

Abstract
Carma1 (also known as caspase recruitment domain [CARD]11, Bimp3) is a CARD-containing membrane-associated guanylate kinase family protein that plays an essential role in antigen receptor–induced nuclear factor κB activation. We investigated the role of Carma1 in the assembly of signaling molecules at the immune synapse using a peptide-specific system. We report that Carma1 is essential for peptide-induced interleukin 2 and interferon γ production, but dispensable for proliferation in T cells. Recruitment and distribution of T cell receptor, lymphocyte function associated 1, lipid rafts, and protein kinase C (PKC)θ to central and peripheral immune synapse regions occur normally in Carma1 − / − T cells. Carma1 controls entry of IκB kinase (IKK) into lipid raft aggregates and the central region of the immune synapse, as well as activation of IKK downstream of PKC. Our data provide the first genetic evidence on a new class of molecular scaffold that controls entry of defined signaling components, IKK, into the central supramolecular activation cluster at T cell–antigen-presenting cell interfaces without having any apparent effect on the overall organization and formation of immune synapses.
Keywords: Carma1/CARD11/Bimp3, MAGUK, T cell, IKK, immune synapse
Introduction
T cell activation depends on contact between TCRs and agonistic peptide displayed by MHC expressed on APCs. TCR-mediated stimulation results in the assembly of antigen receptors, signaling molecules, and lipid rafts to form supramolecular activation clusters (SMACs) at the interface of physical contact between T cells and APCs (1). Based on the structural similarity to synapses in the nervous system, SMACs are also referred to as immune synapses (2–4). SMAC segregate into a central core, the central SMAC (cSMAC), where the TCR and protein kinase C (PKC)θ are colocalized and lipid rafts preferentially accumulate, and a peripheral zone, the peripheral SMAC (pSMAC), which includes LFA-1 and talin (1, 5). The physiological importance of immune synapse formation has been demonstrated by a line of evidence that shows deficiency of proteins such as Vav1 that affect intact immune synapse formation results in impaired T cell activation (6–10).
Membrane-associated guanylate kinase (MAGUK) family proteins control the polarity of membrane domains at epithelial cell junctions and play critical roles in synaptic development and plasticity (11–14). Because immune and neuronal synapses share structural similarities (3, 4), it has been speculated that MAGUK family proteins have a function in the formation and/or organization of the immune synapse in lymphocytes. Carma1/caspase recruitment domain (CARD)11/Bimp3 is a CARD-containing MAGUK family protein that is abundantly expressed in lymphoid tissues. Using gene targeting, we and others have recently demonstrated that Carma1 is essential for TCR-induced activation of T cells (15–18). However, it is not known whether Carma1 is essential to form the immune synapse.
The recognition of pathogens by innate or adaptive immune receptors leads to activation of the NF-κB family of transcription factors (19). NF-κB proteins are present in the cytoplasm in association with inhibitory proteins known as inhibitor of NF-κBs (IκBs). The signals generated by activated receptors result in phosphorylation of IκB proteins. Phosphorylated IκB proteins become targets for ubiquitination and proteasome-mediated degradation, allowing NF-κB to translocate to the nucleus. Phosphorylation of IκB is mediated by the IκB kinase (IKK) complex, consisting of two catalytic subunits, IKK1/α and IKK2/β, and the NEMO/IKKγ regulatory subunit (20,21). Therefore, the understanding of NF-κB regulation and function is tightly linked to the understanding of IKK regulation and function (22).
Activation of NF-κB mediated by TCR and CD28 costimulation is essential for activation, expansion, and effector functions of T cells (19, 23). PKCθ integrates the TCR and CD28 signals that lead to NF-κB activation, and after T cell activation, PKCθ translocates to lipid rafts at the immune synapse (24–26). Importantly, among the several isoenzymes expressed in T cells, it appears that PKCθ is the only PKC isoform that translocates to the immune synapse (1). Genetic approaches have confirmed that PKCθ is a component in the TCR-induced NF-κB activation pathway because T cells from PKCθ−/− mice cannot be activated in response to antigen and display impaired NF-κB activation (27). Other essential components in this pathway are Bcl10, MALT1, and Carma1 (15–18, 28, 29). These three molecules interact with each other and seem to form a signalosome to activate NF-κB (30–32). Recent reports have shown that the IKK complex translocates to lipid rafts at the immune synapse after TCR/CD28 or PMA/ionomycin stimulation and this localization is sufficient to induce NF-κB activation (33, 34). These findings suggested that the recruitment of IKK into the lipid rafts is a key event to connect IKK to upstream components. However, the exact molecular mechanisms that control the recruitment of IKK into lipid rafts are not yet defined. Moreover, despite the similarity in function and phenotype of knockout mice, the mechanistic connections between PKCθ, Carma1, Bcl10, and IKK remain unclear.
Because data on Carma1-regulated NF-κB activation were based on antibody cross-linking studies, we analyzed the role of Carma1 in peptide–APC-induced T cell activation and assembly of the immune synapse. We show that Carma1 − / − T cells can still proliferate but display markedly abrogated cytokine production when activated by specific peptides. This is not due to defects in the immune synapse formation between T cells and APCs, nor due to defective recruitment of PKCθ to the synapse. Moreover, peptide-induced cell adhesion and LFA-1 accumulation at the immune synapse appear normal. Intriguingly, Carma1 was found to be essential for recruitment of IKK into the central region of the immune synapse. Moreover, genetic inactivation of Carma1 completely abolishes PKC-dependent IKK recruitment to aggregated lipid rafts and IKK activation. We also show that Bcl10 and IKK recruitment to lipid rafts is independently regulated. Thus, the MAGUK family protein Carma1 is the first molecular adaptor identified in vivo that couples antigen receptor signaling to the assembly of supramolecular signaling complexes downstream of immune receptor and lipid raft clustering.
Materials and Methods
Mice.
Carma1 − / − mice have been described (16). BALB/c mice and DO11.10 TCR transgenic (Tg; DO11.10-Tg) were purchased from The Jackson Laboratory. DO11.10-Tg/Carma1 + / − mice congenic for I-Ad were made by crossing DO11.10-Tg mice with Carma1 − / − mice and were backcrossed for three generations into a BALB/c background. DO11.10 Tg/Carma1 + / − mice were then intercrossed to generate DO11.10-Tg/Carma1 − / − and control DO11.10-Tg/Carma1 + / + mice. The expression of I-Ad and DO11.10-TCR was determined by flow cytometric analysis of peripheral blood cells using specific antibodies.
T Cell Responses.
Lymph node CD4+ T cells were purified from DO11.10-Tg mice using magnetic beads (Dynal) to remove CD8+, B220+, Mac-1+, and Gr-1+ cells. 5 × 104 purified CD4+ T cells (>95% CD4+ by FACS) were stimulated for 48 h with 10 μg/ml mitomycin c (Sigma-Aldrich) –treated A20 B lymphoma cells (3 × 104; derived from BALB/c mice) as APCs in the presence or absence of various doses of OVA323–339 peptide (ISQAVHAAHAEINEAGR; reference 35) in 96-well plates in RPMI 1640 media supplemented with 10% FCS and 10−5 M β-mercaptoethanol. Cells were stimulated in triplicate followed by an 8-h pulse with 1 μCi per well of [3H]thymidine (Amersham Biosciences) to determine proliferation. Culture supernatants were assayed in triplicate for the production of IL-2, IL-4, and IFN-γ by ELISA (R&D Systems).
Conjugate Formation and Cell Adhesion.
Unless otherwise indicated, A20 B lymphoma cells were loaded with 10−6 M of OVA323–339 peptide for 4 h at 37°C. 106 lymph node CD4+ T cells and peptide-loaded or nonloaded APCs (A20 B cells) were then mixed at a 1:1 ratio, centrifuged briefly to promote conjugate formation, and incubated at 37°C for different times in serum-free medium. The reactions were stopped and cells were fixed by adding 4% paraformaldehyde. Cell conjugates were stained with anti-DO11.10 TCR PE (BD Biosciences) and anti-B220 APC (BD Biosciences) to detect T cells and APCs, respectively. The percentages of T–APC conjugates (DO11.10 TCR and B220 double positive cells) were analyzed by flow cytometry.
Immunofluorescence Confocal Microscopy.
Primary T lymphoblasts were made by culturing CD4+ T cells with 10−6 M peptide-pulsed A20 APCs for 2 d followed by an additional 4-d culture in the presence of 50 U/ml of rmIL-2 (R&D Systems) without antigen stimulation. For peptide-specific stimulation, CD4+ T cells or the primary T lymphoblasts from DO11.10-Tg/Carma1 + / +, Carma1 + / −, and Carma1 − / − mice were stained with 5 μg/ml Alexa Fluor 488 cholera toxin (CTx; Molecular Probes), and then incubated with peptide-pulsed APCs (A20 B cells) as described above. CTx specifically binds to the lipid raft enriched in glycosphingolipid GM-1 (5, 36). For raft cross-linking experiments, Alexa Fluor 488 CTx–stained T cells were incubated with 4 μg/ml goat anti-CTx (Calbiochem) and 50 ng/ml PMA plus 100 ng/ml calcium ionophore for 10 min at 37°C. Cells were fixed with 4% paraformaldehyde, transferred onto slides using cytospin, and permeabilized with 0.1% Triton X-100 in PBS. Cells were then stained with rabbit anti-PKCθ (C-18; Santa Cruz Biotechnology, Inc.), anti-Bcl10 mAb (331.3; Santa Cruz Biotechnology, Inc.), rabbit anti-IKKα/β (H-470; Santa Cruz Biotechnology, Inc.), anti-IKKγ (C73-764; BD Biosciences), anti–LFA-1 (M17/4; BD Biosciences), biotinylated anti-DO11.10 TCR mAb (Caltag), and Texas red–conjugated phalloidin (Molecular Probes). Goat anti–rabbit, anti–rat, or anti–mouse IgG secondary antibodies conjugated to Alexa Fluor 594 or Alexa Fluor 633 (Molecular Probes) were used to visualize the primary antibodies. For biotinylated antibodies, Alexa Fluor 594–conjugated streptavidin (Molecular Probes) was used. Confocal images were obtained using an Olympus 1X-70 inverted microscope equipped with fluorescence optics and Deltavision Deconvolution Microscopy software (Applied Precision). For the detection of cSMACs and pSMACs, fixed cells were stained with rat anti–LFA-1 (M17/4; BD Biosciences) and rabbit anti-PKCθ or rabbit anti-IKKα/β, followed by staining with Alexa Fluor 568–conjugated goat anti–rabbit IgG and Alexa Fluor 633–conjugated goat anti–rat IgG, respectively. Confocal images of T–APC interfaces were obtained and analyzed using confocal scanning microscopy.
Kinase Assays.
Purified lymph node CD4+ T cells were stimulated with 50 ng/ml PMA plus 100 ng/ml calcium ionophore as described above. After immunoprecipitation with anti-IKK (M-280; Santa Cruz Biotechnology, Inc.), IKK activity was assayed using 3 μg of recombinant GST-IκBα (amino acids 1–54) as a substrate as described previously (37).
Flow Cytometry.
Cells were stained with FITC-, PE-, and APC-conjugated antibodies. For analysis of surface antigen expression, the following antibodies were used: anti-CD3 APC, anti-DO11.10 TCR PE, anti-CD4 FITC, anti-CD4 PE, anti-CD28 FITC, and anti–LFA-1 PE. All antibodies were purchased from BD Biosciences. Samples were analyzed by flow cytometry using a FACScan (Becton Dickinson).
Cellular Fractionation.
Cellular fractionations were performed as described previously (33). In brief, 107 purified lymph node T cells were left untreated or stimulated with 50 ng/ml PMA plus 100 ng/ml calcium ionophore. Cells were lysed on ice for 30 min in 1 ml of NP-40 lysis buffer (1% NP-40, 10 mM Tris, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM Na3VO4, and protein inhibitors), and then homogenized with a Dounce homogenizer (20 strokes) and spun at 500 g for 7 min at 4°C. The postnuclear supernatant was centrifuged at 100,000 g for 1 h at 4°C and the supernatant, referred to as the detergent-soluble membrane and cytosolic fraction (S), was collected. The pellet was rinsed once in the NP-40 lysis buffer and solubilized in 100 μl of RIPA buffer (20 mM Tris, pH 7.5, 250 mM NaCl, 10 mM DTT, 10 mM MgCl2, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, and protein inhibitors), and the insoluble material was removed by centrifugation for 10 min at 10,000 g. This preparation was designated as detergent insoluble material (DIM). 25-μl aliquots of each fraction were resolved by SDS-PAGE and analyzed by Western blotting using antibodies specific for anti-IKKγ mAb (14A231; Upstate Biotechnology) and anti-PKCθ (C-18; Santa Cruz Biotechnology, Inc.). For quantification of expression levels, Western blot images were scanned and the intensity of bands was quantified using IPLab Gel (Signal Analytics).
Online Supplemental Material.
Fig. S1 shows defective recruitment of IKK into lipid rafts at the immune synapse in naive Carma1 − / − CD4+ T cells. Fig. S2 shows defective subcellular translocation of IKK from detergent-soluble fractions to detergent-insoluble membrane fractions in Carma1 − / − T cells after PMA plus calcium ionophore stimulation. Fig. S3 shows a schematic model of the role for Carma1 in the TCR-mediated NF-κB pathway. Figs. S1–S3 are available at http://www.jem.org/cgi/content/full/jem.20032246/DC1.
Results
Carma1 Is Essential for Cytokine Production, But Not Proliferation of Peptide-stimulated T Cells.
We and others have previously shown that Carma1 − / − T cells are markedly impaired both in proliferation and IL-2 production in response to anti-CD3ɛ antibody cross-linking and anti-CD3ɛ/CD28 stimulation (14–17). To examine the role of Carma1 in antigen-specific responses of T cells, we bred the Carma1 mutation on DO11.10-Tg mice to generate DO11.10-Tg/Carma1 + / + and DO11.10-Tg/Carma1 − / − mice. The DO11.10 TCR transgene is specific for the OVA323–339 peptide in association with MHC class II (I-Ad). The populations of CD4+ DO11.10 TCR Tg T cells were similar between Carma1 + / + and Carma1 − / − mice (not depicted). Moreover, we could not find any significant changes in the cell surface expression levels of the clonogenic DO11.10 antigen receptor, CD3ɛ, CD4, CD28, or the integrin LFA-1 between DO11.10-Tg/Carma1 + / + and DO11.10-Tg/Carma1 − / − mice (Fig. 1 A). Thus, as described previously for the HY-TCR (16), genetic inactivation of Carma1 has no apparent effect on the development, populations, or expression levels of surface receptors in DO11.10 TCR Tg mice.
Figure 1.

Carma1 is essential for peptide-specific cytokine production, but not proliferation of T cells. (A) Expression levels of CD3, CD4, the clonotypic DO11.10 TCR, LFA-1, and CD28 on lymph node CD4+ T cells purified from DO11.10 Tg/Carma1 + / + and DO11.10 Tg/Carma1 − / − mice. Data from gated CD4+ T cells are shown. (B–D) Proliferation and cytokine production in the absence of Carma1. Purified CD4+ T cells from DO11.10 Tg/Carma1 + / + and DO11.10 Tg/Carma1 − / − were stimulated with A20 APCs pulsed with various concentration of OVA323–339 peptide. Culture supernatants were harvested 40 h after stimulation and levels of (B) IL-2 and (C) IFN-γ production were determined by ELISA. (D) Proliferation was assessed at 48 h by [3H]thymidine incorporation. Values are mean ± SD for triplicate culture. Results are representative of three independent experiments.
To determine the function of Carma1 in antigen-specific responses, purified lymph node CD4+ T cells from DO11.10-Tg/Carma1 + / + and DO11.10-Tg/Carma1 − / − mice were stimulated with A20 B cells as APCs pulsed with different doses of the OVA323–339 peptide. As expected from previous antibody cross-linking data (15, 16), IL-2 and IFN-γ production was completely abrogated in Carma1 − / − T cells (Fig. 1, B and C). IL-4 production could not be detected in both wild-type and Carma1 − / − DO11.10-Tg T cells under the same stimulation conditions. These defects in cytokine production could not be overcome even at high doses of peptide stimulations, indicating that Carma1 is essential for peptide-induced cytokine productions of T cells. Surprisingly, although proliferative responses of DO11.10-Tg/Carma1 − / − T cells were impaired at lower peptide stimulation up to 10−8 M, higher doses of peptide stimulation restored their response to levels that were comparable to that of control DO11.10-Tg/Carma1 + / + T cells (Fig. 1 D). These results indicate that Carma1-mediated signaling is essential for cytokine productions, but dispensable for proliferation of T cells in response to specific peptide plus APCs.
Carma1 Is Not Involved in the Formation of the Immune Synapse.
It has been reported that Carma1 is recruited to the immune synapse and becomes associated with the antigen receptor complex after TCR stimulation (38). Antigen receptor stimulation in T cells results in TCR clustering at the immune synapse, reorganization of the actin cytoskeleton at the site of assembled TCRs, and clustering of integrins (39). In addition, lipid rafts are also assembled at the immune synapse and membrane receptors including TCR and many intracellular signaling molecules are recruited to lipid rafts during T cell activation (36, 40). Thus, similar to the function of other MAGUK family proteins in neuronal synapse, we speculated that lack of Carma1 could affect immune synapse formation in T cells. Therefore, we examined whether loss of Carma1 in T cells results in defective formation of antigen clustering, clustering of cell adhesion receptors, alterations in the cytoskeleton, and/or defective recruitment of downstream effector molecules using confocal imaging of peptide-specific immune synapses between DO11.10-Tg T cells and peptide-loaded APCs. Loss of Carma1 expression had no apparent effect on conjugation of DO11.10-Tg T cells with peptide-pulsed APCs (Fig. 2 A). Lipid raft clustering as well as TCR clustering and recruitment of TCRs to lipid rafts at the T cell–APC interfaces were also comparable between DO11.10-Tg/Carma1 − / − and control DO11.10-Tg/Carma1 + / + T cells (Fig. 2, A and D). Moreover, F-actin (Fig. 2, B and D) as well as LFA-1 clustering (Fig. 2, C and D) at the immune synapses were comparable between Carma1 + / + and Carma1 − / − T cells.
Figure 2.
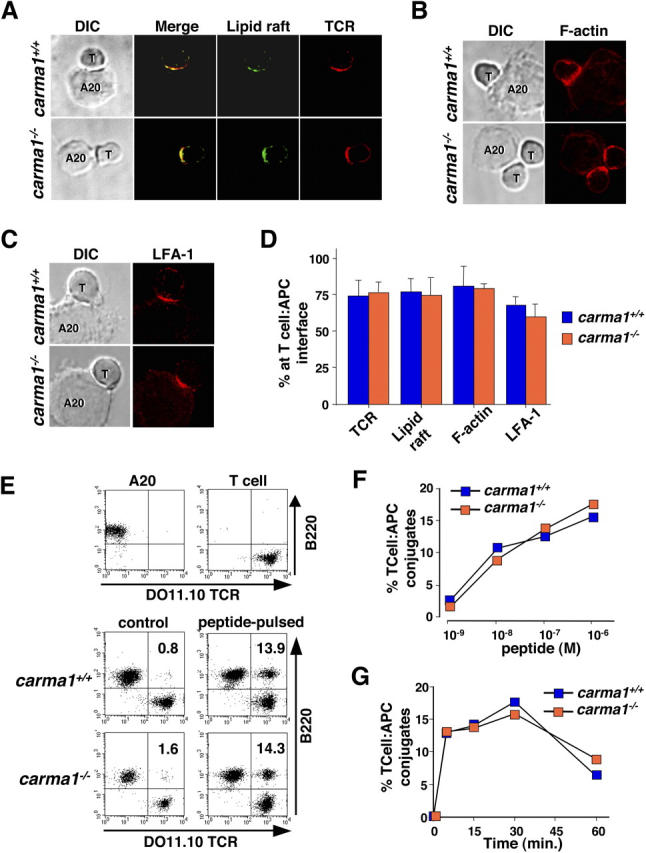
Immune synapse formation and peptide-specific cell adhesion in Carma1−/−T cells. (A) DIC images of T cell–A20-APC conjugates. Localization of lipid rafts (green) and DO11.10 TCR (red) are shown. Yellow in the merged images indicates colocalization of the TCR with lipid raft. (B and C) Localization of (B) polymerized actin (F-actin; visualized with phalloidin) and (C) LFA-1 at the focal contact sites. (D) Percentages (percent ± SD) of DO11.10 TCR, lipid rafts, F-actin, and LFA-1 clustered at the immune synapse. (A–D) Purified lymph node CD4+ T cells from DO11.10 Tg/Carma1 + / + and DO11.10 Tg/Carma1 − / − were labeled with Alexa Fluor 488 CTx, and then incubated with 10−6 M OVA323–339 peptide–pulsed A20 cells for 30 min. At least 100 cells were counted for each sample. Results are representative of three independent experiments. T, T cell; A20, A20 APCs. (E) Peptide-specific cell adhesion assay. A20 APCs and DO11.10-Tg T cells are distinguishable by their expression of B220 and DO11.10 TCR (top). Purified CD4+ T cells from DO11.10 Tg/Carma1+/+ and DO11.10 Tg/Carma1−/− were mixed with 10−6 M OVA323–339 peptide–pulsed or nonpulsed control A20 APCs and incubated for 30 min (middle and bottom), and T cell–APC conjugates were detected as DO11.10 TCR and B220 double positive cells. (F) Percentages of DO11.10 Tg T cells that form conjugates with APCs loaded with various OVA323–339 peptide doses. Conjugate formation was assayed at 30 min. (G) Conjugates were monitored over different time periods using APCs loaded with 10−6 M OVA323–339 peptide. Results are representative of three independent experiments.
To examine whether loss of Carma1 affects peptide-specific cell adhesion of T cells to APCs, CD4+ T cells from DO11.10-Tg/Carma1 + / + and DO11.10-Tg/Carma1 − / − mice were mixed with A20 APCs loaded with OVA peptide, and conjugate formations between T cells and APCs were analyzed by FACS (Fig. 2, E–G). T cell–APC conjugate formation increased in the presence of specific peptide as compared with controls, demonstrating that this peptide-specific induction of cell adhesion is dependent on TCR-mediated signaling. Importantly, T cells from DO11.10-Tg/Carma1 − / − formed conjugates with peptide-pulsed APCs at doses (Fig. 2 F) and time courses (Fig. 2 G) comparable to that of DO11.10-Tg/Carma1 + / + control T cells. Thus, loss of Carma1 has no apparent affect on inside-out signaling and peptide-specific cell adhesion between T cells and APCs. Collectively, our results show that Carma1 is not required for peptide-induced immune synapse formation at the T cell–APC contact sites.
Carma1 Is Required for Recruitment of IKK into the cSMAC.
In neurons, MAGUK family proteins are required for synapse formation and function as scaffolds for the assembly of signaling complexes and cytoskeletal components at cell–cell contact sites (12). It has been shown previously that Carma1 is an essential component in the TCR-mediated NF-κB activation pathway (15, 16, 38, 41). Because immune synapse formation appeared normal in DO11.10-Tg/Carma1 − / − T cells, we determined whether the loss of Carma1 expression affects the recruitment of defined downstream signaling molecules involved in the NF-κB activation pathway to the immune synapse. Moreover, we wanted to determine whether the segregation of the immune synapse into defined cSMAC and pSMAC subregions occurred in the absence of Carma1 expression (1, 2).
Because it has been suggested that NF-κB activation by PKCθ and IKK is regulated by their recruitment into lipid rafts at the immune synapse (25, 33, 34), we first examined the recruitment of these molecules after stimulation with peptide-pulsed APCs (Figs. 3 and 4). When T cells were incubated with control APCs, PKCθ and IKK were localized in the cytoplasm and did not colocalize with GM-1–containing lipid rafts in both Carma1 + / − and Carma1 − / − T cells (Figs. 3 A and 4 A). After conjugation of DO11.10-Tg/Carma1 + / − T cells with OVA peptide–pulsed APCs, PKCθ and IKK translocated to the T cell–APC contact sites (Figs. 3 A and 4 A). Localization experiments with GM-1 rafts to visualize cSMACs (5) and LFA-1 to mark pSMAC (1, 2) regions showed that PKCθ was confined to the cSMAC area and colocalized with the clustered GM-1 lipid rafts. Although IKK can be detected in both cSMAC and pSMAC subregions, IKK preferentially segregated into the cSMAC area and colocalized with the clustered GM-1 lipid rafts (Figs. 3 B and 4, B and C). Similar to control T cells, peptide-mediated stimulation of DO11.10-Tg/Carma1 − / − T cells resulted in the recruitment of PKCθ to the immune synapse (Fig. 3 A). Moreover, in DO11.10-Tg/Carma1 − / − T cells, PKCθ colocalized with GM-1 lipid rafts at the cSMAC of the immune synapse and formed typical cSMAC and pSMAC structure characterized by central PKCθ surrounded by a peripheral LFA-1 ring (Fig. 3 B and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20032246/DC1). Intriguingly, although IKK translocated to the focal contact sites, colocalization of IKK with lipid rafts at the cSMAC of the immune synapse was markedly decreased in DO11.10-Tg/Carma1 − / − T cells (Fig. S1). Z axis analysis of the T cell–APC interface revealed that IKK and lipid rafts were indeed spatially segregated at the contact area of DO11.10-Tg/Carma1 − / − T cells (Fig. 4 B). Moreover, although PKCθ and IKK colocalize at the cSMAC region in Carma1-expressing T cells, IKK was retained in the pSMAC area and did not colocalize with PKCθ at the cSMAC in DO11.10-Tg/Carma1 − / − T cells (Fig. 4, B–D). This segregation of lipid rafts and IKK at immune synapses between Carma1 − / − T cells and APCs followed a typical pattern: clustered lipid rafts and PKCθ at the cSMAC are surrounded by LFA-1 and IKK in the pSMAC areas (Fig. 4, B and C). Together, these results indicate that Carma1 is dispensable for translocation of PKCθ and IKK to the focal contact sites between DO11.10-Tg T cells and peptide-loaded APCs, but Carma1 is essential to recruit the IKK complex to the cSMAC of the immune synapse where PKCθ localizes.
Figure 3.
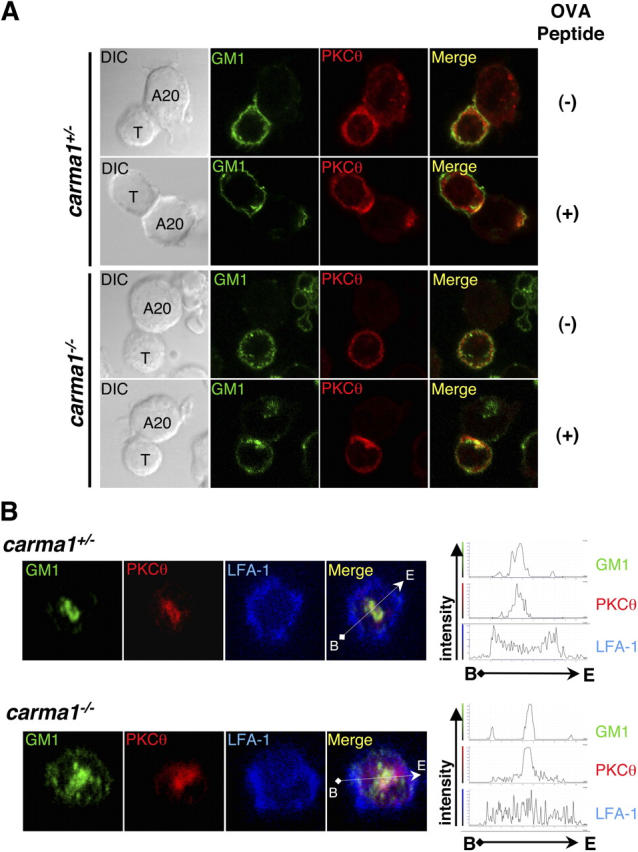
Normal SMAC segregation and recruitment of PKCθ to cSMACs in Carma1−/− T cells. (A and B) CD4+ T cell blasts from DO11.10 Tg/Carma1+/+ and DO11.10 Tg/Carma1−/− were incubated with 10−6 M OVA323–339 peptide–pulsed or nonpulsed control A20 APCs and incubated for 30 min. Images of T cell–A20 conjugates and localization of GM-1–associated lipid rafts (detected by CTx; A and B, green) and PKCθ (A and B, red) and LFA-1 (B, blue) are shown. Yellow in the merged images indicates colocalization of PKCθ with lipid rafts. Localization of PKCθ at the immune synapse interface is also shown in B. (B, right) The histograms of relative fluorescence intensity of indicated molecules by scanning along the white arrows indicated in the merged left panels. T, T cell; A20, A20 APCs.
Figure 4.
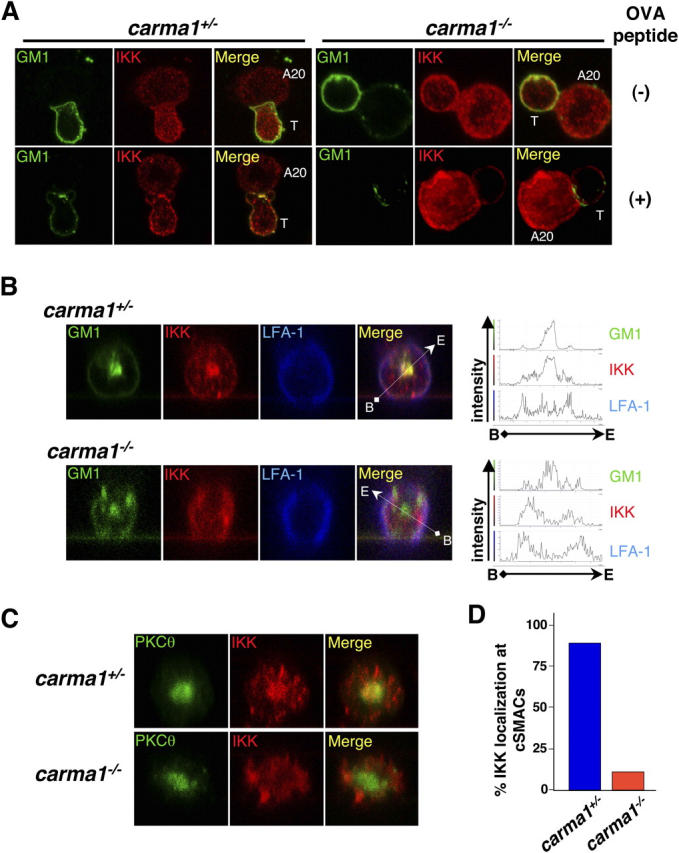
Carma1 is essential for recruitment of IKK into cSMACs and colocalization with PKCθ. (A and B) T cell–APC conjugates were formed as described in Fig. 3. Localization of GM-1–associated lipid rafts (A and B, green) and IKKα/β (A and B, red) and LFA-1 (B, blue) are shown. Yellow in the merged images indicates colocalization of IKK with lipid rafts. Localization of IKKα/β at the immune synapse interface is also shown in B. IKKs localize to the immune synapse, but IKK molecules are excluded from lipid rafts in cSMACs in Carma1−/− T cells. (B, right) Histograms of relative fluorescence intensity of indicated molecules by scanning along the white arrows shown in the merged panels. (C) T cell–APC conjugates were labeled with antibodies for PKCθ (green) and for IKKγ (red). The immune synapse interfaces are shown. Yellow in the merged images indicates colocalization of PKCθ with IKKγ. Note that in Carma1-expressing T cells, IKKα/β as well as IKKγ redistribute to the cSMAC area, whereas in Carma1−/− T cells, IKKα/β and IKKγ molecules move to the contact areas, but are excluded from cSMACs where PKCθ molecules are located. (D) Percentages of the immune synapses that display colocalization of IKKγ with PKCθ at the cSMAC of the immune synapse. At least 100 cells were counted for each sample. Results are representative of three independent experiments.
Carma1 Regulates IKK Recruitment to Lipid Rafts Downstream of PKCs.
Because IKK can be recruited to lipid rafts after PKC stimulation (33, 41, 42) and we observed normal PKCθ but defective IKK recruitment into lipid rafts and cSMACs at immune synapses in Carma1 − / − T cells, we wanted to analyze whether Carma1 mediates IKK recruitment and NF-κB activation downstream of PKCs. We stimulated primary Carma1 + / + and Carma1 − / − T cells with PMA/ionophore and lipid raft cross-linking. It should be noted that PMA/ionophore alone or lipid raft cross-linking alone does not lead to effective raft aggregation nor PKCθ membrane recruitment (not depicted), suggesting that both raft cross-linking and PKC activation are required for efficient raft aggregation in primary T cells. In nonstimulated Carma1 + / + and Carma1 − / − T cells, IKK and PKCθ were located in the cytoplasm (Fig. 5, A and B). In PMA/ionophore-stimulated cells, lipid raft clustering and recruitment of PKCθ to the lipid raft aggregates were comparable between Carma1 + / + and Carma1 − / − T cells (Fig. 5 A). Thus, similar to peptide-mediated immune synapse formation at the T cell–APC interface (Fig. 3), lack of Carma1 does not affect PKCθ recruitment in response to PMA/ionophore stimulation.
Figure 5.
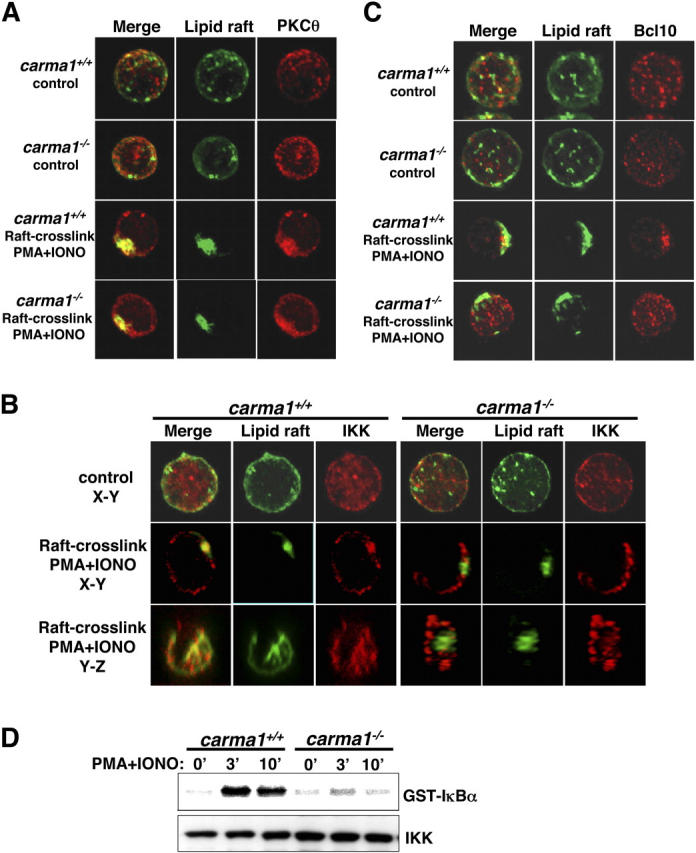
Carma1 controls IKK and Bcl10 recruitment into aggregated lipid rafts and IKK activation downstream of PKCs. (A–C) Activation-induced PKCθ, IKK, and Bcl10 redistribution to aggregated lipid rafts. Purified lymph node T cells from Carma1 + / + and Carma1 − / − mice were labeled with Alexa Flour 488 CTx and left untreated (control) or stimulated with 50 ng/ml PMA/100 ng/ml calcium ionophore plus cross-linking of rafts with 4 μg/ml anti-CTx. Lipid raft (green), PKCθ (A, red), IKKα/β (B, red), and Bcl10 (C, red) localization was analyzed 10 min after stimulation. Yellow in the merged images indicates colocalization of the indicated molecule with GM-1–expressing lipid rafts. Note that IKKα/β molecules are redistributed after stimulation, but are excluded from lipid rafts in Carma1 − / − T cells (x, y, and z axes are shown). IKKα/β localization was determined with an antibody that reacts to both IKKα and IKKβ. Results are representative of three independent experiments. (D) Defective IKK activation in Carma1 − / − T cells. Purified lymph node T cells were stimulated with 50 ng/ml PMA and 50 ng/ml calcium ionophore, and IKK activity was determined by in vitro immunocomplex kinase assays. GST-IκBα (amino acids 1–54) was used as a specific substrate. Levels of total IKKα/β using an antibody reactive to both IKKα and IKKβ are indicated. Results are representative of three independent experiments.
Consistent with a previous report in cell lines and human T cells (33), PMA/ionophore stimulation of Carma1 − / − T cells resulted in redistribution of IKK to the plasma membrane and the formation of IKK clusters (Fig. 5 B). However, in the absence of Carma1, clustered IKK molecules were again excluded from the lipid raft aggregates (Fig. 5 B). Importantly, although almost 100% of Carma1 + / + T cells showed colocalization of IKK with aggregated GM-1–containing lipid rafts, in Carma1 − / − T cells we could not find any cells that colocalize IKK and GM-1–containing lipid rafts by microscopic analysis. Similar to the T cell–APC interface in Carma1 − / − T cells (Fig. 4), z axis analysis of Carma1 − / − T cells showed that IKK aggregates were again spatially segregated from lipid rafts (Fig. 5 B). In both experimental settings, IKK forms cluster and surrounds GM-1–containing lipid raft aggregates, but does not localize inside the GM-1+ lipid rafts.
Because it has been reported that Carma1 − / − T cells are defective in recruitment of Bcl10 to lipid rafts after TCR stimulation and CTx cross-linking (15), we tested whether Bcl10 recruitment is also impaired after PMA/ionophore stimulation. Bcl10 was recruited to the plasma membrane and colocalized with lipid rafts in Carma1 + / + T cells. In contrast, recruitment and redistribution of Bcl10 to clustered lipid rafts did not occur in Carma1 − / − T cells (Fig. 5 C). However, although PMA/ionophore stimulation of Carma1 − / − T cells results in the formation of IKK clusters, albeit clustered IKK molecules are excluded from the lipid rafts (Fig. 5 B), Bcl10 did not translocate to the plasma membrane and remained localized throughout the cytoplasm (Fig. 5 C). These results indicate that Carma1 controls recruitment of both IKK and Bcl10 into lipid rafts, but their recruitment is likely to be regulated differently.
Defective IKK Activation in Carma1−/− T Cells.
To further confirm impaired raft recruitment of IKK in Carma1 − / − T cells, we isolated detergent-soluble membrane and cytosolic (S) and lipid raft–containing DIM fractions (33). Consistent with our confocal microscopy studies, in both Carma1 + / + and Carma1 − / − T cells, PKCθ translocated from the S fraction into DIM fractions after PMA/ionophore stimulation, and the ratio of DIM/S fractions was increased by three to four times (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20032246/DC1). IKK was detected in S fractions as well as DIM fractions at similar levels in unstimulated Carma1 + / + and Carma1 − / − T cells. After stimulation of Carma1 + / + T cells, the amount of IKK in the S fractions was decreased and consequently the DIM/S ratio was markedly increased, indicating that IKK translocates from S into DIM fractions. In Carma1 − / − T cells, however, the amount of IKK in S fractions did not change after stimulation (Fig. S2). Because it has been shown that IKK recruitment to lipid rafts is important for activation (33, 34) and our results indicated that IKK lipid raft recruitment is defective in Carma1 − / − T cells, we evaluated IKK function by in vitro kinase activity. In parallel with defective recruitment of IKK to the lipid rafts, IKK activation after PMA/ionophore stimulation was significantly impaired in Carma1 − / − T cells (Fig. 5 D). Thus, genetic inactivation of Carma1 abolishes stimulation-dependent IKK recruitment to lipid rafts and IKK activation.
Discussion
It has been shown previously that T cells from Carma1 − / − mice have a severe defect in both proliferation and IL-2 production when stimulated with CD3ɛ/CD28 cross-linking or PMA/ionophore (15–17). Interestingly, our results here showed that Carma1 is dispensable for proliferation when T cells are stimulated with APCs pulsed with high peptide doses, although at the same peptide concentration Carma1 is essential for cytokine production. Stimulation of T cells using CD3ɛ/CD28 cross-linking is critically different from that of peptide-pulsed APCs. For instance, APCs express a variety of costimulatory and adhesion molecules and produce cytokines that promote T cell–APC interaction and enhance TCR signaling (43–45). Moreover, cognate interactions between T cells and APCs increase and induce the expression of molecules on both cells that then amplify antigen receptor signaling.
Our data using a peptide–APC system show that Carma1 does not simply determine an all or nothing response as previously reported, but under physiological stimulation conditions Carma1 sets the threshold for T cell proliferation. Importantly, on the other hand, signals required for cytokine production definitely depend on Carma1 expression. This differential requirement of Carma1 may explain the in vivo T cell phenotype in Carma1 − / − mice. Carma1-deficient mice show normal numbers and development of thymocytes and normal populations of peripheral T cells in lymph nodes and spleen (15–17), indicating that loss of Carma1 does not affect thymocyte expansion during development, thymocyte selection, or homeostatic expansion of peripheral T cells in vivo. In contrast, Carma1 − / − mice show impaired T-dependent immunity in vivo, which is consistent with the abrogated cytokine production of Carma1 − / − T cells observed in our study. Thus, our findings provide a novel aspect of Carma1-mediated TCR signaling in T cell homeostasis and T cell activation. Whether compensatory cytokine production in the absence of Carma1 could drive proliferation of these cells needs to be determined. Moreover, it would be interesting to elucidate whether Carma1 might control localized cytokine secretion at the focal contact site.
The nervous system and immune system use specialized cell surface contacts to transduce signals between their constituent cell populations (4). These two synaptic junctions in neurons and lymphocytes share common structural features. In neurons and epithelial cells, MAGUK family proteins such as CASK, DLG, or PDS-95 have been shown to assemble receptors, cytoskeletal components, and signaling molecules at sites of cell–cell contact, including synapses, cellular junctions, and polarized membrane domains (13, 46). Carma1 is a lymphocyte-specific member of the MAGUK family of proteins and is an essential component required for TCR- or B cell receptor–induced lymphocyte activation. Thus, Carma1 was predicted to be a candidate molecule for the assembly and/or organization of supramolecular signaling complexes in lymphocytes.
Using the DO11.10 immune synapse formation system, our data show that loss of Carma1 does not typically affect features of immune synapses at the T cell–APC interface. DO11.10-Tg/Carma1 − / − T cells display normal antigen receptor clustering, lipid raft clustering, integrin clustering, as well as reorganization of filamentous actin at the focal contact sites. Importantly, inactivation of Carma1 also did not affect the normal segregation of the immune synapse into cSMACs and pSMACs. Consistent with normal surface receptor clustering and cytoskeletal reorganization, conjugate formation and peptide-induced adhesion between T cells and APCs were also found to be comparable between DO11.10-Tg/Carma1 − / − and DO11.10-Tg/Carma1 + / + T cells. Our experiments refute the hypothesis that Carma1 functions as a scaffold that assembles multiple surface receptors and the actin cytoskeleton at the synapse. Another MAGUK family protein, the human lymphocyte homologue of the Drosophila discs large tumor suppressor protein, has also been implicated in T cell activation and is recruited to SMACs upon CD2 cross-linking (47). Whether MAGUK family proteins other than Carma1 are indeed essential for immune synapse formation needs to be determined. Importantly, our data provide the first genetic evidence on a new class of molecular scaffold essential for the selective recruitment of IKK into lipid rafts and spatial segregation of IKK into the cSMAC at immune synapses.
PKCθ, Carma1, Bcl10, and IKK are all essential components in the TCR-induced NF-κB activation pathway (15–18, 27–29). Although understanding of this pathway has progressed rapidly, the molecular and structural relationship between these molecules still remains unsolved. It has been suggested that PKCθ, Bcl10, and IKK are located in the cytoplasm in resting cells and these molecules translocate to lipid rafts at the immune synapse after TCR activation (33, 34, 38). Carma1 is located in both the cytoplasm and lipid rafts in resting T cells and the amount of Carma1 in lipid rafts is increased after antigen receptor activation. Moreover, kinase activation of IKK is regulated by the recruitment into lipid rafts, but the molecule(s) that mediates the recruitment was not clear. Because previous reports showed that PKCθ physically associates with the IKK complex in lipid rafts after TCR stimulation (33), the same molecular mechanism that regulates PKCθ recruitment was a potential candidate pathway to also mediate the recruitment of IKK. However, our data clearly show that Carma1 acts downstream of PKCs in the recruitment of IKK into aggregated lipid rafts. In addition, Carma1 is essential for the recruitment of IKK, but not PKCθ, into lipid rafts at the immune synapse. Consistent with this notion, a recent report has shown that Carma1 can physically interact with IKKγ (48). Thus, Carma1 is an essential molecular adaptor that controls IKK recruitment into lipid rafts and cSMACs downstream of immune synapse formation and PKC activation. Our results also demonstrate that activation-induced recruitment of PKCθ and IKK into lipid rafts are separately regulated.
It has been proposed that Bcl10 is the molecule that acts upstream of IKK and may recruit IKK to membranes (38). Our data here and a recent report by Egawa et al. (15) have shown that Carma1 is indeed essential for the recruitment of Bcl10 into lipid rafts. Importantly, although Bcl10 does not translocate to plasma membrane at all and remains distributed in the cytoplasm after TCR activation in Carma1 − / − T cells, IKKs translocate to the membrane region after stimulation and form aggregates at the immune synapse or PMA/ionophore-induced receptor caps, but does not enter into the lipid rafts. Thus, although the same molecule, Carma1, regulates the recruitment of both IKK and Bcl10 to lipid rafts, our data suggest that the underlying mechanisms of recruitment are different. Carma1 not only regulates translocation of Bcl10 to the membrane, but also lipid raft recruitment of Bcl10. In contrast, translocation of IKK from the cytoplasm to the focal contact sites at the immune synapse does occur in Carma1 − / − T cells, but Carma1 is essential for the recruitment of the IKK complex into aggregated GM-1 lipid rafts and the cSMAC. Based on our results, we propose that Bcl10 is required for IKK activation in the lipid rafts. Alternatively, Bcl10 recruitment into lipid rafts through Carma1 might be the prerequisite for the translocation of the IKK complex into GM-1–containing rafts and cSMAC. For a model of Carma1-regulated IKK and Bcl10 recruitment and activation, see Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20032246/DC1.
In conclusion, we have found that Carma1 has no apparent role in the formation of the immune synapse, PKCθ recruitment, lipid raft aggregations, cSMAC and pSMAC segregation, or activation-dependent cell adhesion. Carma1 acts downstream of immune synapse formation and PKC activation and controls stimulation-dependent IKK recruitment into the cSMAC of the immune synapse, thereby regulating IKK activation. Moreover, Carma1 was found to be essential for cytokine production, but set the activation threshold for proliferation in peptide-activated T cells. Our data establish the molecular hierarchies by which different components of the NF-κB activation pathway are recruited into defined areas of the immune synapse. Moreover, these results elucidate the structural complexities by which the adaptor Carma1 couples antigen receptor signaling to IKK and NF-κB activation in T cells.
Acknowledgments
We thank K. Kuba, T. Nakashima, N. Joza, M. Cheng, M. Rangachari, A. Atfield, U. Eriksson, L. Zhang, U. Danylczyk, A. Oliviera-dos-Santos, K. Bachmaier, H. Jones, C. Vorbach, E. Griffith, K. Krawczyk, T. Yokosuka, N. Suzuki, S. Suzuki, A. Takeuchi, and S. Yamasaki for comments, and I. Kozieradzki, Y. Nakashima, S. Arya, M. Nghiem, R. Sarao, A. Furuno, W. Kobayashi, and J. Suzuki for technical support.
J.M. Penninger is supported by grants from IMBA, The Austrian Academy of Sciences, The Austrian Ministry of Science and Education, and grants from the 6th EU Science framework program. This work is also supported by grants from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
The authors have no conflicting financial interests.
Abbreviations used in this paper: CARD, caspase recruitment domain; cSMAC, central supramolecular activation cluster; CTx, cholera toxin; DIM, detergent insoluble material; IKK, IκB kinase; MAGUK, membrane-associated guanylate kinase; PKC, protein kinase C; pSMAC, peripheral supramolecular activation cluster; SMAC, supramolecular activation cluster; Tg, transgenic.
References
- 1.Monks, C.R.F., B.A. Freiberg, H. Kupfer, N. Sciaky, and A. Kupfer. 1998. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 395:82–86. [DOI] [PubMed] [Google Scholar]
- 2.Grakoui, A., S.K. Bromley, C. Sumen, M.M. Davis, A.S. Shaw, P.M. Allen, and M.L. Dustin. 1999. The immunological synapse: a molecular machine controlling T cell activation. Science. 285:221–227. [DOI] [PubMed] [Google Scholar]
- 3.Donnadieu, E., P. Revy, and A. Trautmann. 2001. Imaging T-cell antigen recognition and comparing immunological and neuronal synapses. Immunology. 103:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dustin, M.L., and D.R. Colman. 2002. Neural and immunological synaptic relations. Science. 25:785–789. [DOI] [PubMed] [Google Scholar]
- 5.Burack, W.R., K.H. Lee, A.D. Holdorf, M.L. Dustin, and A.S. Shaw. 2002. Quantitative imaging of raft accumulation in the immunological synapse. J. Immunol. 169:2837–2841. [DOI] [PubMed] [Google Scholar]
- 6.Dustin, M.L., M. Olszowy, A.D. Holdorf, J. Li, S. Bromley, N. Desai, P. Widder, F. Rosenberger, P.A. van der Merwe, P.M. Allen, and A.S. Shaw. 1998. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell. 94:667–677. [DOI] [PubMed] [Google Scholar]
- 7.Fischer, K.D., Y.Y. Kong, H. Nishina, K. Tedford, L.E. Marengere, I. Kozieradzki, T. Sasaki, M. Starr, G. Chan, S. Gardener, et al. 1998. Vav is a regulator of cytoskeletal reorganization mediated by the T-cell receptor. Curr. Biol. 8:554–562. [DOI] [PubMed] [Google Scholar]
- 8.Holsinger, L.J., I.A. Graef, W. Swat, T. Chi, D.M. Bautista, L. Davidson, R.S. Lewis, F.W. Alt, and G.R. Crabtree. 1998. Defects in actin-cap formation in Vav-deficient mice implicate an actin requirement for lymphocyte signal transduction. Curr. Biol. 8:563–572. [DOI] [PubMed] [Google Scholar]
- 9.Zhang, J., A. Shehabeldin, L.A. da Cruz, J. Butler, A.K. Somani, M. McGavin, I. Kozieradzki, A.O. dos Santos, A. Nagy, S. Grinstein, et al. 1999. Antigen receptor–induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein–deficient lymphocytes. J. Exp. Med. 190:1329–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanui, T., A. Inayoshi, M. Noda, E. Iwata, M. Oike, T. Sasazuki, and Y. Fukui. 2003. DOCK2 is essential for antigen-induced translocation of TCR and lipid rafts, but not PKC-theta and LFA-1, in T cells. Immunity. 19:119–129. [DOI] [PubMed] [Google Scholar]
- 11.Tejedor, F.J., A. Bokhari, O. Rogero, M. Gorczyca, J. Zhang, E. Kim, M. Sheng, and V. Budnik. 1997. Essential role for dlg in synaptic clustering of Shaker K+ channels in vivo. J. Neurosci. 17:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Migaud, M., P. Charlesworth, M. Dempster, L.C. Webster, A.M. Watabe, M. Makhinson, Y. He, M.F. Ramsay, R.G. Morris, J.H. Morrison, T.J. O'Dell, and S.G. Grant. 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396:433–439. [DOI] [PubMed] [Google Scholar]
- 13.Fanning, A.S., and J.M. Anderson. 1999. Protein modules as organizers of membrane structure. Curr. Opin. Cell Biol. 11:432–439. [DOI] [PubMed] [Google Scholar]
- 14.Hong, Y., B. Stronach, N. Perrimon, L.Y. Jan, and Y.N. Jan. 2001. Drosophila Stardust interacts with Crumbs to control polarity of epithelia but not neuroblasts. Nature. 414:634–638. [DOI] [PubMed] [Google Scholar]
- 15.Egawa, T., B. Albrecht, B. Favier, M.J. Sunshine, K. Mirchandani, W. O'Brien, M. Thome, and D.R. Littman. 2003. Requirement for CARMA1 in antigen receptor-induced NF-κB activation and lymphocyte proliferation. Curr. Biol. 13:1252–1258. [DOI] [PubMed] [Google Scholar]
- 16.Hara, H., T. Wada, C. Bakal, I. Kozieradzki, S. Suzuki, N. Suzuki, M. Nghiem, E.K. Griffiths, C. Krawczyk, B. Bauer, et al. 2003. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. 18:763–775. [DOI] [PubMed] [Google Scholar]
- 17.Jun, J.E., L.E. Wilson, C.G. Vinuesa, S. Lesage, M. Blery, L.A. Miosge, M.C. Cook, E.M. Kucharska, H. Hara, J.M. Penninger, et al. 2003. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 18:751–762. [DOI] [PubMed] [Google Scholar]
- 18.Newton, K., and V.M. Dixit. 2003. Mice lacking the CARD of CARMA1 exhibit defective B lymphocyte development and impaired proliferation of their B and T lymphocytes. Curr. Biol. 13:1247–1251. [DOI] [PubMed] [Google Scholar]
- 19.Li, Q., and I.M. Verma. 2002. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2:725–734. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin, A.S., Jr. 1996. The NF-κB and IκB proteins: new discoveries and insights. Annu. Rev. Immunol. 14:649–683. [DOI] [PubMed] [Google Scholar]
- 21.Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18:621–663. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-κB puzzle. Cell. 109:S81–S96. [DOI] [PubMed] [Google Scholar]
- 23.Kane, L.P., J. Lin, and A. Weiss. 2002. It's all Rel-ative: NF-κB and CD28 costimulation of T-cell activation. Trends Immunol. 23:413–420. [DOI] [PubMed] [Google Scholar]
- 24.Monks, C.R., H. Kupfer, I. Tamir, A. Barlow, and A. Kupfer. 1997. Selective modulation of protein kinase C-θ during T-cell activation. Nature. 385:83–86. [DOI] [PubMed] [Google Scholar]
- 25.Bi, K., Y. Tanaka, N. Coudronniere, K. Sugie, S. Hong, M.J. van Stipdonk, and A. Altman. 2001. Antigen-induced translocation of PKC-θ to membrane rafts is required for T cell activation. Nat. Immunol. 2:556–563. [DOI] [PubMed] [Google Scholar]
- 26.Isakov, N., and A. Altman. 2002. Protein kinase Cθ in T cell activation. Annu. Rev. Immunol. 20:61–94. [DOI] [PubMed] [Google Scholar]
- 27.Sun, Z., C.W. Arendt, W. Ellmeier, E.M. Schaeffer, M.J. Sunshine, L. Gandhi, J. Annes, D. Petrzilka, A. Kupfer, P.L. Schwartzberg, and D.R. Littman. 2000. PKC-θ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature. 404:402–407. [DOI] [PubMed] [Google Scholar]
- 28.Ruland, J., G.S. Duncan, A. Elia, I. del Barco Barrantes, L. Nguyen, S. Plyte, D.G. Millar, D. Bouchard, A. Wakeham, P.S. Ohashi, and T.W. Mak. 2001. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-κB and neural tube closure. Cell. 104:33–42. [DOI] [PubMed] [Google Scholar]
- 29.Ruefli-Brasse, A.A., D.M. French, and V.M. Dixit. 2003. Regulation of NF-κB-dependent lymphocyte activation and development by paracaspase. Science. 302:1581–1584. [DOI] [PubMed] [Google Scholar]
- 30.Bertin, J., L. Wang, Y. Guo, M.D. Jacobson, J.L. Poyet, S.M. Srinivasula, S. Merriam, P.S. DiStefano, and E.S. Alnemri. 2001. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-κB. J. Biol. Chem. 276:11877–11882. [DOI] [PubMed] [Google Scholar]
- 31.Gaide, O., F. Martinon, O. Micheau, D. Bonnet, M. Thome, and J. Tschopp. 2001. Carma1, a CARD-containing binding partner of Bcl10, induces Bcl10 phosphorylation and NF-κB activation. FEBS Lett. 11:121–127. [DOI] [PubMed] [Google Scholar]
- 32.Lucas, P.C., M. Yonezumi, N. Inohara, L.M. McAllister-Lucas, M.E. Abazeed, F.F. Chen, S. Yamaoka, M. Seto, and G. Nunez. 2001. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-κB signaling pathway. J. Biol. Chem. 276:19012–19019. [DOI] [PubMed] [Google Scholar]
- 33.Khoshnan, A., D. Bae, C.A. Tindell, and A.E. Nel. 2000. The physical association of protein kinase Cθ with a lipid raft-associated inhibitor of κB factor kinase (IKK) complex plays a role in the activation of the NF-κB cascade by TCR and CD28. J. Immunol. 165:6933–6940. [DOI] [PubMed] [Google Scholar]
- 34.Weil, R., K. Schwamborn, A. Alcover, C. Bessia, V. Di Bartolo, and A. Israel. 2003. Induction of the NF-κB cascade by recruitment of the scaffold molecule NEMO to the T cell receptor. Immunity. 18:13–26. [DOI] [PubMed] [Google Scholar]
- 35.Sette, A., S. Buus, S. Colon, J.A. Smith, C. Miles, and H.M. Grey. 1987. Structural characteristics of an antigen required for its interaction with Ia and recognition by T cells. Nature. 328:395–399. [DOI] [PubMed] [Google Scholar]
- 36.Viola, A., S. Schroeder, Y. Sakakibara, and A. Lanzavecchia. 1999. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 283:680–682. [DOI] [PubMed] [Google Scholar]
- 37.May, M.J., F. D'Acquisto, L.A. Madge, J. Glockner, J.S. Pober, and S. Ghosh. 2000. Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science. 289:1550–1554. [DOI] [PubMed] [Google Scholar]
- 38.Gaide, O., B. Favier, D.F. Legler, D. Bonnet, B. Brissoni, S. Valitutti, C. Bron, J. Tschopp, and M. Thome. 2002. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-κB activation. Nat. Immunol. 3:836–843. [DOI] [PubMed] [Google Scholar]
- 39.Penninger, J.M., and G.R. Crabtree. 1999. The actin cytoskeleton and lymphocyte activation. Cell. 96:9–12. [DOI] [PubMed] [Google Scholar]
- 40.Cherukuri, A., M. Dykstra, and S.K. Pierce. 2001. Floating the raft hypothesis: lipid rafts play a role in immune cell activation. Immunity. 14:657–660. [DOI] [PubMed] [Google Scholar]
- 41.Wang, D., Y. You, S.M. Case, L.M. McAllister-Lucas, L. Wang, P.S. DiStefano, G. Nunez, J. Bertin, and X. Lin. 2002. A requirement for CARMA1 in TCR-induced NF-κB activation. Nat. Immunol. 3:830–835. [DOI] [PubMed] [Google Scholar]
- 42.Su, T.T., B. Guo, Y. Kawakami, K. Sommer, K. Chae, L.A. Humphries, R.M. Kato, S. Kang, L. Patrone, R. Wall, et al. 2002. PKC-β controls IκB kinase lipid raft recruitment and activation in response to BCR signaling. Nat. Immunol. 3:780–786. [DOI] [PubMed] [Google Scholar]
- 43.Wasik, M.A., and C. Morimoto. 1990. Differential effects of cytokines on proliferative response of human CD4+ T lymphocyte subsets stimulated via T cell receptor-CD3 complex. J. Immunol. 144:3334–3340. [PubMed] [Google Scholar]
- 44.Watts, T.H., and M.A. DeBenedette. 1999. T cell co-stimulatory molecules other than CD28. Curr. Opin. Immunol. 11:286–293. [DOI] [PubMed] [Google Scholar]
- 45.Miceli, M.C., M. Moran, C.D. Chung, V.P. Patel, T. Low, and W. Zinnanti. 2001. Co-stimulation and counter-stimulation: lipid raft clustering controls TCR signaling and functional outcomes. Semin. Immunol. 13:115–128. [DOI] [PubMed] [Google Scholar]
- 46.Caruana, G. 2002. Genetic studies define MAGUK proteins as regulators of epithelial cell polarity. Int. J. Dev. Biol. 46:511–518. [PubMed] [Google Scholar]
- 47.Hanada, T., L. Lin, E.V. Tibaldi, E.L. Reinherz, and A.H. Chishti. 2000. GAKIN, a novel kinesin-like protein associates with the human homologue of the Drosophila discs large tumor suppressor in T lymphocytes. J. Biol. Chem. 275:28774–28784. [DOI] [PubMed] [Google Scholar]
- 48.Stilo, R., D. Liguoro, B.D. Jeso, S. Formisano, E. Consiglio, A. Leonardi, and P. Vito. 2004. Physical and functional interaction of CARMA1 and CARMA3 with IKKγ-NEMO. J Biol. Chem. 279:34323–34331. [DOI] [PubMed] [Google Scholar]