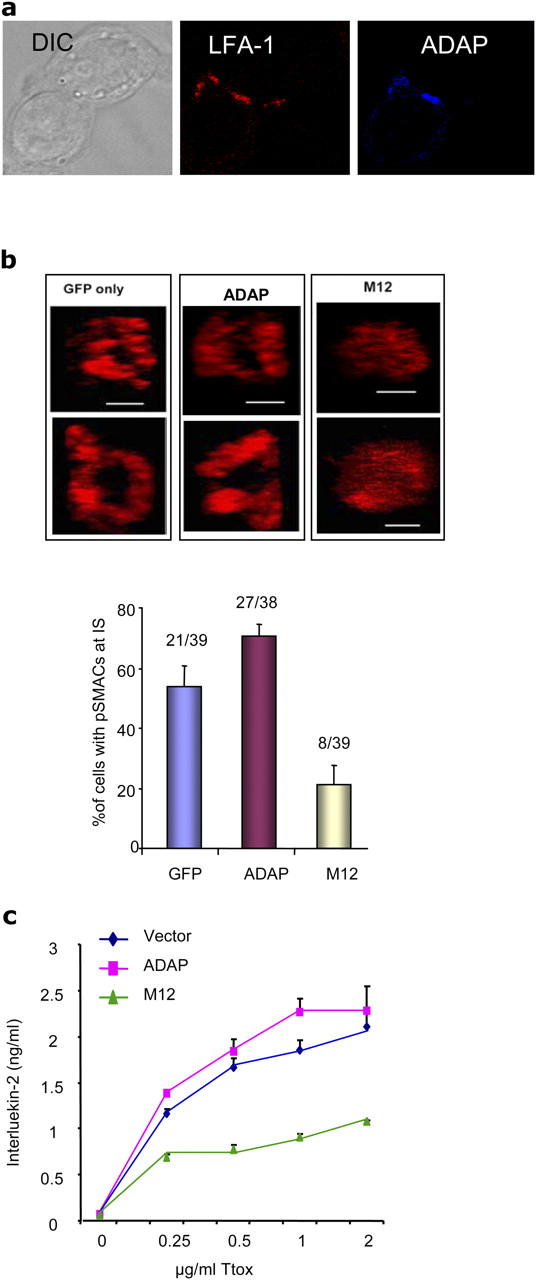
M12 acts as a DN to inhibit pSMAC formation and IL-2 production. (a) Conjugates of GFP, ADAP-GFP, and M12-GFP–infected T8.1 cells were stained with LFA-1 and ADAP. (b) LFA-1 staining of conjugates was examined by laser-scanning microscopy. Samples of pSMACs at the immunological synapse visualized by z-section analysis are shown (top). Histogram showed the percentage of pSMACs formation at the immunological synapse (bottom). (c) GFP, ADAP-GFP, and M12-GFP–infected T8.1 cells were cocultured with L625.7 cells in the presence of various concentrations of Ttox peptide and followed by a measurement of IL-2 production by ELISA.
Given that M12 abrogates pSMAC formation, an opportunity arose to test if disruption of the pSMAC correlated with an effect on T cell function. Thus, we assessed the effect of M12 on TCR induction of IL-2 production (Fig. 5 c). The potent effect of M12 on SMAC formation provided a potential window into the role of SMAC formation in TCR induction of IL-2 production. We showed previously that ADAP expression had a slight effect in increasing IL-2 production, although the underlying basis was not clear (43). Initially, using the same concentration of Ttox peptide (at 2 μg/ml) as used in the analysis of pSMAC formation, retroviral expression of ADAP increased cytokine production slightly at levels comparable to that observed on pSMAC formation (Fig. 5 c). Similarly, M12 acted as a potent DN in blocking IL-2 production by >50%. This was shown over a range of peptide concentrations (i.e., 0–2 μg/ml) followed by a measurement of IL-2 production using an ELISA assay (Fig. 5 c). Although the production of IL-2 increased with increasing amounts of peptide antigen, and ADAP increased this at each concentration, M12 consistently acted as DN in the inhibition of cytokine production below that of GFP vector–infected cells. Overall, our findings indicate that, although M12 failed to support an increase in conjugate formation, its most potent influence was observed at the level of the subsequent events that lead to pSMAC formation and IL-2 production. The similarity in the degree of inhibition of pSMAC formation and IL-2 production is consistent with a model whereby proper SMAC formation is needed for the full induction of IL-2 production in T cells.
Discussion
Despite the marked nature of receptor rearrangements at the immunological synapse, an outstanding question has concerned the identity and nature (i.e., intermolecular binding) of signaling events that regulate SMAC formation. Two candidates are talin and RapL that colocalize with LFA-1 in the pSMAC (1, 18). In this work, we have identified SLP-76 SH2 domain binding to ADAP (i.e., via EVYDDV motifs) as a key event that bridges the TCR complex via inside-out signaling to LFA-1 clustering, adhesion, T cell–APC conjugation, and formation of the pSMAC. Loss of SLP-76 binding abrogated ADAP potentiation of LFA-1 adhesion/clustering, and as such is likely to account for the previous findings that ADAP expression is needed for these events (53, 54). Our work further uncovered a difference in the reliance of pSMAC formation versus general LFA-1 clustering/conjugation on the ADAP–SLP-76 interaction. The DN effect of M12 on SMAC, but not on general LFA-1 clustering/adhesion, pointed to a special connection between ADAP–SLP-76 binding and the higher order events that lead to the segregation of receptors in SMAC formation. This close connection is supported by the colocalization of ADAP with LFA-1 at the immunological synapse. Furthermore, our work documents a correlation between the DN effect of M12 inhibition on SMAC formation and subsequent IL-2 production, thereby supporting a model where SMAC formation contributes to cytokine production in T cells.
ADAP joins a number of mediators such as VAV-1, WASP, WIP, Rap1/RapL, and SKAP-55 that regulate conjugation (59, 66–69). This was observed in two models with retroviral gene transfer in T8.1 cells that respond to Ttox peptide, and in DO11.10 primary T cells that respond to OVA peptide (Figs. 1 and 3). We previously identified the two ExYDDV motifs as sites of SLP-76 domain binding (45, 46), a finding confirmed with BLNK–SLP-65 SH2 binding to a related site DxYDDV in HPK-1 (47). Although our work focused on ADAP-120, the higher M r isoform ADAP-130 also has the motifs (44), and can potentiate conjugation (unpublished data). Specificity was shown by the finding that mutations in the putative ADAP EVH1 binding sites (52) had no detrimental effect on conjugation. VASP acts to prevent the termination of growing actin filaments by competing for the binding of capping proteins (70). Therefore, VASP–ADAP binding may play other roles in conjugation, distinct from the regulation of LFA-1–ICAM-1 binding. Actin branching is generally associated with lateral extensions at the cell surface (i.e., lamellapodia) and, therefore, ADAP–VASP may extend the T cell–APC interface after LFA-1 binding, or play a supplementary nonessential role in ADAP function. In a similar vein, class II myosin modulates the cytoskeleton and motility, but not synapse formation (71). In either case, this involvement of the ADAP–VASP interaction was clearly distinct from that of the ADAP–SLP-76 interaction. One a simple level, the ADAP–SLP-76 interaction would be expected to operate downstream of TCR-p56lck-ZAP-70–SLP-76–VAV (with the possible intermediate involvement of LAT and GADS) and be integrated downstream with its other binding partner SKAP-55. We showed previously that the loss of the SKAP-55 SH3 domain that binds to ADAP abrogated the ability of SKAP-55 to enhance T cell–APC conjugation (59). The pathway is further complicated by the involvement of p59fyn. p59fyn (and not p56lck) mediates phosphorylation of ADAP on sites needed for SLP-76 SH2 domain binding (46), and p59fyn-deficient T cells show a major loss of ADAP phosphorylation (43).
Information on the intracellular process that regulates SMAC formation has been missing, as has information on the relationship between general LFA-1 clustering and the subsequent events that segregate receptors in the SMAC. TCR-CD3 clustering is controlled by WASP (72), whereas VAV-1 has been reported to regulate both TCR-CD3 and LFA-1 clustering (66). The involvement of Vav1 and RapL appears to occur at the level of general LFA-1 clustering, before the segregation of receptors leading to SMAC formation clustering (18, 66). Constitutively active Rap1 enhances LFA-1 clustering (17), whereas DN RapL blocks general clustering and conjugation (18). The proximity of SLP-76 to TCR-CD3 signaling would place ADAP–SLP-76 upstream of RapL and talin, perhaps operating in conjunction with SLP-76 binding to VAV1 and NCK during actin remodelling (36, 37). At the same time, the special DN effect of M12 on pSMAC formation and the colocalization of ADAP with LFA-1 underscores an additional close downstream connection to the SMAC (and possibly RapL and talin). Although ADAP does not coprecipitate LFA-1 (unpublished data), it must be central to this process because a single protein with two mutations essentially eliminated >50–60% of SMAC formation (Fig. 5 b). Future studies will need to be conducted to clarify the connection between ADAP and RapL/talin in the SMAC. An additional connection to SKAP-55 has also been implied by SKAP-55 binding to ADAP and its regulation of LFA-1 adhesion/clustering and T cell–APC conjugation (59).
Our findings showing a similarity in the degree of inhibition of pSMAC formation and IL-2 production suggest a role for SMAC formation in the full induction of IL-2 production in T cells (Fig. 5). Previous studies have been mixed in their support for a connection between these events, most likely due to the fact that both the clustering of receptors (i.e., a positive signal) and receptor internalization (i.e., probable termination signal) occur in the SMAC (9). SMAC formation occurs during a decrease in the optimal levels of tyrosine phosphorylation (9), and early signaling can occur outside the SMAC (4, 7, 8). T cells from CD2-associated protein-deficient mice fail to form stable SMACs and yet hyperproliferate to antigen (9). Despite this, continuous T cell receptor signaling promotes synapse formation and T cell effector function (73). The DN effect of M12 on pSMAC formation, but not conjugation, allowed for a distinction to be made between these events and IL-2 production. M12 blocked both SMAC formation and IL-2 production by >50%, and occasionally, as high as 80% (unpublished data). In this scenario, the limited numbers of agonist peptide–MHC complexes and continuous receptor degradation in in vivo responses would be offset by more effective receptor aggregation and signaling leading to enhanced cytokine production. The use of intracellular mutants and genetics may eventually be the most effective way of uncovering a role for the SMAC in T cell function.
Lastly, the connection between ADAP–SLP-76 and SMAC formation/IL-2 production provides a molecular basis to explain previous results showing that SLP-76 can exert more than one signaling function in T cells. Although NH2-terminal phosphorylation and the GADS binding sites are needed for the activation of PLCγ1, the SH2 domain has been found previously to be needed for proliferation, but can be dispensed for PLCγ1 phosphorylation (39, 40). Unlike in the case of the NH2-terminal tyrosines, the SH2 domain is also not required for progression of thymocytes beyond the pre-TCR stage (CD44+CD25+) of thymic differentiation (39, 40). The requirement for SLP-76 SH2 domain binding to ADAP in conjugation and SMAC formation provides a basis to account for its role in T cell proliferation.
Acknowledgments
We thank Dr. C. Casimir (Imperial College London) for helpful discussions regarding approaches to efficient retroviral infection and Drs. H. Schneider and B. Wilkinson (Imperial College London) for critical reading of this work.
This work was supported by National Institutes of Health grant no. A139021 and a grant from the Wellcome Trust, London (C.E. Rudd is a Principal Research Fellow of the Wellcome Trust).
The authors have no conflicting financial interests.
Abbreviations used in this paper: ADAP, adaptor adhesion and degranulation promoting adaptor protein; DN, dominant negative; HA, hemaglutinin; ICAM, intercellular adhesion molecule; LFA-1, leukocyte function-associated antigen-1; pSMAC, peripheral SMAC; SMAC, supramolecular activation cluster; Ttox, tetanus toxoid; WASP, Wiskot Aldrich Syndrome protein.
References
- 1.Monks, C.R., B.A. Freiburg, H. Kupher, N. Sciaky, and A. Kupfer. 1998. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 395:82–86. [DOI] [PubMed] [Google Scholar]
- 2.Dustin, M.L., and A.S. Shaw. 1999. Costimulation: building an immunological synapse. Science. 283:649–650. [DOI] [PubMed] [Google Scholar]
- 3.Davis, D.M. 2002. Assembly of the immunological synapse for T cells and NK cells. Trends Immunol. 23:356–363. [DOI] [PubMed] [Google Scholar]
- 4.Freiberg, B.A., H. Kupfer, W. Maslanik, J. Delli, J. Kappler, D.M. Zaller, and A. Kupfer. 2002. Staging and resetting T cell activation in SMACs. Nat. Immunol. 3:911–917. [DOI] [PubMed] [Google Scholar]
- 5.Chakraborty, A.K., M.L. Dustin, and A.S. Shaw. 2003. In silico models for cellular and molecular immunology: successes, promises and challenges. Nat. Immunol. 4:933–936. [DOI] [PubMed] [Google Scholar]
- 6.Davis, D.M., and M.L. Dustin. 2004. What is the importance of the immunological synapse? Trends Immunol. 25:323–327. [DOI] [PubMed] [Google Scholar]
- 7.Lee, K.H., A.D. Holdorf, M.L. Dustin, A.C. Chan, P.M. Allen, and A.S. Shaw. 2002. T cell receptor signaling precedes immunological synapse formation. Science. 295:1539–1542. [DOI] [PubMed] [Google Scholar]
- 8.Costello, P., M. Gallagher, and D. Cantrell. 2002. Sustained and dynamic inositol lipid metabolism inside and outside the immunological synapse. Nat. Immunol. 3:1082–1089. [DOI] [PubMed] [Google Scholar]
- 9.Lee, K.H., A.R. Dinner, C. Tu, G. Campi, S. Raychaudhuri, R. Varma, T.N. Sims, W.R. Burack, H. Wu, J. Wang, et al. 2003. The immunological synapse balances T cell receptor signaling and degradation. Science. 302:1218–1222. [DOI] [PubMed] [Google Scholar]
- 10.Igakura, T., J.C. Stinchcombe, P.K. Goon, G.P. Taylor, J.N. Weber, G.M. Griffiths, Y. Tanaka, M. Osame, and C.R. Bangham. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science. 299:1713–1716. [DOI] [PubMed] [Google Scholar]
- 11.Purbhoo, M.A., D.J. Irvine, J.B. Huppa, and M.M. Davis. 2004. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 5:524–530. [DOI] [PubMed] [Google Scholar]
- 12.Kupfer, A., and H. Kupfer. 2003. Imaging immune cell interactions and functions: SMACs and the immunological synapse. Semin. Immunol. 15:295–300. [DOI] [PubMed] [Google Scholar]
- 13.Dustin, M.L., T.G. Bivona, and M.R. Philips. 2004. Membranes as messengers in T cell adhesion signaling. Nat. Immunol. 5:363–372. [DOI] [PubMed] [Google Scholar]
- 14.Shimaoka, M., T. Xiao, J.H. Liu, Y. Yang, Y. Dong, C.D. Jun, A. McCormack, R. Zhang, A. Joachimiak, J. Takagi, et al. 2003. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 112:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hogg, N., M. Laschinger, K. Giles, and A. McDowall. 2003. T-cell integrins: more than just sticking points. J. Cell Sci. 116:4695–4705. [DOI] [PubMed] [Google Scholar]
- 16.Zhang, W., Y. Shao, D. Fang, J. Huang, M.S. Jeon, and Y.C. Liu. 2003. Negative regulation of T cell antigen receptor-mediated Crk-L-C3G signaling and cell adhesion by Cbl-b. J. Biol. Chem. 278:23978–23983. [DOI] [PubMed] [Google Scholar]
- 17.Sebzda, E., M. Bracke, T. Tugal, N. Hogg, and D.A. Cantrell. 2002. Rap1A positively regulates T cells via integrin activation rather than inhibiting lymphocyte signaling. Nat. Immunol. 3:251–258. [DOI] [PubMed] [Google Scholar]
- 18.Katagiri, K., A. Maeda, M. Shimonaka, and T. Kinashi. 2003. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 4:741–748. [DOI] [PubMed] [Google Scholar]
- 19.Yan, B., D.A. Calderwood, B. Yaspan, and M.H. Ginsberg. 2001. Calpain cleavage promotes talin binding to the beta 3 integrin cytoplasmic domain. J. Biol. Chem. 276:28164–28170. [DOI] [PubMed] [Google Scholar]
- 20.Sampath, R., P.J. Gallagher, and F.M. Pavalko. 1998. Cytoskeletal interactions with the leukocyte integrin beta2 cytoplasmic tail. Activation-dependent regulation of associations with talin and alpha-actinin. J. Biol. Chem. 273:33588–33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiosses, W.B., S.J. Shattil, N. Pampori, and M.A. Schwartz. 2001. Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat. Cell Biol. 3:316–320. [DOI] [PubMed] [Google Scholar]
- 22.Giagulli, C., E. Scarpini, L. Ottoboni, S. Narumiya, E.C. Butcher, G. Constantin, and C. Laudanna. 2004. RhoA and zeta PKC control distinct modalities of LFA-1 activation by chemokines: critical role of LFA-1 affinity triggering in lymphocyte in vivo homing. Immunity. 20:25–35. [DOI] [PubMed] [Google Scholar]
- 23.Wilkinson, B., H. Wang, and C.E. Rudd. 2004. Positive and negative adaptors in T-cell signalling. Immunology. 111:368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samelson, L.E. 1999. Adaptor proteins and T-cell antigen receptor signaling. Prog. Biophys. Mol. Biol. 71:393–403. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, W.J., J. Sloan-Lancaster, J. Kitchen, R.P. Trible, and L.E. Samelson. 1998. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell. 92:83–92. [DOI] [PubMed] [Google Scholar]
- 26.Weber, J.R., S. Orstavik, K.M. Torgersen, N.C. Danbolt, S.F. Berg, J.C. Ryan, K. Tasken, J.B. Imboden, and J.T. Vaage. 1998. Molecular cloning of the cDNA encoding pp36, a tyrosine-phosphorylated adaptor protein selectively expressed by T cells and natural killer cells. J. Exp. Med. 187:1157–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackman, J.K., D.G. Motto, Q. Sun, M. Tanemoto, C.W. Turck, G.A. Peltz, G.A. Koretzky, and P.R. Findell. 1995. Molecular cloning of SLP-76, a 76-kDa tyrosine phosphoprotein associated with Grb2 in T cells. J. Biol. Chem. 270:7029–7032. [DOI] [PubMed] [Google Scholar]
- 28.Yablonski, D., M.R. Kuhne, T. Kadlecek, and A. Weiss. 1998. Uncoupling of nonreceptor tyrosine kinases from PLC-γ1 in a SLP-76-deficient T cell. Science. 218:413–416. [DOI] [PubMed] [Google Scholar]
- 29.Zhang, W., B.J. Irvin, R.P. Trible, R.T. Abraham, and L.E. Samelson. 1999. Functional analysis of LAT in TCR-mediated signaling pathways using a LAT-deficient Jurkat cell line. Int. Immunol. 11:943–950. [DOI] [PubMed] [Google Scholar]
- 30.Raab, M., A.J. da Silva, P.R. Findell, and C.E. Rudd. 1997. Regulation of Vav-SLP-76 binding by ZAP-70 and its relevance to TcRζ/CD3 induction of interleukin-2. Immunity. 6:1–11. [DOI] [PubMed] [Google Scholar]
- 31.Wardenburg, J.B., C. Fu, J.K. Jackman, H. Flotow, S.E. Wilkinson, D.H. Williams, R. Johnson, G. Kong, A.C. Chan, and P.R. Findell. 1996. Phosphorylation of SLP-76 by the ZAP-70 protein-tyrosine kinase is required for T-cell receptor function. J. Biol. Chem. 271:19641–19644. [DOI] [PubMed] [Google Scholar]
- 32.Schneider, H., B. Guerette, C. Guntermann, and C.E. Rudd. 2000. Resting lymphocyte kinase (Rlk/Txk) targets lymphoid adaptor SLP-76 in the cooperative activation of interleukin-2 transcription in T-cells. J. Biol. Chem. 275:3835–3840. [DOI] [PubMed] [Google Scholar]
- 33.Bunnell, S.C., M. Diehn, M.B. Yaffe, P.R. Findell, L.C. Cantley, and L.J. Berg. 2000. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem. 275:2219–2230. [DOI] [PubMed] [Google Scholar]
- 34.Wu, J., D.G. Motto, G.A. Koretsky, and A. Weiss. 1996. Vav and SLP-76 interact and functionally cooperate in IL-2 gene activation. Immunity. 4:593–602. [DOI] [PubMed] [Google Scholar]
- 35.Onodera, H., D.G. Motto, G.A. Koretzky, and D.M. Rothstein. 1996. Differential regulation of activation-induced tyrosine phosphorylation and recruitment of SLP-76 to Vav by distinct isoforms of the CD45 protein-tyrosine phosphatase. J. Biol. Chem. 271:22225–22230. [DOI] [PubMed] [Google Scholar]
- 36.Wardenburg, J.B., R. Pappu, J.Y. Bu, B. Mayer, J. Chernoff, D. Straus, and A.C. Chan. 1998. Regulation of PAK activation and the T cell cytoskeleton by the linker protein SLP-76. Immunity. 9:607–616. [DOI] [PubMed] [Google Scholar]
- 37.Zeng, R., J. Cannon, R. Abraham, M. Way, D. Billadeau, J. Bubeck-Wardenberg, and J. Burkhardt. 2003. SLP-76 coordinates Nck-dependent Wiskott-Aldrich syndrome protein recruitment with Vav-1-Cdc42-dependent Wiskott-Aldrich syndrome protein activation at the T cell-APC contact site. J. Immunol. 171:1360–1368. [DOI] [PubMed] [Google Scholar]
- 38.Higgs, H.N., and T.D. Pollard. 2001. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu. Rev. Biochem. 70:649–676. [DOI] [PubMed] [Google Scholar]
- 39.Myung, P.S., G.S. Derimanov, M.S. Jordan, J.A. Punt, Q.H. Liu, B.A. Judd, E.E. Meyers, C.D. Sigmund, B.D. Freedman, and G.A. Koretzky. 2001. Differential requirement for SLP-76 domains in T cell development and function. Immunity. 15:1011–1026. [DOI] [PubMed] [Google Scholar]
- 40.Kumar, L., V. Pivniouk, M.A. de la Fuente, D. Laouini, and R.S. Geha. 2002. Differential role of SLP-76 domains in T cell development and function. Proc. Natl. Acad. Sci. USA. 99:884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.da Silva, A.J., Z. Li, C. de Vera, E. Canto, P. Findell, and C.E. Rudd. 1997. Cloning of a novel T-cell protein FYB that binds FYN and SH2-domain-containing leukocyte protein 76 and modulate interleukin-2 production. Proc. Natl. Acad. Sci. USA. 94:7493–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musci, M.A., L.R. Hendricks-Taylor, D.G. Motto, M. Paskind, J. Kamens, C.W. Turck, and G.A. Koretzky. 1997. Molecular cloning of SLAP-130, an SLP-76-associated substrate of the T cell antigen receptor-stimulated protein tyrosine kinases. J. Biol. Chem. 272:11674–11677. [DOI] [PubMed] [Google Scholar]
- 43.da Silva, A.J., J.M. Rosenfield, I. Mueller, A. Bouton, H. Hirai, and C.E. Rudd. 1997. Biochemical analysis of p120/130: a protein-tyrosine kinase substrate restricted to T and myeloid cells. J. Immunol. 158:2007–2016. [PubMed] [Google Scholar]
- 44.Veale, M., M. Raab, Z. Li, A.J. da Silva, S.K. Kraeft, S. Weremowicz, C.C. Morton, and C.E. Rudd. 1999. Novel isoform of lymphoid adaptor FYN-T-binding protein (FYB-130) interacts with SLP-76 and up-regulates interleukin-2 production. J. Biol. Chem. 274:28427–28435. [DOI] [PubMed] [Google Scholar]
- 45.Geng, L., M. Raab, and C.E. Rudd. 1999. Cutting edge: SLP-76 cooperativity with FYB/FYN-T in the up-regulation of TCR-driven IL-2 transcription requires SLP-76 binding to FYB at Tyr595 and Tyr651. J. Immunol. 163:5753–5757. [PubMed] [Google Scholar]
- 46.Raab, M., H. Kang, A. da Silva, X. Zhu, and C.E. Rudd. 1999. FYN-FYB-SLP-76 interactions define a TcRζ/CD3 mediated tyrosine phosphorylation pathway that regulates interleukin-2 transcription in T-cells. J. Biol. Chem. 274:21170–21179. [DOI] [PubMed] [Google Scholar]
- 47.Sauer, K., J. Liou, S. Singh, D. Yablonski, A. Weiss, and R. Perlmutter. 2001. Hematopoietic progenitor kinase 1 associates physically and functionally with the adaptor proteins B cell linker protein and SLP-76 in lymphocytes. J. Biol. Chem. 276:45207–45216. [DOI] [PubMed] [Google Scholar]
- 48.Boerth, N.J., B.A. Judd, and G.A. Koretzky. 2000. Functional association between SLAP-130 and SLP-76 in Jurkat T cells. J. Biol. Chem. 275:5143–5152. [DOI] [PubMed] [Google Scholar]
- 49.Liu, J., H. Kang, M. Raab, A. da Silva, S.-K. Kraeft, and C.E. Rudd. 1998. FYB (FYN binding protein) serves as a binding partner for lymphoid protein and FYN kinase substrate SKAP55 and a SKAP55-related protein in T cells. Proc. Natl. Acad. Sci. USA. 95:8779–8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marie-Cardine, A., L.R. Hendricks-Taylor, N.J. Boerth, H. Zhao, B. Schraven, and G.A. Koretzky. 1998. Molecular interaction between the Fyn-associated protein SKAP55 and the SLP-76-associated phosphoprotein SLAP-130. J. Biol. Chem. 273:25789–25795. [DOI] [PubMed] [Google Scholar]
- 51.Kang, H., C. Freund, J.S. Duke-Cohan, A. Musacchio, G. Wagner, and C.E. Rudd. 2000. SH3 domain recognition of a proline-independent tyrosine-based RKxxYxxY motif in immune cell adaptor SKAP55. EMBO J. 19:2889–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krause, M., M.S. Sechi, M. Konradt, D. Monner, F.B. Gertler, and J. Wehland. 2000. Fyn-binding protein (Fyb)/SLP-76-associated protein, Vasodilator-stimulated phosphoprotein (VASP) proteins and the Arp2/3 complex link TCR signaling to the actin cytoskeleton. J. Cell Biol. 149:181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Griffiths, E.K., C. Krawczyk, Y.-Y. Kong, M. Rabb, S.J. Hyduk, D. Bouchard, V.S. Chan, I. Kozieradski, A.J. Oliveira-dos-Santos, A. Wakeham, et al. 2001. Positive regulation of T cells activation and integrin adhesion by the adapter Fyb/Slap. Science. 293:2260–2263. [DOI] [PubMed] [Google Scholar]
- 54.Peterson, E.J., M.L. Woods, S.A. Dmowski, G. Derimanov, M.S. Jordan, J.N. Wu, P.S. Myung, Q.-H. Liu, J.T. Pribila, B.D. Freedman, et al. 2001. Coupling of the TCR to integrin activation by SLAP-130/Fyb. Science. 293:2263–2265. [DOI] [PubMed] [Google Scholar]
- 55.Hunter, A.J., N. Ottoson, N. Boerth, G.A. Koretzky, and Y. Shimizu. 2000. A novel function for the Slap-130/Fyb adapter protein in Beta-1 integrin signalling and T cell migration. J. Immunol. 164:1143–1147. [DOI] [PubMed] [Google Scholar]
- 56.Geng, L., S. Pfister, S.K. Kraeft, and C.E. Rudd. 2001. Adaptor FYB (Fyn-binding protein) regulates integrin-mediated adhesion and mediator release: differential involvement of the FYB SH3 domain. Proc. Natl. Acad. Sci. USA. 98:11527–11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geng, L., and C.E. Rudd. 2001. Adaptor ADAP (adhesion and degranulation promoting adaptor protein) regulates β-1 integrin clustering on mast cells. Biochem. Biophys. Res. Commun. 289:2042–2050. [DOI] [PubMed] [Google Scholar]
- 58.Fujii, Y., S. Wakahara, T. Nakao, T. Hara, H. Ohtake, T. Komurasaki, K. Kitamura, A. Tatsuno, N. Fujiwara, N. Hozumi, et al. 2003. Targeting of MIST to Src-family kinases via SKAP55-SLAP-130 adaptor complex in mast cells. FEBS Lett. 540:111–116. [DOI] [PubMed] [Google Scholar]
- 59.Wang, H., E.Y. Moon, A. Azouz, X. Wu, A. Smith, H. Schneider, N. Hogg, and C.E. Rudd. 2003. SKAP-55 regulates integrin adhesion and formation of T cell-APC conjugates. Nat. Immunol. 4:366–374. [DOI] [PubMed] [Google Scholar]
- 60.Pear, W.S., G.P. Nolan, M.L. Scott, and D. Baltimore. 1993. Production of high-tite helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 90:8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCann, F.E., B. Vanherberghen, K. Eleme, L.M. Carlin, R.J. Newsam, D. Goulding, and D.M. Davis. 2003. The size of the synaptic cleft and distinct distributions of filamentous actin, ezrin, CD43, and CD45 at activating and inhibitory human NK cell immune synapses. J. Immunol. 170:2862–2870. [DOI] [PubMed] [Google Scholar]
- 62.Michel, F., and O. Acuto. 1996. Induction of T cell adhesion by antigen stimulation and modulation by the coreceptor CD4. J. Immunol. 173:165–175. [DOI] [PubMed] [Google Scholar]
- 63.Van Parijs, L., Y. Refaeli, A.K. Abbas, and D. Baltimore. 1999. Autoimmunity as a consequence of retrovirus-mediated expression of C-FLIP in lymphocytes. Immunity. 11:763–770. [DOI] [PubMed] [Google Scholar]
- 64.Prehoda, K.E., D.J. Lee, and W.A. Lim. 1999. Structure of the enabled/VASP homology 1 domain-peptide complex: a key component in the spatial control of actin assembly. Cell. 97:471–480. [DOI] [PubMed] [Google Scholar]
- 65.van Kooyk, Y., and C. Figdor. 2000. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr. Opin. Cell Biol. 12:542–547. [DOI] [PubMed] [Google Scholar]
- 66.Krawczyk, C., A. Oliveira-dos-Santos, T. Sasaki, E. Griffiths, P.S. Ohashi, S. Snapper, F. Alt, and J.M. Penninger. 2002. Vav1 controls integrin clustering and MHC/peptide-specific cell adhesion to antigen-presenting cells. Immunity. 16:331–343. [DOI] [PubMed] [Google Scholar]
- 67.Cannon, J.L., and J.K. Burkhardt. 2002. The regulation of actin remodeling during T-cell-APC conjugate formation. Immunol. Rev. 186:90–99. [DOI] [PubMed] [Google Scholar]
- 68.Katagiri, K., M. Hattori, N. Minato, and T. Kinashi. 2002. Rap1 functions as a key regulator of T-cell and antigen-presenting cell interactions and modulates T-cell responses. Mol. Cell. Biol. 22:1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anton, I.M., M. de la Fuente, T.N. Sims, S. Freeman, N. Ramesh, J.H. Hartwig, M.L. Dustin, and R.S. Geha. 2002. WIP deficiency reveals a differential role for WIP and the actin cytoskeleton in T and B cell activation. Immunity. 16:193–204. [DOI] [PubMed] [Google Scholar]
- 70.Bear, J.E., T.M. Svitkina, M. Krause, D.A. Schafer, J.J. Loureiro, G.A. Strasser, I.V. Maly, O.Y. Chaga, J.A. Copper, and F.B. Gertler. 2002. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 109:509–521. [DOI] [PubMed] [Google Scholar]
- 71.Jacobelli, J., S.A. Chmura, D.B. Buxton, M.M. Davis, and M.F. Krummel. 2004. A single class II myosin modulates T cell motility and stopping, but not synapse formation. Nat. Immunol. 5:531–538. [DOI] [PubMed] [Google Scholar]
- 72.Snapper, S.B., F.S. Rosen, E. Mizoguchi, P. Cohen, W. Khan, C.H. Liu, T.L. Hagemann, S.P. Kwan, R. Ferrini, L. Davidson, et al. 1998. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 9:81–91. [DOI] [PubMed] [Google Scholar]
- 73.Huppa, J.B., and M.M. Davis. 2003. T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 3:973–983. [DOI] [PubMed] [Google Scholar]