

Abstract
Ataxia telangiectasia mutated (ATM) kinase is critical for initiating the signaling pathways that lead to cell cycle checkpoints and DNA double strand break repair. In the absence of ATM, humans and mice show a primary immunodeficiency that includes low serum antibody titers, but the role of ATM in antigen-driven immunoglobulin gene diversification has not been defined. Here, we show that although ATM is dispensable for somatic hypermutation, it is required for efficient class switch recombination (CSR). The defect in CSR is not due to alterations in switch region transcription, accessibility, DNA damage checkpoint protein recruitment, or short-range intra-switch region recombination. Only long-range inter-switch recombination is defective, indicating an unexpected role for ATM in switch region synapsis during CSR.
Keywords: class switch recombination, somatic hypermutation, activation-induced, cytidine deaminase, ATM, DNA repair
Introduction
In mature B cells responding to antigen, antibody genes are diversified by somatic hypermutation (SHM; 1) and class switch recombination (CSR; 2, 3). Both of these reactions are initiated by activation-induced cytidine deaminase (AID; 4, 5), an enzyme that deaminates cytidines in single stranded DNA (6–13). SHM alters the antigen binding specificity of antibodies by introducing point mutations in Ig variable regions (1). CSR is a region-specific deletional recombination reaction that joins large repetitive switch region sequences located upstream of each Ig constant region (2, 3). CSR replaces the upstream constant region (Cμ) with a downstream CH gene (γ, ɛ, or α), thereby producing an Ig with a new set of effector functions while retaining the original antigen-binding specificity (2, 3).
Specific switch regions are targeted for recombination by cytokine-induced transcription from intronic (I) promoters located 5′ of each switch region. These promoters are essential for CSR (14–19), possibly because they facilitate switch region accessibility to AID and expose single stranded DNA during the transcription reaction (9–13, 20, 21). AID has been proposed to initiate CSR by creating dU:dG mismatches that are processed by uracyl-DNA glycosylase (6–8) and mismatch repair enzymes (22–27) to produce DNA double strand break (DSB) intermediates. However, the exact mechanism by which these lesions are generated remains unclear (28). Ultimately, DNA breaks occurring in both donor and acceptor S regions are repaired by nonhomologous end joining (29–32), and the intervening DNA sequences are released as a circular episome (33–35).
Switch region DNA DSBs become associated with phosphorylated histone H2AX (γ-H2AX; 32, 36, 37). This modified histone forms foci at sites of CSR that are believed to facilitate the focal accumulation of several additional DNA repair factors (36) like 53BP1, which is required for CSR (38, 39). Although the role of these foci in CSR has not been determined, efficient switching requires H2AX (32, 36, 37).
H2AX is one of many substrates of the phosphatydil-inositol-3 kinase-like kinase (PIKK) family of proteins that includes ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR), and DNA-dependent protein kinase (DNA-PK; 40). These kinases are the prototype transducers of the DNA damage signal (40), and DNA-PK (consisting of DNA-PK catalytic subunit [DNA-PKcs], Ku80 and Ku70 subunits) is required for CSR (29–32), but the role of ATM and ATR in CSR has not been determined. ATM deficiency in humans and mice causes multi-organ pathology, including a primary immunodeficiency characterized by low serum levels of switched antibodies (41–45). Here, we report that ATM is required for efficient joining of distal switch regions.
Materials and Methods
Mice and Immunizations.
WT (C57BL/6), ATM−/− (43), Ku80−/− carrying prerearranged heavy and light chains (31), and H2AX−/− (37) mice were bred and maintained under specific pathogen-free conditions. Age-matched 8–10-wk-old mice were immunized by footpad injection with 50 μg NP-CGG (Biosearch Technologies) in complete Freund's adjuvant. All work with mice was performed under Rockefeller University Institutional Animal Care and Use Committee–approved protocols.
Lymphocyte Cultures and Cell Sorting.
B lymphocytes were isolated from the spleen using CD43 microbeads (Miltenyi Biotec), labeled with 5 μM CFDA-SE for 10 min at 37°C (Molecular Probes), and cultured (106 cells/ml) with 25 μg/ml LPS alone or in combination with 5 ng/ml IL-4 for 5 d. Lymph nodes were dissected before or after immunization. Germinal center B cells were stained with APC anti-B220, FITC anti-GL7, and PE anti-FAS monoclonal antibodies (BD Biosciences). In all cell sorting experiments, 0.5 μg/ml propidium iodide was added immediately before laser excitation to exclude dead cells. Cell sorting was performed on a FACSVantage (Becton Dickinson), and an aliquot of each of the sorted fractions was reanalyzed for purity on a FACSCalibur (Becton Dickinson).
Hybridoma Analysis.
B cells were stimulated with LPS and IL-4 for 72 h and fused to the SP2/0Ag-14 myeloma cell line. IgM-secreting clones were selected by ELISA for further analysis. Genomic DNA was prepared and Southern blot analysis was performed as described previously (32).
PCR, Mutation Analysis, and Quantitative Real-Time RT-PCR.
Genomic DNA was amplified by PCR using Pfu Turbo DNA polymerase (Stratagene) from 5,000 sorted cell equivalents in four independent reactions that were pooled for cloning experiments. Total RNA was extracted with TRIzol (Invitrogen) and reverse transcribed with random hexamers and superscript II reverse transcriptase (Invitrogen). First strand cDNA was used for SYBR Green fluorogenic dye real-time PCR (Applied Biosystems). The primers and PCR conditions that were used have been described (32).
53BP1 Focus Analysis.
After 48 h of stimulation, B cells were spun onto coverslips and processed for immunocytochemistry-FISH as described previously (36). Rabbit polyclonal anti-53BP1 antibodies (provided by J. Chen, Mayo Clinic, Rochester, MN) were used at a 1:1,000 dilution. The IgH-specific BAC probe spanned from Cγ1 to 3′ of Cα (36).
Results
ATM Is Required for CSR, But Not SHM.
To determine whether ATM is required for CSR, we stimulated ATM−/− and WT B cells in vitro with LPS alone or in combination with IL-4. Cells were labeled with CFSE to follow cell division, and CSR was measured by cell surface expression of IgG1, IgG2b, and IgG3 (Fig. 1, A–C). Despite the reported growth defect in ATM−/− mice (42–44), we found that the rates of cell division and death in ATM−/− B cells were indistinguishable from WT (Fig. 1 A, top, and not depicted). Nevertheless, ATM−/− B cells showed an average threefold reduction of CSR in all three isotypes analyzed (Fig. 1, A–C, bottom; n = 7 experiments). We conclude that ATM is required for normal CSR, but not for B cell proliferation in response to LPS or LPS and IL-4.
Figure 1.

ATM is required for efficient CSR, but not for SHM. Flow cytometry analysis of WT and ATM−/− B cells stimulated with LPS plus IL-4 for 4 d (A) or LPS alone (B and C) for 5 d. Cell division as measured by CFSE dye dilution is shown on the top. The percentage of cells expressing (A) IgG1, (B) IgG2b, and (C) IgG3 after a specific number of cell divisions is indicated on the bottom. Cells were stained with Topro-3 before acquisition and analysis was performed on live B cells (Topro-3−). (D) Mutation analysis in the JH4 intron (reference 46) of WT and ATM−/− germinal center B cells (B220+ Fas+ GL-7+) obtained from the lymph nodes of immunized mice. Segment sizes in the pie charts are proportional to the number of sequences carrying the number of mutations indicated in the periphery of the charts. The frequency of mutations per basepair sequenced and the total number of independent sequences analyzed is indicated underneath and in the center of each chart, respectively. Statistical significance was determined by a two-tailed t test assuming unequal variance and comparing to WT (P = 0.914). Percent nucleotide substitutions adjusted for base composition is shown to the right of each pie chart. Percentage of mutations within hotspot motifs (references 81–83) is indicated underneath each panel. The total number of mutations analyzed was as follows: WT, 88 mutations/28,120 bp; ATM−/−, 63 mutations/20,920 bp.
To determine whether ATM is required for SHM, we immunized ATM−/− and WT control mice with NP-CGG and cloned and sequenced the JH4 intron (46) from sorted germinal center B cells (the B220+ Fas+ GL-7+ population). We found no differences in mutation frequencies (WT: 3.6 × 10−3 vs. ATM−/−: 3.0 × 10−3; P = 0.914) or in the proportion of mutated clones (Fig. 1 D). Furthermore, there was no significant bias in the nucleotide substitution patterns found in JH4 sequences cloned from ATM−/− germinal center B cells (Fig. 1 D). Thus, ATM is dispensable for SHM.
Switch Region Transcription and Mutation.
Switch region transcription plays an essential role in CSR (2, 3). To determine whether ATM deficiency alters switch region transcription, we measured Sμ and Sγ1 sterile transcripts in B cells stimulated with LPS and IL-4 by real-time RT-PCR (Fig. 2 A). We found that Sμ and Sγ1 sterile transcripts were similar in ATM−/− and control B cells (Fig. 2 A). However, the IgG1 post-switch transcripts produced by the circular episomes created by productive CSR (47) were reduced an average of 2.3-fold in ATM-deficient B cells (Fig. 2 A; n = 4 experiments). Thus, although sterile transcription of IgM and IgG1 switch regions is not altered in the absence of ATM, post-switch transcripts are reduced in proportion to the reduced frequency of CSR.
Figure 2.

Switch region accessibility in the absence of ATM. (A) Real-time RT-PCR for μ sterile transcript (μ ST), γ1 sterile transcript (γ1 ST), and post-switch γ1 circle transcript (γ1 CT) in WT (closed bars) and ATM−/− (open bars) B cells stimulated with LPS and IL-4 for 3 d. Mean results from four independent cultures are expressed as fold induction relative to WT. (B) Mutations in Sμ determined in WT and ATM−/− B cells sorted for five cell divisions and expressing IgM. The number of mutations was as follows: WT, 31 mutations/83,773 bp; ATM−/−, 14 mutations/55,069 bp. Pie charts are as in Fig. 1. Statistical significance was determined by a two-tailed t test assuming unequal variance and comparing to background (resting B cells from WT mice) or WT. P-values are indicated below each pie chart.
DNA sequences located upstream of the Ig switch regions are mutated by an AID-dependent mechanism in B cells undergoing CSR (32, 36, 48, 49), and these mutations have been used to measure AID targeting to switch region DNA. To determine whether ATM is required for Sμ mutation, we analyzed mutation frequencies in B cells induced to switch in vitro with LPS and IL-4. The analysis was performed on sorted cells that were IgM+ and had completed five cell divisions. We found similar levels of Sμ mutation in ATM−/− and WT B cells (WT: 3.7 × 10−4 vs. ATM−/−: 2.5 × 10−4; P = 0.55; Fig. 2 B). We conclude that switch regions are accessible to and targeted by AID in the absence of ATM.
CSR Junctions.
Although CSR is inefficient in the absence of ATM, some cells do switch to IgG1 in response to LPS and IL-4 (Fig. 1 A). To determine whether these CSR junctions are normal, we cloned and sequenced IgG1 CSR junctions from WT (n = 40) and ATM−/− (n = 32) B cells. Analysis of the ATM−/− switch junctions revealed no significant differences in the extent of donor/acceptor homology at the junctions or the average length of overlap (1.9 bp in ATM−/− and 2 bp in controls; Fig. 3 A). The only notable difference was a slightly lower mutation frequency in the vicinity of the junctions (± 50 bp) in ATM−/− B cells (WT: 3.2 × 10−2 vs. ATM−/−: 2.0 × 10−2; P = 0.01; Fig. 3 B). We conclude that switch region joining is not significantly altered in the absence of ATM.
Figure 3.

CSR junctions in the absence of ATM. (A) Histogram depicting the percentage of sequences with the indicated length of microhomologies at Sμ/Sγ1 junctions in WT (closed bars) and ATM−/− (open bars) B cells. Overlap was determined by identifying the longest region at the switch junction of perfect uninterrupted donor/acceptor identity. (B) Mutations in the vicinity of the junctions obtained from WT and ATM−/− B cells. Pie charts are as in Fig. 1. Statistical significance was determined by a two-tailed t test assuming unequal variance and comparing to WT. P-values are indicated below each pie chart.
Intra-Switch Region Recombination.
B cells stimulated to undergo CSR suffer frequent internal deletions in Sμ region DNA (15, 50–52). These deletions are AID dependent (32, 53) and repaired like bona fide CSR through the nonhomologous end joining pathway (32). To determine whether ATM is required for this intra-switch region recombination, we examined Sμ and Sγ1 switch regions from IgM-expressing hybridomas derived from LPS plus IL-4–stimulated ATM−/− B cells. By Southern blotting, 11 out of 54 ATM−/− hybridomas showed internal deletions in Sμ (Fig. 4 A), and only 1 of these also showed an internal deletion in Sγ1 (Fig. 4 B). The frequency of internal deletions in Sμ (20%) and Sγ1 (2%) in the ATM−/− hybridomas was similar to that found in hybridomas derived from WT B cells (32). We conclude that in the absence of ATM, intra-switch recombination proceeds normally. Furthermore, because both intra-switch recombination and CSR junctions appeared to be normal in ATM−/− B cells, we conclude that switch DNA end joining is not impaired in the absence of ATM. Consistent with this, ATM−/− cells also support normal DNA end joining in V(D)J recombination (45, 54, 55).
Figure 4.

Intra-switch region recombination in the absence of ATM. Southern blot analysis of (A) Sμ and (B) Sγ1 regions in IgM-secreting hybridomas derived from ATM−/− B cells. Restriction enzymes and probes used are indicated in the top panels. Δ, deletions. Control digests performed on tail DNA (ATM−/−) and the SP2/0Ag-14 (SP2) fusion partner were loaded on lanes 1 and 2. The SP2 cell line has a deletion in Cμ and no hybridization is observed. The same deletions in Sμ were found using an Eμ probe (not depicted). Number of deletions over hybridomas screened is indicated below each panel. Molecular weight markers in kilobase pairs are indicated on the left side of each panel.
ATM and DNA Repair Focus Formation.
53BP1 is a DNA repair factor that like H2AX, is an ATM substrate (56, 57) and is required for CSR (38, 39). In irradiated cells, 53BP1 forms H2AX-dependent foci in areas of DNA damage (37, 58, 59). To determine whether 53BP1 forms CSR-associated foci and whether these are ATM dependent, we examined B cells undergoing CSR using antibodies to 53BP1 and a DNA probe spanning the IgH locus (36). After 2 d of stimulation with LPS and IL-4, 18% of B cells in a given optical section contained at least one 53BP1 focus and a signal from one or both IgH alleles (n = 1,028 cells examined). Coincidence of these 53BP1 foci with one or both IgH alleles was detected in 69% of the cells (n = 185 cells examined; Fig. 5). In contrast, 53BP1 foci were absent from H2AX−/− B cells (n = 1,014 cells examined; Fig. 5), which is in agreement with the observation that phosphorylation of H2AX is required for 53BP1 focus formation in response to γ irradiation (37, 58, 59). Thus, 53BP1 does accumulate at sites of DNA damage during CSR and this accumulation is H2AX dependent.
Figure 5.
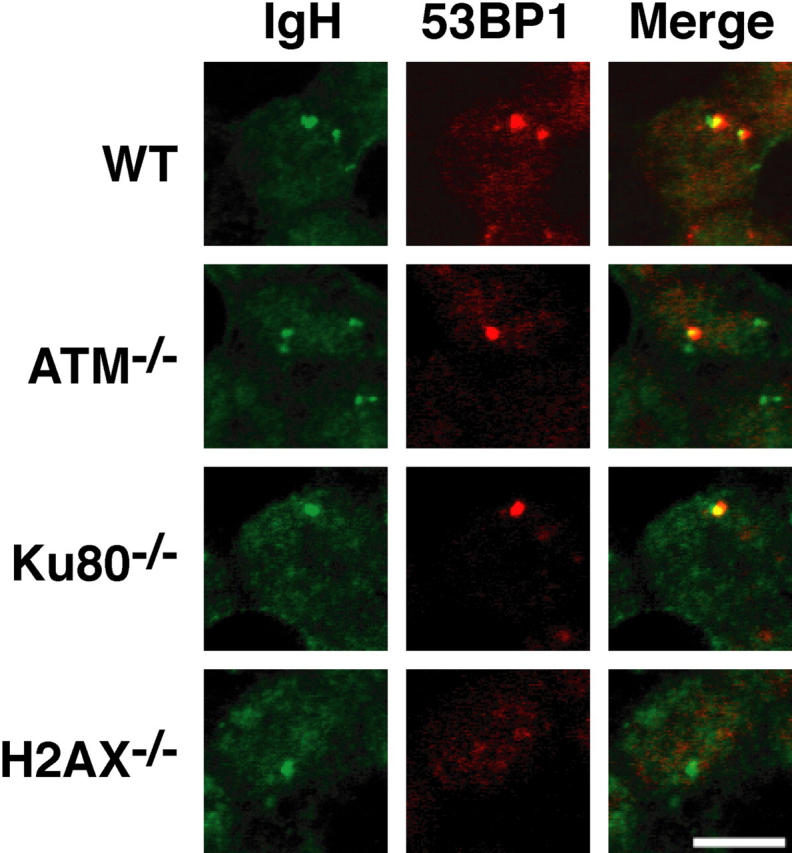
Activation of 53BP1 in response to CSR-associated DSBs. Distribution of 53BP1 in activated B cells from WT, ATM−/−, Ku80−/−, and H2AX−/− mice. B cells were stained with anti-53BP1 antibodies followed by DNA FISH detection of the CH region. Fluorescent images represent a single optical section.
To determine whether CSR foci are ATM or DNA-PK dependent, we assayed for 53BP1 focus formation at the IgH locus in ATM−/− and Ku80−/− B cells. In the absence of Ku80, the catalytic subunit of DNA-PK is not targeted to DSBs, leaving the DNA-dependent kinase inactive (60). In contrast to H2AX−/− B cells, ATM−/− and Ku80−/− B cells assembled CSR-associated 53BP1 foci (Fig. 5). The percentage of cells containing 53BP1/IgH foci that showed colocalization was 57% in ATM−/− B cells (n = 1,145 cells examined) and 66% in Ku80−/− B cells (n = 1,273 cells examined). We conclude that despite the requirement for both ATM and DNA-PK in CSR, neither of these enzymes is essential for CSR-associated repair protein focus formation.
Discussion
We found that ATM, a central regulator of the DNA damage response, is required for efficient CSR. ATM does not regulate transcription of donor and acceptor switch regions, nor switch region accessibility to AID. It is not required for DNA repair protein focus formation or intra-switch region recombination. Analysis of switch recombination junctions revealed only a small decrease in the mutation frequency in the vicinity of CSR junctions, which was also found in ATM-deficient human B cells (61). Therefore, ATM is entirely dispensable for targeting, lesion formation, and intra-switch region recombination during CSR. Our results are consistent with a role for ATM in promoting joining between distant switch regions.
CSR is a deletional recombination reaction with double strand DNA break intermediates resolved by the nonhomologous end joining DNA repair pathway (29–32). However, DNA damage in CSR is not limited to DSBs and is thought to include U:G mismatches, single strand DNA produced during lesion processing, R loops, and single strand nicks (2, 3, 6–8, 21, 62). Although evidence for the involvement of base excision repair and mismatch repair enzymes in the processing of AID-induced lesions has been provided (7, 22–27), it remains unclear how DSBs are generated (28). Single and double strand DNA breaks trigger repair pathways that depend on activation of PIKK kinases ATM and ATR. These kinases recognize DNA damage in conjunction with additional DNA binding proteins: replication protein A, a single strand DNA binding protein that recruits ATR (63) and has been implicated in targeting AID to Ig genes (64), and the complex of Mre11, Rad50, and Nbs1 (MRN), which is required for maximal activation of ATM (65). Consistent with the idea that ATM is dependent on MRN, mutations in Nbs1 or Mre11 produce human disease phenotypes similar to mutations of ATM (66–68).
Once activated, PIKK kinases phosphorylate numerous cellular substrates (40), including histone H2AX and 53BP1 (56, 57, 69–71). H2AX and 53BP1 play important roles in DSB repair because deletion of either gene leads to irradiation sensitivity, genomic instability, increased tumor susceptibility, and a decrease in CSR (32, 36, 37, 72–75). γ-H2AX spreads over megabase domains that flank the break facilitating DNA repair by remodeling chromatin and tethering additional repair factors such as MRN and 53BP1 to damaged DNA (76, 77). In the absence of γ-H2AX, these factors recognize and bind damaged DNA, but they are not retained at the site of DNA damage and do not appear to spread along the chromosome as evidenced by lack of DNA damage–associated focus formation (58, 59). Thus, DNA damage–associated chromatin remodeling is impaired in the absence of H2AX. Aberrant chromatin remodeling may also account for the specific defect in joining of distant switch regions in the absence of H2AX (32).
We have found that CSR-associated 53BP1 foci are dependent on γ-H2AX, but form independently of ATM and DNA-PK activity. Therefore, during CSR, H2AX must be phosphorylated by a PIKK kinase with overlapping specificity, which could be ATM, DNA-PK, or ATR (39). Indeed, in irradiated fibroblasts, ATM and DNA-PK function redundantly in γ-H2AX focus formation (78). If γ-H2AX is phosphorylated by a redundant kinase and 53BP1 is recruited, why is there a defect in distal switch region joining in the absence of ATM? One possibility is that ATM, like DNA-PKcs, has a structural role in CSR in addition to its catalytic function. DNA-PKcs catalytic mutants have a milder CSR phenotype than complete deletion of DNA-PKcs (79, 80). A second possibility is that additional ATM kinase substrates involved in CSR are uniquely phosphorylated by ATM and are required for switch region synapsis. In conclusion, ATM deficiency leads to a primary defect in CSR that may account for the low serum antibody levels seen in A-T patients.
Acknowledgments
We thank members of the Michel and André Nussenzweig laboratories and E. Besmer for discussions, K. Velinzon for cell sorting, F. Weiss-Garcia for hybridoma fusions, and P. Stavropoulos for screening and maintenance of hybridomas.
This work was supported in part by grants from National Institutes of Health to M.C. Nussenzweig. M.C. Nussenzweig is a Howard Hughes Medical Institute (HHMI) investigator. B. Reina-San-Martin is an HHMI postdoctoral associate.
The authors have no conflicting financial interests.
Abbreviations used in this paper: AID, activation-induced cytidine deaminase; ATM, ataxia telangiectasia mutated; ATR, ATM and Rad3 related; CSR, class switch recombination; DNA-PK, DNA-dependent protein kinase; DNA-PKcs, DNA-PK catalytic subunit; DSB, double strand breaks; MRN, complex of Mre11, Rad50, and Nbs1; PIKK, phosphatydil-inositol-3 kinase-like kinase; SHM, somatic hypermutation.
References
- 1.McKean, D., K. Huppi, M. Bell, L. Staudt, W. Gerhard, and M. Weigert. 1984. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA. 81:3180–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stavnezer, J. 1996. Immunoglobulin class switching. Curr. Opin. Immunol. 8:199–205. [DOI] [PubMed] [Google Scholar]
- 3.Manis, J.P., M. Tian, and F.W. Alt. 2002. Mechanism and control of class-switch recombination. Trends Immunol. 23:31–39. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 5.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 6.Petersen-Mahrt, S.K., R.S. Harris, and M.S. Neuberger. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–103. [DOI] [PubMed] [Google Scholar]
- 7.Rada, C., G.T. Williams, H. Nilsen, D.E. Barnes, T. Lindahl, and M.S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755. [DOI] [PubMed] [Google Scholar]
- 8.Di Noia, J., and M.S. Neuberger. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 419:43–48. [DOI] [PubMed] [Google Scholar]
- 9.Bransteitter, R., P. Pham, M.D. Scharff, and M.F. Goodman. 2003. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA. 100:4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramiro, A.R., P. Stavropoulos, M. Jankovic, and M.C. Nussenzweig. 2003. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4:452–456. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhuri, J., M. Tian, C. Khuong, K. Chua, E. Pinaud, and F.W. Alt. 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 422:726–730. [DOI] [PubMed] [Google Scholar]
- 12.Dickerson, S.K., E. Market, E. Besmer, and F.N. Papavasiliou. 2003. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 197:1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larson, E.D., and N. Maizels. 2004. Transcription-coupled mutagenesis by the DNA deaminase AID. Genome Biol. 5:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stavnezer-Nordgren, J., and S. Sirlin. 1986. Specificity of immunoglobulin heavy chain switch correlates with activity of germline heavy chain genes prior to switching. EMBO J. 5:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu, H., Y.R. Zou, and K. Rajewsky. 1993. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 73:1155–1164. [DOI] [PubMed] [Google Scholar]
- 16.Jung, S., K. Rajewsky, and A. Radbruch. 1993. Shutdown of class switch recombination by deletion of a switch region control element. Science. 259:984–987. [DOI] [PubMed] [Google Scholar]
- 17.Seidl, K.J., J.P. Manis, A. Bottaro, J. Zhang, L. Davidson, A. Kisselgof, H. Oettgen, and F.W. Alt. 1999. Ponatsition-dependent inhibition of class-switch recombination by PGK-neor cassettes inserted into the immunoglobulin heavy chain constant region locus. Proc. Natl. Acad. Sci. USA. 96:3000–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu, L., B. Gorham, S.C. Li, A. Bottaro, F.W. Alt, and P. Rothman. 1993. Replacement of germ-line epsilon promoter by gene targeting alters control of immunoglobulin heavy chain class switching. Proc. Natl. Acad. Sci. USA. 90:3705–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang, J., A. Bottaro, S. Li, V. Stewart, and F.W. Alt. 1993. A selective defect in IgG2b switching as a result of targeted mutation of the I gamma 2b promoter and exon. EMBO J. 12:3529–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nambu, Y., M. Sugai, H. Gonda, C.G. Lee, T. Katakai, Y. Agata, Y. Yokota, and A. Shimizu. 2003. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 302:2137–2140. [DOI] [PubMed] [Google Scholar]
- 21.Yu, K., F. Chedin, C.L. Hsieh, T.E. Wilson, and M.R. Lieber. 2003. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 4:442–451. [DOI] [PubMed] [Google Scholar]
- 22.Ehrenstein, M.R., and M.S. Neuberger. 1999. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. EMBO J. 18:3484–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schrader, C.E., W. Edelmann, R. Kucherlapati, and J. Stavnezer. 1999. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 190:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehrenstein, M.R., C. Rada, A.M. Jones, C. Milstein, and M.S. Neuberger. 2001. Switch junction sequences in PMS2-deficient mice reveal a microhomology-mediated mechanism of Ig class switch recombination. Proc. Natl. Acad. Sci. USA. 98:14553–14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schrader, C.E., J. Vardo, and J. Stavnezer. 2002. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J. Exp. Med. 195:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin, A., Z. Li, D.P. Lin, P.D. Bardwell, M.D. Iglesias-Ussel, W. Edelmann, and M.D. Scharff. 2003. Msh2 ATPase activity is essential for somatic hypermutation at A-T basepairs and for efficient class switch recombination. J. Exp. Med. 198:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bardwell, P.D., C.J. Woo, K. Wei, Z. Li, A. Martin, S.Z. Sack, T. Parris, W. Edelmann, and M.D. Scharff. 2004. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat. Immunol. 5:224–229. [DOI] [PubMed] [Google Scholar]
- 28.Honjo, T., M. Muramatsu, and S. Fagarasan. 2004. AID: how does it aid antibody diversity? Immunity. 20:659–668. [DOI] [PubMed] [Google Scholar]
- 29.Rolink, A., F. Melchers, and J. Andersson. 1996. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 5:319–330. [DOI] [PubMed] [Google Scholar]
- 30.Manis, J.P., Y. Gu, R. Lansford, E. Sonoda, R. Ferrini, L. Davidson, K. Rajewsky, and F.W. Alt. 1998. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 187:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casellas, R., A. Nussenzweig, R. Wuerffel, R. Pelanda, A. Reichlin, H. Suh, X.F. Qin, E. Besmer, A. Kenter, K. Rajewsky, and M.C. Nussenzweig. 1998. Ku80 is required for immunoglobulin isotype switching. EMBO J. 17:2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reina-San-Martin, B., S. Difilippantonio, L. Hanitsch, R.F. Masilamani, A. Nussenzweig, and M.C. Nussenzweig. 2003. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 197:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwasato, T., A. Shimizu, T. Honjo, and H. Yamagishi. 1990. Circular DNA is excised by immunoglobulin class switch recombination. Cell. 62:143–149. [DOI] [PubMed] [Google Scholar]
- 34.Matsuoka, M., K. Yoshida, T. Maeda, S. Usuda, and H. Sakano. 1990. Switch circular DNA formed in cytokine-treated mouse splenocytes: evidence for intramolecular DNA deletion in immunoglobulin class switching. Cell. 62:135–142. [DOI] [PubMed] [Google Scholar]
- 35.von Schwedler, U., H.M. Jack, and M. Wabl. 1990. Circular DNA is a product of the immunoglobulin class switch rearrangement. Nature. 345:452–456. [DOI] [PubMed] [Google Scholar]
- 36.Petersen, S., R. Casellas, B. Reina-San-Martin, H.T. Chen, M.J. Difilippantonio, P.C. Wilson, L. Hanitsch, A. Celeste, M. Muramatsu, D.R. Pilch, et al. 2001. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 414:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Celeste, A., S. Petersen, P.J. Romanienko, O. Fernandez-Capetillo, H.T. Chen, O.A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M.J. Difilippantonio, et al. 2002. Genomic instability in mice lacking histone H2AX. Science. 296:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward, I.M., B. Reina-San-Martin, A. Olaru, K. Minn, K. Tamada, J.S. Lau, M. Cascalho, L. Chen, A. Nussenzweig, F. Livak, et al. 2004. 53BP1 is required for class switch recombination. J. Cell Biol. 165:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manis, J.P., J.C. Morales, Z. Xia, J.L. Kutok, F.W. Alt, and P.B. Carpenter. 2004. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol. 5:481–487. [DOI] [PubMed] [Google Scholar]
- 40.Shiloh, Y. 2003. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 3:155–168. [DOI] [PubMed] [Google Scholar]
- 41.Savitsky, K., A. Bar-Shira, S. Gilad, G. Rotman, Y. Ziv, L. Vanagaite, D.A. Tagle, S. Smith, T. Uziel, S. Sfez, et al. 1995. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 268:1749–1753. [DOI] [PubMed] [Google Scholar]
- 42.Xu, Y., T. Ashley, E.E. Brainerd, R.T. Bronson, M.S. Meyn, and D. Baltimore. 1996. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 10:2411–2422. [DOI] [PubMed] [Google Scholar]
- 43.Barlow, C., S. Hirotsune, R. Paylor, M. Liyanage, M. Eckhaus, F. Collins, Y. Shiloh, J.N. Crawley, T. Ried, D. Tagle, and A. Wynshaw-Boris. 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 86:159–171. [DOI] [PubMed] [Google Scholar]
- 44.Elson, A., Y. Wang, C.J. Daugherty, C.C. Morton, F. Zhou, J. Campos-Torres, and P. Leder. 1996. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc. Natl. Acad. Sci. USA. 93:13084–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borghesani, P.R., F.W. Alt, A. Bottaro, L. Davidson, S. Aksoy, G.A. Rathbun, T.M. Roberts, W. Swat, R.A. Segal, and Y. Gu. 2000. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc. Natl. Acad. Sci. USA. 97:3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jolly, C.J., N. Klix, and M.S. Neuberger. 1997. Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res. 25:1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kinoshita, K., M. Harigai, S. Fagarasan, M. Muramatsu, and T. Honjo. 2001. A hallmark of active class switch recombination: transcripts directed by I promoters on looped-out circular DNAs. Proc. Natl. Acad. Sci. USA. 98:12620–12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagaoka, H., M. Muramatsu, N. Yamamura, K. Kinoshita, and T. Honjo. 2002. Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin Smu region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J. Exp. Med. 195:529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schrader, C.E., S.P. Bradley, J. Vardo, S.N. Mochegova, E. Flanagan, and J. Stavnezer. 2003. Mutations occur in the Ig Smu region but rarely in Sgamma regions prior to class switch recombination. EMBO J. 22:5893–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hummel, M., J.K. Berry, and W. Dunnick. 1987. Switch region content of hybridomas: the two spleen cell Igh loci tend to rearrange to the same isotype. J. Immunol. 138:3539–3548. [PubMed] [Google Scholar]
- 51.Winter, E., U. Krawinkel, and A. Radbruch. 1987. Directed Ig class switch recombination in activated murine B cells. EMBO J. 6:1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bottaro, A., F. Young, J. Chen, M. Serwe, F. Sablitzky, and F.W. Alt. 1998. Deletion of the IgH intronic enhancer and associated matrix-attachment regions decreases, but does not abolish, class switching at the mu locus. Int. Immunol. 10:799–806. [DOI] [PubMed] [Google Scholar]
- 53.Dudley, D.D., J.P. Manis, A.A. Zarrin, L. Kaylor, M. Tian, and F.W. Alt. 2002. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proc. Natl. Acad. Sci. USA. 99:9984–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pergola, F., M.Z. Zdzienicka, and M.R. Lieber. 1993. V(D)J recombination in mammalian cell mutants defective in DNA double-strand break repair. Mol. Cell. Biol. 13:3464–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsieh, C.L., C.F. Arlett, and M.R. Lieber. 1993. V(D)J recombination in ataxia telangiectasia, Bloom's syndrome, and a DNA ligase I-associated immunodeficiency disorder. J. Biol. Chem. 268:20105–20109. [PubMed] [Google Scholar]
- 56.Anderson, L., C. Henderson, and Y. Adachi. 2001. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol. Cell. Biol. 21:1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rappold, I., K. Iwabuchi, T. Date, and J. Chen. 2001. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell Biol. 153:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernandez-Capetillo, O., H.T. Chen, A. Celeste, I. Ward, P.J. Romanienko, J.C. Morales, K. Naka, Z. Xia, R.D. Camerini-Otero, N. Motoyama, et al. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4:993–997. [DOI] [PubMed] [Google Scholar]
- 59.Celeste, A., O. Fernandez-Capetillo, M.J. Kruhlak, D.R. Pilch, D.W. Staudt, A. Lee, R.F. Bonner, W.M. Bonner, and A. Nussenzweig. 2003. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5:675–679. [DOI] [PubMed] [Google Scholar]
- 60.Finnie, N.J., T.M. Gottlieb, T. Blunt, P.A. Jeggo, and S.P. Jackson. 1995. DNA-dependent protein kinase activity is absent in xrs-6 cells: implications for site-specific recombination and DNA double-strand break repair. Proc. Natl. Acad. Sci. USA. 92:320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pan, Q., C. Petit-Frere, A. Lahdesmaki, H. Gregorek, K.H. Chrzanowska, and L. Hammarstrom. 2002. Alternative end joining during switch recombination in patients with ataxia-telangiectasia. Eur. J. Immunol. 32:1300–1308. [DOI] [PubMed] [Google Scholar]
- 62.Tian, M., and F.W. Alt. 2000. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J. Biol. Chem. 275:24163–24172. [DOI] [PubMed] [Google Scholar]
- 63.Zou, L., D. Liu, and S.J. Elledge. 2003. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA. 100:13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chaudhuri, J., C. Khuong, and F.W. Alt. 2004. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 430:992–998. [DOI] [PubMed] [Google Scholar]
- 65.Lee, J.H., and T.T. Pull. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 304:93–96. [DOI] [PubMed] [Google Scholar]
- 66.Carney, J.P., R.S. Maser, H. Olivares, E.M. Davis, M. Le Beau, J.R. Yates III, L. Hays, W.F. Morgan, and J.H. Petrini. 1998. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 93:477–486. [DOI] [PubMed] [Google Scholar]
- 67.Varon, R., C. Vissinga, M. Platzer, K.M. Cerosaletti, K.H. Chrzanowska, K. Saar, G. Beckmann, E. Seemanova, P.R. Cooper, N.J. Nowak, et al. 1998. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 93:467–476. [DOI] [PubMed] [Google Scholar]
- 68.Stewart, G.S., R.S. Maser, T. Stankovic, D.A. Bressan, M.I. Kaplan, N.G. Jaspers, A. Raams, P.J. Byrd, J.H. Petrini, and A.M. Taylor. 1999. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 99:577–587. [DOI] [PubMed] [Google Scholar]
- 69.Rogakou, E.P., D.R. Pilch, A.H. Orr, V.S. Ivanova, and W.M. Bonner. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 70.Paull, T.T., E.P. Rogakou, V. Yamazaki, C.U. Kirchgessner, M. Gellert, and W.M. Bonner. 2000. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10:886–895. [DOI] [PubMed] [Google Scholar]
- 71.Burma, S., B.P. Chen, M. Murphy, A. Kurimasa, and D.J. Chen. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276:42462–42467. [DOI] [PubMed] [Google Scholar]
- 72.Bassing, C.H., K.F. Chua, J. Sekiguchi, H. Suh, S.R. Whitlow, J.C. Fleming, B.C. Monroe, D.N. Ciccone, C. Yan, K. Vlasakova, et al. 2002. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA. 99:8173–8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ward, I.M., K. Minn, J. van Deursen, and J. Chen. 2003. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23:2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morales, J.C., Z. Xia, T. Lu, M.B. Aldrich, B. Wang, C. Rosales, R.E. Kellems, W.N. Hittelman, S.J. Elledge, and P.B. Carpenter. 2003. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J. Biol. Chem. 278:14971–14977. [DOI] [PubMed] [Google Scholar]
- 75.Fernandez-Capetillo, O., A. Lee, M.C. Nussenzweig, and A. Nussenzweig. 2004. H2AX: the histone guardian of the genome. DNA Repair (Amst.). 3:959–967. [DOI] [PubMed] [Google Scholar]
- 76.Downs, J.A., N.F. Lowndes, and S.P. Jackson. 2000. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 408:1001–1004. [DOI] [PubMed] [Google Scholar]
- 77.Fernandez-Capetillo, O., S.K. Mahadevaiah, A. Celeste, P.J. Romanienko, D.R. Camerini-Otero, W. Bonner, K. Manova, P.S. Burgoyne, and A. Nussenzweig. 2003. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male meiosis. Dev. Cell. 4:497–508. [DOI] [PubMed] [Google Scholar]
- 78.Stiff, T., M. O'Driscoll, N. Rief, K. Iwabuchi, M. Lobrich, and P.A. Jeggo. 2004. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 64:2390–2396. [DOI] [PubMed] [Google Scholar]
- 79.Bosma, G.C., J. Kim, T. Urich, D.M. Fath, M.G. Cotticelli, N.R. Ruetsch, M.Z. Radic, and M.J. Bosma. 2002. DNA-dependent protein kinase activity is not required for immunoglobulin class switching. J. Exp. Med. 196:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manis, J.P., D. Dudley, L. Kaylor, and F.W. Alt. 2002. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 16:607–617. [DOI] [PubMed] [Google Scholar]
- 81.Rogozin, I.B., and N.A. Kolchanov. 1992. Somatic hypermutagenesis in immunoglobulin genes. II. Influence of neighbouring base sequences on mutagenesis. Biochim. Biophys. Acta. 1171:11–18. [DOI] [PubMed] [Google Scholar]
- 82.Shapiro, G.S., K. Aviszus, D. Ikle, and L.J. Wysocki. 1999. Predicting regional mutability in antibody V genes based solely on di- and trinucleotide sequence composition. J. Immunol. 163:259–268. [PubMed] [Google Scholar]
- 83.Pham, P., R. Bransteitter, J. Petruska, and M.F. Goodman. 2003. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 424:103–107. [DOI] [PubMed] [Google Scholar]