

Abstract
Class I and class II MHC-restricted T cells specific for proteins present in myelin have been shown to be involved in autoimmunity in the central nervous system (CNS). It is not yet known whether CD1d-restricted T cells reactive to myelin-derived lipids are present in the CNS and might be targeted to influence the course of autoimmune demyelination. Using specific glycolipid-CD1d tetramers and cloned T cells we have characterized a T cell population reactive to a myelin-derived glycolipid, sulfatide, presented by CD1d. This population is distinct from the invariant Vα14+ NK T cells, and a panel of Vα3/Vα8+ CD1d-restricted NK T cell hybridomas is unable to recognize sulfatide in the presence of CD1d+ antigen-presenting cells. Interestingly, during experimental autoimmune encephalomyelitis a model for human multiple sclerosis, sulfatide-reactive T cells but not invariant NK T cells are increased severalfold in CNS tissue. Moreover, treatment of mice with sulfatide prevents antigen-induced experimental autoimmune encephalomyelitis in wild-type but not in CD1d-deficient mice. Disease prevention correlates with the ability of sulfatide to suppress both interferon-γ and interleukin-4 production by pathogenic myelin oligodendrocyte glycoprotein-reactive T cells. Since recognition of sulfatide by CD1d-restricted T cells has now been shown both in mice and humans, study of murine myelin lipid-reactive T cells may form a basis for the development of intervention strategies in human autoimmune demyelinating diseases.
Keywords: sulfatides, CD1d, EAE, NK T cells, glycolipids
Introduction
The discovery of CD1-dependent antigen presentation pathways has provided a mechanism by which T cells can also recognize lipid antigens (1–3). CD1 molecules are MHC class I–like, β2-microglobulin–associated nonpolymorphic proteins that can be categorized into two groups: group 1, comprised of CD1a, CD1b, and CD1c, is present in humans; and group 2, the CD1d molecule, is present in most species examined so far (4, 5). Crystal structures of CD1d and CD1b molecules have revealed a deep hydrophobic groove well suited for the binding of long alkyl chains of amphipathic lipids and thus allow carbohydrate or other hydrophilic residues of the lipid to interact with the TCR (6, 7). Consistent with this, crystal structure of CD1a complexed with sulfatide suggests binding of the sphingosine and fatty acid chains within the hydrophobic grooves with the exposed head group available for TCR recognition (8). CD1-reactive T cells are thought to be diverse and include the invariant population that uses conserved CDR3 regions, Vα14-Jα18+ in mice and Vα24-Jα15+ in humans (3, 9, 10). Most of the detailed understanding of the phenotype, development, and modulation of immune responses by lipid-reactive T cells has been derived from the use of a surrogate antigen, the marine sponge-derived glycolipid, α-galactosyl ceramide (α-GalCer), which is recognized by the invariant NK T cell population in the context of CD1d molecules. Unlike α-GalCer, most mammalian glycolipids have the carbohydrate moiety attached in a β-linkage.
CD1d-restricted T cells are thought to be self-reactive, but with the exception of two studies, one suggesting recognition of phospholipids (11) and another indicating expansion of T cells reactive to a tumor-derived GD3 molecule (12), self-glycolipids that stimulate CD1d-reactive T cells have not been described. In an attempt to characterize the phenotype and function of mammalian self-glycolipid–reactive T cells, we have focused on myelin in the nervous system for several reasons. It is rich in glycolipids that are formed from a common precursor ceramide that has a potential CD1d-binding domain similar to the well-characterized α-GalCer. One fifth of the total galactolipid in myelin occurs in the form of sulfatide (or 3′-sulfogalactosyl ceramide) in which the 3′-OH moiety on the galactose is sulfated. Furthermore, sulfatide has been shown to be a promiscuous CD1 ligand and can be presented by human CD1a, CD1b, and CD1c molecules (13). The myelin sheath is the target of an autoimmune inflammatory process in multiple sclerosis (MS) (14) and in its animal model experimental autoimmune encephalomyelitis (EAE), which is characterized by inflammation and demyelination in the central nervous system (CNS) accompanied by paralysis (15). Recently, myelin-glycolipid–reactive T cells have been found with increased frequency in peripheral blood of MS patients (16). Although class I and class II MHC-restricted myelin protein-reactive T cells have been shown to be involved in autoimmune pathology (14, 17, 18), it is not known whether CD1d-reactive T cells recognizing myelin lipids are part of the mature T cell repertoire and might be targeted to influence the course of demyelination or paralysis. Here we show that sulfatide-reactive CD1d-restricted T cells represent a distinct population of T cells that infiltrate into the CNS during EAE and can be appropriately activated to prevent disease.
Materials and Methods
Mice.
C57BL/6 (BL/6) mice were from Jackson Labs. CD1d1−/− and Jα18−/− mutant BL/6 mice originally generated in the laboratories of Dr. Luc Van Kaer (Vanderbilt University, Nashville, TN) and Dr. M. Taniguchi (Chiba University, Chiba, Japan) were provided by Dr. Mitch Kronenberg (LIAI). BL/6.CGT−/− mice were provided by Dr. Brian Popko (University of North Carolina, Chapel Hill, NC). All mice were bred and maintained in specific pathogen-free conditions in facilities at the LIAI and TPIMS.
Disease Model.
EAE was induced in BL/6 mice with MOG35–55 peptide emulsified in complete Freund's adjuvant followed by injection with the pertussis toxin (MOG35–55/CFA/PT), essentially as described (19).
Reagents.
Synthetic α-GalCer and β-GalCer were provided by Dr. Koezuka (Kirin Brewery Co., Tokyo, Japan). Purified (>99%) myelin-derived lipids, sulfatide, sphingomyelin, and the gangliosides mono-GM1, GD1a, and GD1b were purchased from Fluka, Sigma-Aldrich, or Maitreya. All lipids were dissolved in vehicle (0.5% polysorbate 20 and 0.9% NaCl solution) and diluted with PBS for in vitro proliferation and in vivo injections. The Vα3+/Vα8+ and Vα14+ hybridomas were provided by Drs. S. Cardell (University of Lund), A. Bendelac (University of Chicago, Chicago, IL) and M. Kronenberg (LIAI), respectively. All monoclonal antibodies were purchased from BD Biosciences.
Measurement of Proliferative and Cytokine Responses.
For proliferative responses, 8 × 105 splenocytes were cultured in vitro in the presence of graded concentrations of glycolipids, (1–50 ng/ml for α-GalCer and 0.5–50 μg/ml for other lipids). Quantitation of these responses was accomplished by measuring incorporation of [3H]-thymidine as described earlier (19). For comparison of the proliferative and cytokine secretion profiles, splenocytes from naive animals were cultured in the presence of graded concentrations of sulfatide. Supernatants from 16- or 48-h cultures were used in ELISA assays to measure cytokine secretion. Standard sandwich ELISA and ELISPOT were used to measure cytokine levels and the frequency of cytokine-secreting cells, using reagents from BD Biosciences as described (20). In hybridoma assays, IL-2 secretion was determined in culture supernatants after 16 h incubation with APC and different glycolipids.
Cellular ELISPOT Analysis.
IFN-γ– and IL-4–producing cells were enumerated from glycolipid-pulsed lymphocytes isolated from immunized mice by cellular ELISPOT assay as described earlier (20). Briefly, splenocytes (5 × 106 cells/ml) were cultured for 48 h in 24-well plates either with medium alone or with sulfatide (40 μg/ml). Millititer HA nitrocellulose plates (Millipore) were coated overnight at 4°C with anti–IFN-γ or anti–IL-4 Abs. After blocking coated plates, antigen-stimulated cells were added at graded concentrations for 24 h at 37°C. The wells were then incubated with biotin-conjugated anti–IFN-γ or anti–IL-4 mAbs followed by incubation with avidin peroxidase (Vector Laboratories). Spots were developed by the addition of 3-amino-9-ethylcarbazole substrate (Sigma-Aldrich) and counted using a computerized image analysis system (Ligh-tools Research) and the image analyzer program, NIH Image 1.61.
Isolation of Mononuclear Cells from the Liver and CNS.
Single cell suspensions were prepared from liver, brain, and spinal cords using discontinuous density gradient centrifugation as described previously (19). Briefly, after whole body perfusion with chilled PBS infiltrating cells from brain and spinal cord were suspended in 70% Percoll (Amersham Biosciences) in HBSS and overlaid with equal volumes of 37 and 30% Percoll, and the gradient was centrifuged at 500 g for 15 min. Mononuclear cells were harvested from the 37–70% interface and washed in HBSS.
Tetrameric mCD1d–lipid Complexes.
PE- or Cy-chrome–labeled mCD1d1–α-GalCer tetrameric complexes were made in a baculovirus expression system as described previously (19, 21). “Unloaded” mCD1d1 tetramers were prepared by preincubating biotinylated mCD1d protein with vehicle only. To produce self-lipid–CD1d tetramers, biotinylated mCD1d protein was incubated with sulfatide at a molar ratio of 1:6 for 18–20 h.
Intracellular Cytokine Analysis.
For intracellular staining, anti–FcR-γ and neutravidin-treated CNS-infiltrating mononuclear cells were cultured for 5 h in the presence of Brefeldin A and permeabilized using Cytofix/Cytoperm Plus™ (BD Biosciences) and stained using either FITC-labeled anti–IL-4 clone BVD4–1D11 or FITC-labeled anti–IFN-γ clone XMG1.2 (BD Biosciences). Staining was done in conjunction with anti-TCRβ chain antibody (H57–597; BD Biosciences) that reacts with a common epitope of the β-chain of the receptor complex on α-β TCR-expressing T cells of all mouse strains tested, according to the manufacturer's protocol.
Flow Cytometry.
Mononuclear cells suspended in PBS buffer containing 0.001% NaN3 (wt/vol) and 5% BSA (vol/vol) were first treated with antibodies against FcR-γ (2.4G2) and then labeled with the indicated antibodies. For mCD1d–lipid tetramer staining, cells were also treated with neutravidin (Molecular Probes) and then incubated with tetramers for 1 h as described (19, 21). All FITC-, PE-, Cy-Chrome, and allophycocyanin-conjugated mAbs were acquired from BD Biosciences. For flow cytometry staining, 2.5 μg of tetramerized mCD1d was used. Flow cytometric analysis was performed on a FACSCalibur instrument using CellQuest software (Becton Dickinson). In each flow cytometric staining profile, the percentage of the gated population is shown in the top right corner.
Statistical Analysis.
Statistical analyses were performed using χ2 and student's t tests as appropriate. p-values <0.05 were considered statistically significant.
Results
A CD1d-dependent Immune Response to a Myelin-derived Glycolipid, Sulfatide.
To assess the nature of the response to self-glycolipids and their dependence on the CD1d antigen presentation pathway, splenocytes from naive wild-type BL/6 or CD1d−/− mice were cultured with graded doses of glycolipids and assayed for proliferation and cytokine secretion (Fig. 1, A and B). Splenic cells from WT but not from CD1d−/− mice proliferate and produce both IFN-γ and IL-4 in response to sulfatide. Proliferation and cytokine secretion in response to sulfatide were lower in magnitude than to α-GalCer, possibly indicating a lower cell number or lower TCR avidity of sulfatide-reactive T cells in comparison to the α-GalCer–reactive Vα14 invariant population. The failure of sulfatide to elicit a response in CD1d−/− mice indicates the requirement for CD1d-restricted presentation. There were no detectable responses to other self-glycolipids capable of binding to CD1d molecules, including β-GalCer, sphingomyelin, and GM1.
Figure 1.

A distinct CD1d-restricted sulfatide-reactive T cell population in naive mice. (A) Proliferative response of splenocytes from naive BL/6 or BL/6.CD1d−/− mice in response to an in vitro stimulation with indicated self-lipids (10 μg/ml) or α-GalCer (10 ng/ml) suspended in PBS/vehicle. Each symbol represents response of an individual animal in each group (n = 4–11), with the mean proliferative response indicated by a horizontal bar. *P = 0.004. (B) In vitro cytokine and proliferative response of splenocytes from naive BL/6 (▪) or BL/6.CD1d−/− (○) mice to a titrated dose of sulfatide. One of three representative experiments is shown. (C) FACS® analysis of lymphocytes from the liver, spleen, and thymus from BL/6 (CD1d+/+) or BL/6.CD1d−/− (CD1d−/−) mice after staining with fluoresceinated tetramers or anti-NK1.1 mAbs in conjunction with the anti-TCRβ chain antibody. One of three representative experiments is shown. (D) Flow cytometric profile of splenocytes from BL/6 mice stained simultaneously with the CyChrome-labeled α-GalCer/CD1d tetramers and with the PE-labeled sulfatide/CD1d tetramers. (E) Flow cytometric profile of splenocytes from BL/6 and BL/6.Jα18−/− mice stained with α-GalCer/CD1d tetramers or sulfatide/CD1d tetramers.
Sulfatide/mCD1d Tetramer+ Cells Are Part of the Naive T Cell Repertoire.
To define the cell population responsible for the proliferative and cytokine responses to sulfatide, tetrameric mCD1d molecules loaded with sulfatide were used to stain T cells in the thymus, liver, and spleen (Fig. 1 C). Sulfatide/CD1d tetramer+ T cells are predominantly CD4+CD8− (∼90%) and comprise 5–7% of mononuclear cells in liver, 0.3–0.5% in thymus, and ∼0.3–0.7% in spleen and thus represent ∼1/3–1/4 the number of α-GalCer–reactive iNK T cells in BL/6 mice. We found no staining with CD1d complexes loaded with GM1 (Fig. 1 C), GD1a, GD1b, β-GalCer, or with unloaded tetramers. There was no significant staining of cells in liver and spleen from CD1d−/− mice with sulfatide or α-GalCer tetramers (Fig. 1 C, right). Although most sulfatide/CD1d tetramer+ cells in the thymus do not express NK1.1, 25–28% are NK1.1+ in spleen and liver. The relatively lower level of staining with sulfatide tetramer may indicate a lower TCR avidity of sulfatide-reactive cells in comparison to the higher avidity Vα14 TCR (22) of the α-GalCer–reactive population and/or the presence of diverse alkyl chains within the myelin-derived sulfatide preparation used here. Indeed the higher molar requirement of lipid to stimulate sulfatide-reactive T cells compared with Vα14+ T cells (Fig. 2, C and D) is consistent with the notion that these cells bear lower avidity TCR.
Figure 2.
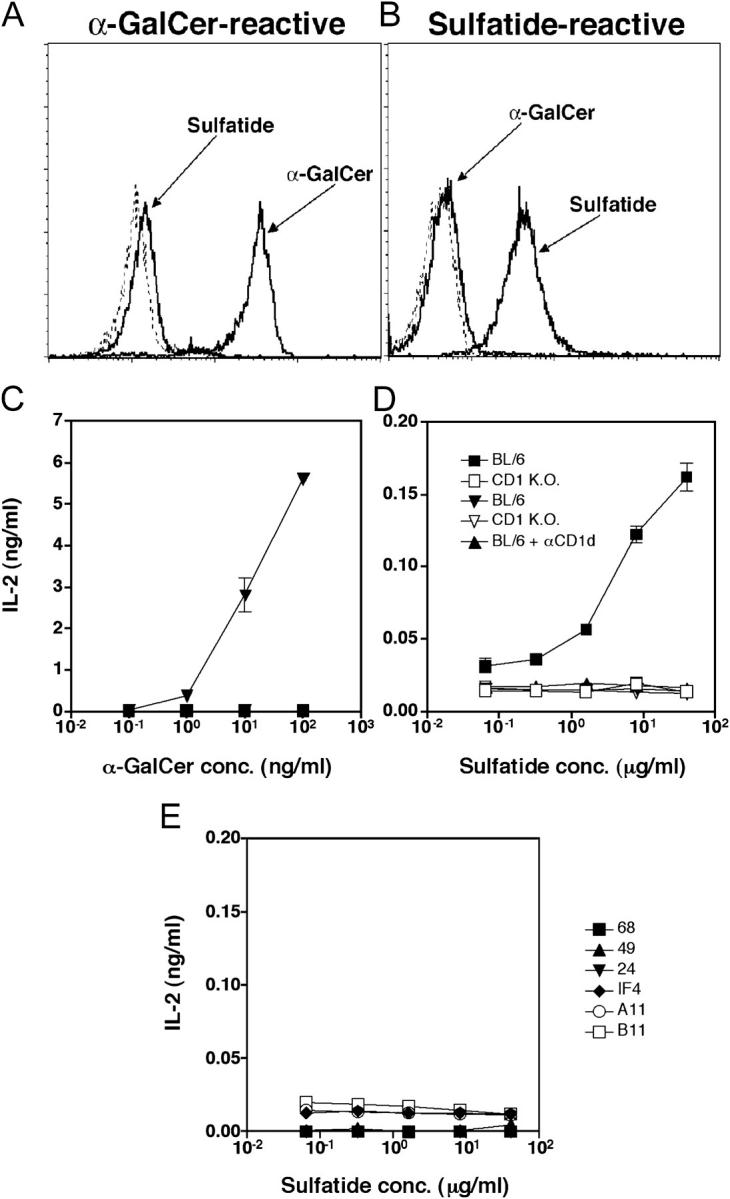
Sulfatide-reactive T cell hyridomas stain with sulfatide/CD1d tetramer and secrete IL-2 in response to sulfatide in a CD1d-dependent manner. A representative T cell hybridoma reactive to sulfatide (Hy19.3) (B and D) and a α-GalCer–reactive hybridoma (Hy1.2) (A and C) were stained with sulfatide/CD1d or α-GalCer/CD1d tetramers and analyzed by FACS®. Unloaded tetramer or GM1-filled tetramer staining is shown by dotted lines. For IL-2 secretion assay, hybridomas (50,000 cells/well) were incubated with irradiated (3000R) splenocytes (400,000 cells/well) from wild-type C57BL/6 (BL/6) or CD1d−/− BL/6 (CD1KO) mice in the absence or presence of graded concentrations of α-GalCer or sulfatide, as shown. For blocking of CD1d-dependent presentation, 10 μg/ml of anti-CD1d mAb (1B1 [47]) or an isotype-matched mAb (IgG2b; BD Biosciences) was added during the incubation period. IL-2 secretion levels by Hy19.3 in response to plate-bound anti-CD3 were 1/8–1/10 that of Hy1.2. Concentration of IL-2 in the supernatants is shown. (E) The CD1d-restricted Vα3+ (A11, 24, IF4), Vα8+ (B11, 49), and Vα4+ (68) T cell hybridomas were incubated with irradiated CD1d+ splenocytes in the presence of graded concentrations of sulfatide, as above. Concentration of IL-2 in the supernatants is shown. All of the hybridomas responded well to plate-bound anti-CD3.
Sulfatide-reactive T Cells Are Distinct and Do Not Overlap with the α-GalCer–reactive Invariant T Cell Population.
To determine whether sulfatide-reactive T cells are part of the Vα14 iNK T cell population or represent a distinct CD1d-restricted T cell repertoire, splenic cells were simultaneously stained with α-GalCer/CD1d tetramer conjugated to CyChrome and sulfatide/CD1d tetramer conjugated to PE, and analyzed by flow cytometry (Fig. 1 D). A clearly distinct and nonoverlapping staining profile with the two tetramers indicates that sulfatide-reactive cells belong to a different subset of T cells. The difference in T cell repertoire between the sulfatide- and α-GalCer–reactive T cells is further confirmed by the presence of sulfatide/CD1d but not α-GalCer/CD1d tetramer+ cells in Jα18−/− mice (Fig. 1 E). These mice lack α-GalCer–reactive iNK T cells owing to the absence of the Jα18 gene segment used by the invariant Vα14 TCR (23).
CD1d-restricted Vα3/Vα8+ NK T Cells Do Not Recognize Sulfatide.
A minor autoreactive CD1d-dependent NK T cell population has been shown to utilize semiinvariant TCRs encoded by the TCR Vα3 or Vα8 gene segments (24, 25). Although sulfatide-reactive T cells are distinct from the invariant Vα14+ T cell population, we wanted to examine whether the Vα3 or Vα8+ subsets of NK T cells are part of the sulfatide-reactive population. To avoid background staining problems and nonavailability of an anti-TCR mAb capable of staining Vα8+ T cell subset, we have directly analyzed several Vα3+ and Vα8+ NK T cell hybridomas for their ability to secrete IL-2 in response to sulfatide. As shown in Fig. 2 E, none of these hybridomas secretes IL-2 upon stimulation in the presence of CD1d+ APC and sulfatide, although they are able to secrete IL-2 in response to the plate-bound anti-CD3. None of the Vα3+ or Vα8+ hybridomas stained with the sulfatide/CD1d tetramer.
Sulfatide-reactive T Cell Hybridomas Stain with Sulfatide/CD1d Tetramer and Secrete IL-2 in a CD1d-dependent Manner.
To further examine the presence of sulfatide-reactive T cells at the single cell level, newly established T cell hybridomas and CD1d-restricted autoreactive hybridomas generated earlier (24, 25) were analyzed using staining with specific CD1d tetramers and IL-2 secretion in response to glycolipids. A representative sulfatide-reactive T cell hybridoma (Hy19.3, Vα14−) stains with sulfatide/CD1d tetramer and not α-GalCer tetramer. In contrast, a α-GalCer–reactive T cell hybridoma (Hy1.2, Va14+) stains with α-GalCer/CD1d tetramer but not with sulfatide tetramer (Fig. 2, A and B). These hybridomas do not show any significant staining with either unloaded or GM1-loaded tetramer. Furthermore, the sulfatide-reactive hybridoma secretes IL-2 in the presence of syngenic CD1d+ APC (BL/6) pulsed with sulfatide but not with α-GalCer. In contrast, the α-GalCer–reactive hybridoma responds to α-GalCer but not to sulfatide.
To further examine the MHC restriction of sulfatide-reactive T cells, IL-2 secretion by the T cell hybridoma was analyzed in the presence of sulfatide-pulsed APC from CD1d−/− mice and in the presence of a blocking anti-CD1 mAb. As shown in Fig. 2, C and D, IL-2 secretion was not detected when APC from CD1d−/− (CD1KO) mice were used. Additionally, cytokine secretion was almost completely blocked in the presence of blocking anti-CD1d mAb and sulfatide-pulsed APC from CD1d+/+ (BL/6) mice. Furthermore, IL-2 secretion was also detected when CD1d-transfected A20 cells were used as APC (unpublished data). These data clearly show that Vα14− T cells recognize sulfatide in the context of CD1d molecules.
Absence of Endogenous Sulfatide Results in a Severalfold Increase in Sulfatide/CD1d Tetramer+ Cells.
CD1d-reactive Vα14 invariant T cell development is thymus dependent (26). Recent studies suggest that maturation of these cells is subject to positive selection and that the early steps of this process might be similar to those proposed for the classical MHC I and II–restricted T cells (10). Because CD1d self-glycolipid ligands have not been well defined, it is not yet known whether CD1d-restricted T cells are also subject to thymic negative selection. To determine whether the sulfatide-reactive T cell repertoire is altered in the absence of the endogenous glycolipid, thymocytes from mice functionally deficient in the gene encoding the enzyme UDP-galactose ceramide galactosyltransferase (CGT) (27), which is required for myelin galactolipid synthesis, were analyzed using lipid tetramers (Fig. 3). These mice lack all galactolipids, including sulfatide, and exhibit tremor and paralysis and do not survive beyond 3–6 wk of age. Sulfatide/CD1d- but not α-GalCer/CD1d-tetramer+ T cells are increased >10-fold in the thymus of CGT−/− mice (3.21%) compared with WT mice (0.28%). Although, sulfatide/CD1d tetramer+ cells appear to be TCRlow, their CD4 and CD8 expression is similar to those of α-GalCer/CD1d tetramer+ cells. These data suggest that sulfatide-reactive T cells may be subject to negative selection in the thymus. This is consistent with recent reports suggesting negative selection of classical Vα14+ iNK T cells using the nonmammalian glycolipid α-GalCer (28, 29).
Figure 3.
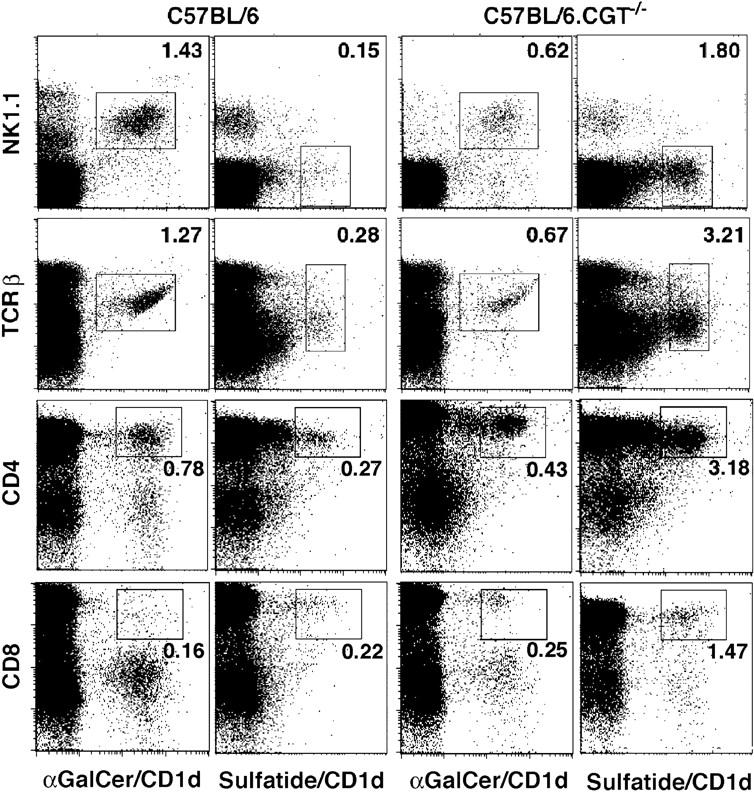
Sulfatide/CD1d tetramer+ cells are increased severalfold in mice lacking the CGT. Thymocytes pooled from three age-matched, naive BL/6 or BL/6.CGT−/− mice were stained with α-GalCer/CD1d or sulfatide/CD1d tetramers in conjunction with anti-NK1.1, anti-TCRβ, anti-CD4, or anti-CD8 mAb and analyzed by FACS®. The CD4 and CD8 staining profiles of thymocytes were similar in both sets of mice. This is representative of two independent experiments.
Selective Enrichment of IFN-γ–secreting Sulfatide-reactive T Cells in the CNS During EAE.
Although sulfatide is present in most membranes, it is particularly enriched in the myelin sheath, which becomes the target of autoimmune inflammation during EAE. To determine whether sulfatide-reactive T cells are present in CNS-infiltrating lymphocytes, mononuclear cells isolated from the brain and spinal cord of BL/6 mice with ongoing disease were analyzed following staining with tetramers and compared with cells in liver and spleen (Fig. 4, A and D). Sulfatide-reactive T cells accumulate in the CNS (3.8 ± 1.4%) selectively during clinical EAE and are not detectable in naive mice, or of particular interest, in immunized mice that do not develop clinical paralysis. In contrast, α-GalCer–reactive T cells constitute only a minor part (0.96 ± 0.13%) of the CNS infiltrate in diseased animals. A similar increase in the number of sulfatide/CD1d tetramer+ T cells is also found in the CNS tissue from B10.PL mice with myelin basic protein (MBP)-induced EAE (unpublished data).
Figure 4.
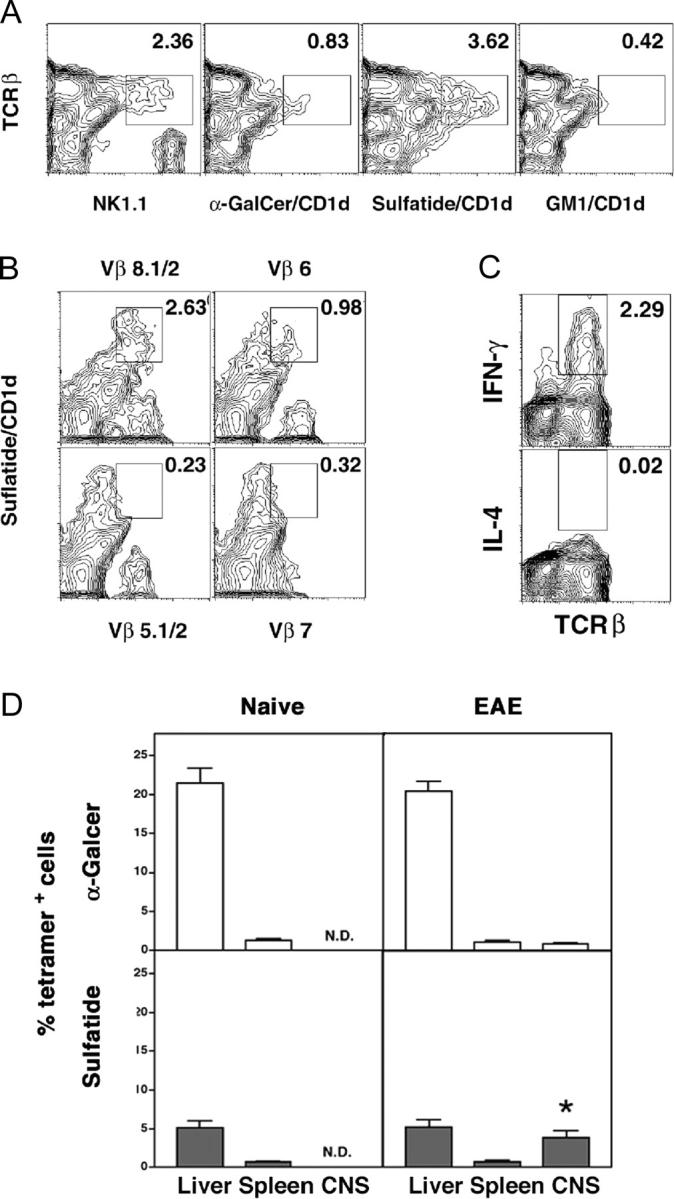
Sulfatide-reactive T cells infiltrate the CNS during the course of EAE. (A) Mononuclear cells from the CNS of diseased BL/6 mice (n = 7) were pooled and stained with the indicated lipid/CD1d tetramers and analyzed by FACS®. A representative of two experiments is shown. (B) To analyze the TCR Vβ gene usage, sulfatide/CD1d tetramer+ cells were stained with various anti-Vβ mAbs. A typical positive and negative staining profile is shown. (C) Ex vivo intracytoplasmic staining of the CNS-infiltrating cells for IFN-γ and IL-4. (D) The percentages of α-GalCer– or sulfatide-reactive T cells in spleen, liver, and the CNS of naive animals or mice with EAE were determined by FACS® analysis. The plot represents the averaged data of three independent experiments (a total of 19 mice in each group). *P < 0.0001.
To determine their TCR Vβ repertoire, sulfatide/CD1d tetramer+ T cells were also stained with a set of available anti-TCR Vβ-chain mAbs (BD Biosciences). Sulfatide-reactive T cells infiltrating CNS predominantly utilize TCR Vβ8 and Vβ6 and not the Vβ2 or Vβ7 gene segments used by iNK T cells (Fig. 4 B). There was no significant staining with other Vβ-specific antibodies analyzed (see Materials and Methods). Thus, only TCR Vβ8 usage is shared between α-GalCer– and sulfatide-reactive T cells in BL/6 mice.
Since sulfatide-reactive T cells accumulate in the CNS in diseased animals and show an activated phenotype, we next determined the secretion of type 1 (IFN-γ) or type 2 (IL-4) cytokines by the infiltrating cells directly ex vivo using intracytoplasmic staining in the absence of in vitro stimulation with sulfatide. Fig. 4 C shows that the CNS-infiltrating cells secrete only IFN-γ, with undetectable levels of IL-4. This may be related to the proinflammatory milieu of CNS in diseased mice.
Increase in Sulfatide-reactive T Cells After Immunization with Sulfatide.
Myelin protein–reactive class I or class II–restricted T cells have been shown to expand in the periphery after immunization with myelin-derived proteins, for example, MBP. A sensitive ELISPOT assay has been successfully used to examine the frequency of these cells under different conditions of priming (20, 30). Since sulfatide-reactive T cells are present in small numbers, especially in spleen, we have used cytokine ELISPOT assays to examine the frequency of these T cells in splenocytes after immunization with sulfatide. As shown in Fig. 5, immunization of mice with sulfatide expands a T cell population in splenocytes that produces IFN-γ and IL-4 in response to an in vitro recall with sulfatide. The frequency of sulfatide-reactive T cells is three- to fourfold higher in sulfatide-immunized mice compared with mice immunized with mono-GM1 or PBS. These data indicate an expansion of peripheral myelin-glycolipid–reactive T cells after immunization with the self-glycolipid and parallels that of myelin protein–reactive T cells after challenge with myelin-derived proteins.
Figure 5.

Increased frequency of cytokine-secreting, sulfatide-reactive lymphocytes after immunization with sulfatide. Groups of mice (two in each) were immunized in complete Freund's adjuvant with 20 μg of sulfatide or with 20 μg of mono-GM1 or with PBS/vehicle alone. 10 d later, splenocytes were isolated, recalled in vitro with sulfatide, and subjected to ELISPOT analysis as described in Materials and Methods. The average number of spots/106 spleen cells for both IFN-γ and IL-4 in each group is shown.
CD1d-dependent Prevention of EAE by Sulfatide.
i.v. delivery of α-GalCer results in the rapid activation of iNK T cells followed by the disappearance of α-GalCer tetramer+ cells and significant protection from antigen-induced EAE (19, 31). To determine whether in vivo activation of sulfatide-reactive T cells influences the course of EAE, both wild-type and CD1d−/− mice were given 20 μg of sulfatide in vehicle or vehicle only and at the same time challenged with MOG35–55/CFA/PT for the induction of EAE (Fig. 6 and Table I). Coinjection with sulfatide results in significant protection from disease that is dependent on the presence of the CD1d molecules, since CD1d-deficient animals are not protected. In contrast, injection with other self-glycolipids has no effect on the course of EAE (Table I). Mice injected with sulfatide alone in the presence of CFA/PT immunization do not develop clinical signs of disease and lack detectable numbers of sulfatide/CD1d tetramer+ cells in the CNS. Similarly, a single injection of sulfatide into B10.PL mice also prevents MBPAc1-9–induced EAE in a CD1d-dependent fashion (not depicted). Timing of injection with α-GalCer in relation to the immunization with MBP or MOG has been shown to influence the efficacy of protection from EAE (19). To examine this issue for sulfatide, mice were injected either 1 wk before or 1 wk after the MOG/CFA/PT immunization for the induction of disease. As shown in Table I, in both immunization protocols mice were significantly protected from EAE.
Figure 6.
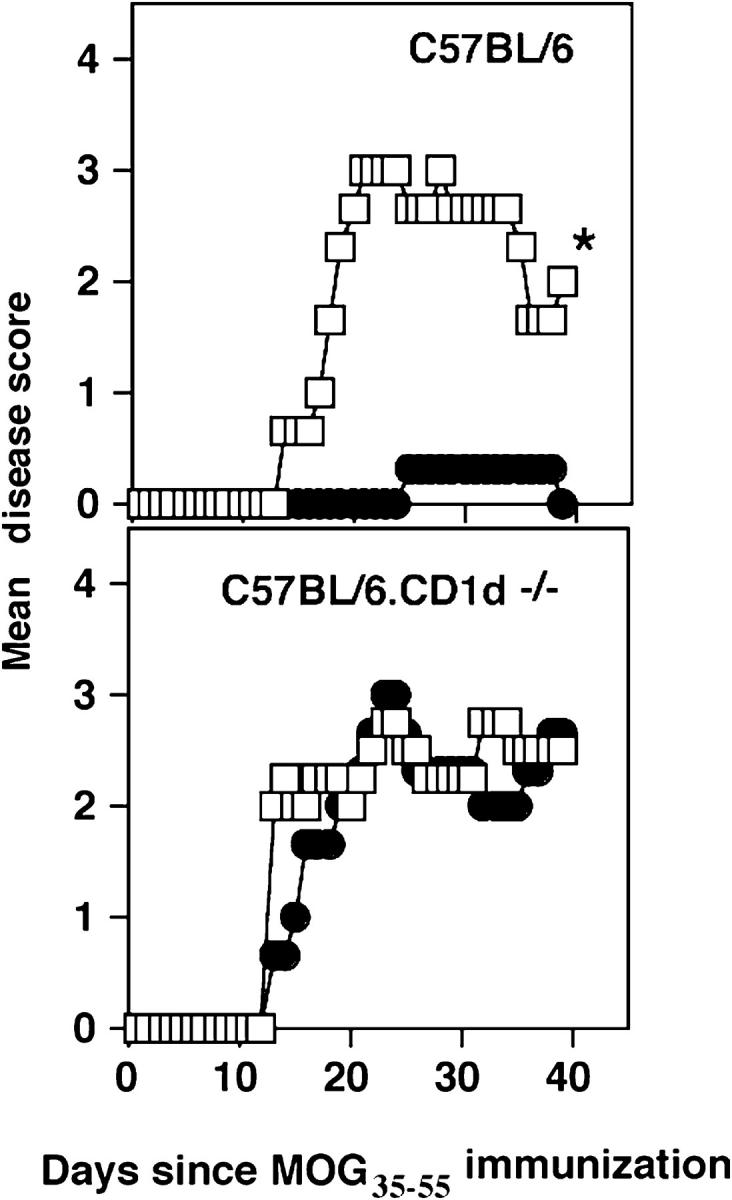
Immunization of mice with sulfatide protects mice from EAE. In a representative experiment, groups of female BL/6 or BL6.CD1−/− mice were immunized i.p. with 20 μg of sulfatide (•) (n = 4) or with PBS/vehicle alone (□) (n = 4) at the time of disease induction with MOG35–55/CFA/PT. The mean clinical disease course of mice in each group is shown. The summary of all independent experiments is provided in Table I. *P < 0.0001.
Table I.
Specificity of Protection from EAE After Injection with Myelin-derived Sulfatide a
| Treatment | MOG35-55/CFA/PT | Incidence (no. of mice [maximum score]) |
Mean maximum score | Mean day of onset |
|---|---|---|---|---|
| C57BL/6 | ||||
| PBS | + | 24/24 [2(2), 7(3), 12(4), 3(5)] |
3.7 | 12.9 |
| Sulfatide (d 0) | + | 13/23b[10(0), 8(2), 1(3), 1(4)] | 1.8 | 18 |
| Sulfatide (d − 7) | + | 4/6 [1(2), 3(3)] |
1.9 | 15.4 |
| Sulfatide (d + 7) | + | 2/6 [2(2)] |
0.7 | 16.1 |
| GM1 | + | 11/11 [2(2), 5(3), 4(4)] |
3.2 | 16 |
| Sphingomyelin | + | 4/4 [2(2), 2(4)] |
3.0 | 13.3 |
| β-GalCer | + | 6/6 [1(2), 1(3), 2(4), 2(5)] |
4.0 | 14.3 |
| Sulfatide | − | 0/5 | ||
| C57BL/6 – CD1dKO | ||||
| PBS | + | 7/7 [2(2), 2(3), 2(4), 1(5)] |
2.9 | 13 |
| Sulfatide | + | 8/8 [1(2), 3(3), 3(4)] |
3.0 | 12.8 |
Groups of mice were injected i.p. with 20 μg of sulfatide, mono-GM1, sphingomyelin, β-GalCer, or PBS/vehicle only. On the same day (d 0), injected mice were immunized s.c. with MOG35-55/CFA/PT for the induction of EAE. Sulfatide was also injected either 7 d prior to (d − 7) or after (d + 7) the immunization with MOG35-55/CFA/PT. The incidence, mean maximum score, and mean day of onset of clinical EAE is shown.
P < 0.0001.
Protection from Disease Correlates with Inhibition of Cytokine Secretion by MOG-reactive T Cells.
The mechanism of iNK T cell–mediated protection from autoimmune diseases (19, 31–35) has been attributed either to immune deviation or to inhibition of the differentiation of effector functions of pathogenic self-protein–reactive T cells. To gain insight into the mechanism of sulfatide-induced modulation of EAE, levels and frequency of both IFN-γ– and IL-4–secreting MOG-reactive T cells were determined in draining LN cells using ELISA and ELISPOT analysis, respectively, in groups of MOG35–55-immunized animals coinjected either with sulfatide or PBS only (Fig. 7). In sulfatide-injected mice, the numbers of both IFN-γ– and IL-4–secreting cells and levels of cytokine secretion by MOG35–55-reactive T cells are diminished significantly. There is no significant difference in response to a polyclonal stimulus, protein-purified derivative of mycobacterium.
Figure 7.

Inhibition of cytokine secretion by MOG-reactive T cells after coinjection with sulfatide. Groups of female BL/6 mice were coinjected i.p. with either 20 μg of sulfatide in PBS/vehicle or PBS/vehicle alone at the time of s.c. challenge with 100 μg MOG35–55 in CFA. 10 d later, LN cells were cultured with graded concentrations of MOG35–55 or protein-purified derivative of Mycobacterium (PPD) for the measurement of proliferation and cytokine secretion by ELISA and ELISPOT analysis. Averaged responses within the group are plotted. A representative of three independent experiments is shown. *P-value < 0.004.
Discussion
This study demonstrates that sulfatide is a self-glycolipid ligand recognized by a population of CD1d-restricted T cells that is distinct from the invariant Vα14+ NK T cell population and is part of the mature naive T cell repertoire. Sulfatide-reactive T cells also appear to be distinct from other CD1d-restricted T cell populations expressing semiinvariant Vα3.2-Jα9 or Vα8 TCRs. Sulfatide-reactive but not α-GalCer–reactive T cells are increased severalfold within the CNS during EAE. Notably, treatment of mice with sulfatide prevents antigen-induced EAE by inhibiting the effector function of self-protein–reactive pathogenic T cells.
Several observations demonstrate the distinctiveness of sulfatide-reactive T cells from the α-GalCer–reactive Vα14+ iNK T lymphocytes. (a) These are two nonoverlapping populations, as revealed by simultaneous staining with sulfatide/CD1d− and α-GalCer/CD1d tetramers; (b) sulfatide/CD1d tetramer+ cells are present in Jα18−/− mice, which lack iNK T cells, showing that sulfatide-reactive T cells do not use the invariant TCR using the Vα14-Jα18 gene segment. This is further confirmed using sulfatide-reactive T cell hybridomas, which are also Vα14 negative. (c) There is a differential increase in the frequency of these cells in the CNS during EAE and in galactolipid-deficient mice; (d) though these cells share the TCR Vβ8 usage, sulfatide-reactive T cells do not use theVβ2 or Vβ7 chains predominantly used by the classical Vα14+ iNK T cells; (e) higher percentages of sulfatide- but not α-GalCer–reactive T cells in CGT−/− mice lacking self-galactolipids indicate that these two populations may recognize different lipids in vivo and that at least some CD1d-restricted T cells, like the classical MHC I– or II–restricted T cells, may be subject to central tolerance; (f) α-GalCer–reactive Vα14+ T cell hybridomas do not secrete IL-2 in response to sulfatide, whereas sulfatide-reactive Vα14− hybridomas respond to sulfatide and are nonresponsive to α-GalCer or to other self-glycolipids, such as β-GalCer, sphingomyelin, or GM1. Furthermore, sulfatide-reactive T cells are also different from the other CD1d-restricted T cells that use semiinvariant TCR. These studies show the diverse nature of the CD1d-restricted T cell repertoire and demonstrate the feasibility of using self-glycolipid–CD1 tetramers as a powerful means for investigating their involvement in a variety of autoimmune and infectious diseases.
Lack of detectable immune reactivity to other myelin-derived lipids such as mono-GM1, GD1a, and GD1b may reflect their low availability for a CD1d-dependent display or their cryptic T cell repertoire. Consistent with studies in humans (13), our data suggest that the sulfate moiety and not simply the negative charge in sulfatide is required for interaction with the TCR. Thus, we have found no response to β-GalCer lacking the sulfate group or to mono-GM1, which contains a negatively charged residue sialic acid, despite the fact that both lipids are able to bind to CD1d molecules (36). Further studies using synthetic lipids are required to determine the precise role of saturated versus unsaturated acyl chains of different lengths (C16–24) present in myelin-derived sulfatides in binding either to CD1d molecules or to the TCR.
Accumulation of sulfatide-reactive T cells during the process of autoimmune demyelination in CNS tissue, which is enriched in this glycolipid antigen, suggests that after local inflammation sulfatide may be presented by resident or circulating CD1d+ APC. Increased levels of CD1d expression in the macrophage/microglia fraction of the CNS during EAE in mice (unpublished data) and CD1d-like proteins in guinea pigs with EAE have been found recently (37). Similarly up-regulation of CD1b in chronic-active MS lesions but not in silent lesions of the brain has been reported (38). Thus, after myelin destruction owing to either viral infection or demyelination, APCs in the CNS may present both glycolipids and proteins (39). Our findings demonstrating the enrichment of sulfatide-reactive T cells in target tissue further suggest that determinant spreading (40) during chronic autoimmune responses may involve T cells reactive not only to different protein antigens but also to glycolipids. Consistent with our data in EAE, the frequency of sulfatide-reactive, CD1-restricted T cells secreting IFN-γ has been shown to be significantly higher in active MS patients compared with normal individuals (Duda et al., personal communication).
It is becoming increasingly clear that similar to pathogen-derived protein antigens, microbial lipids can be presented in the context of CD1 molecules, for example, after Mycobacterial infection (5, 41). Consistent with this, expansion of CD1d-reactive invariant Vα14+ T cells (42) or a noninvariant CD1d-restricted T cell population (43) in murine models of colitis and acute hepatitis, respectively, has been reported recently. Additionally, the presence of sulfatide-reactive T cells in the mature naive repertoire and their enrichment in a target tissue also mimic insulin and myelin proteolipid protein-reactive T cells in diabetes and EAE, respectively, (44, 45) and may further indicate parallels between classical MHC-restricted and CD1d-restricted T cells. This is also reflected in data showing increased frequency of sulfatide-reactive T cells in peripheral lymphoid organs after immunization of sulfatide.
Even though a detailed mechanism of sulfatide-mediated modulation of EAE remains to be determined, our study demonstrates that early administration of sulfatide results in the inhibition of both type 1 and 2 cytokine secretion by pathogenic myelin protein–reactive T cells and is sufficient to prevent clinical disease symptoms. This may be similar to the inhibition of diabetogenic T cell differentiation by the invariant Vα14+ T cell population in the absence of activation by α-GalCer (35) and is distinct from the proposed immune deviation of pathogenic T cells after their in vivo activation with α-GalCer resulting in protection from diabetes (32, 33) and EAE (19, 31, 46). These earlier studies suggested that Th2 cytokines, such as IL-4 and IL-10, are likely to be involved in α-GalCer–mediated modulation of autoimmune disease. In contrast, our preliminary data suggest that these Th2 cytokines are not involved in sulfatide-mediated protection from EAE, since administration of anti–IL-4 mAb had no effect on this modulation (unpublished data). It is notable that injection of sulfatide at different times relative to the immunization with MOG peptide results in significant protection from EAE (Table I). Furthermore our preliminary experiments suggest that an appropriate dose of sulfatide injected during ongoing EAE is effective in reversing disease. These findings, along with the studies showing an increased frequency of sulfatide-reactive CD1-restricted T cells in MS patients, demonstrate that in addition to myelin protein–reactive class I or II MHC-restricted T cells, myelin-glycolipid–reactive, CD1d-restricted T cells represent potential targets for intervention in human autoimmune demyelinating diseases.
Acknowledgments
We thank Mitch Kronenberg and Randle Ware for critical review of the manuscript, Olga Naidenko (LIAI), Masaru Taniguchi, and Brian Popko for providing the construct for mCD1d expression and Jα18−/− and CGT−/− mice.
V. Kumar is supported by grants from the National Institutes of Health and the National Multiple Sclerosis Society, USA.
Abbreviations used in this paper: CGT, UDP-galactose ceramide galactosyltransferase; CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; GalCer, galactosyl ceramide; MBP, myelin basic protein; MS, multiple sclerosis.
References
- 1.Porcelli, S., C.T. Morita, and M.B. Brenner. 1992. CD1b restricts the response of human CD4-8− T lymphocytes to a microbial antigen. Nature. 360:593–597. [DOI] [PubMed] [Google Scholar]
- 2.Sieling, P.A., D. Chatterjee, S.A. Porcelli, T.I. Prigozy, R.J. Mazzaccaro, T. Soriano, B.R. Bloom, M.B. Brenner, M. Kronenberg, P.J. Brennan, et al. 1995. CD1-restricted T cell recognition of microbial lipoglycan antigens. Science. 269:227–230. [DOI] [PubMed] [Google Scholar]
- 3.Bendelac, A., M.N. Rivera, S.H. Park, and J.H. Roark. 1997. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu. Rev. Immunol. 15:535–562. [DOI] [PubMed] [Google Scholar]
- 4.Porcelli, S.A., B.W. Segelke, M. Sugita, I.A. Wilson, and M.B. Brenner. 1998. The CD1 family of lipid antigen-presenting molecules. Immunol. Today. 19:362–368. [DOI] [PubMed] [Google Scholar]
- 5.Moody, D.B., and S.A. Porcelli. 2003. Intracellular pathways of CD1 antigen presentation. Nat. Rev. Immunol. 3:11–22. [DOI] [PubMed] [Google Scholar]
- 6.Zeng, Z., A.R. Castano, B.W. Segelke, E.A. Stura, P.A. Peterson, and I.A. Wilson. 1997. Crystal structure of mouse CD1: an MHC-like fold with a large hydrophobic binding groove. Science. 277:339–345. [DOI] [PubMed] [Google Scholar]
- 7.Gadola, S.D., N.R. Zaccai, K. Harlos, D. Shepherd, J.C. Castro-Palomino, G. Ritter, R.R. Schmidt, E.Y. Jones, and V. Cerundolo. 2002. Structure of human CD1b with bound ligands at 2.3 A, a maze for alkyl chains. Nat. Immunol. 3:721–726. [DOI] [PubMed] [Google Scholar]
- 8.Zajonc, D.M., M.A. Elsliger, L. Teyton, and I.A. Wilson. 2003. Crystal structure of CD1a in complex with a sulfatide self antigen at a resolution of 2.15 A. Nat. Immunol. 4:808–815. [DOI] [PubMed] [Google Scholar]
- 9.Godfrey, D.I., K.J. Hammond, L.D. Poulton, M.J. Smyth, and A.G. Baxter. 2000. NKT cells: facts, functions and fallacies. Immunol. Today. 21:573–583. [DOI] [PubMed] [Google Scholar]
- 10.Kronenberg, M., and L. Gapin. 2002. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2:557–568. [DOI] [PubMed] [Google Scholar]
- 11.Gumperz, J.E., C. Roy, A. Makowska, D. Lum, M. Sugita, T. Podrebarac, Y. Koezuka, S.A. Porcelli, S. Cardell, M.B. Brenner, and S.M. Behar. 2000. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity. 12:211–221. [DOI] [PubMed] [Google Scholar]
- 12.Wu, D.Y., N.H. Segal, S. Sidobre, M. Kronenberg, and P.B. Chapman. 2003. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J. Exp. Med. 198:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamshiev, A., H.J. Gober, A. Donda, Z. Mazorra, L. Mori, and G. De Libero. 2002. Presentation of the same glycolipid by different CD1 molecules. J. Exp. Med. 195:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinman, L. 1996. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 85:299–302. [DOI] [PubMed] [Google Scholar]
- 15.Paterson, P.Y. 1980. Autoimmune diseases of myelin. Prog. Clin. Biol. Res. 49:19–36. [PubMed] [Google Scholar]
- 16.Shamshiev, A., A. Donda, I. Carena, L. Mori, L. Kappos, and G. De Libero. 1999. Self glycolipids as T-cell autoantigens. Eur. J. Immunol. 29:1667–1675. [DOI] [PubMed] [Google Scholar]
- 17.Kumar, V., D.H. Kono, J.L. Urban, and L. Hood. 1989. The T-cell receptor repertoire and autoimmune diseases. Annu. Rev. Immunol. 7:657–682. [DOI] [PubMed] [Google Scholar]
- 18.Kuchroo, V.K., A.C. Anderson, H. Waldner, M. Munder, E. Bettelli, and L.B. Nicholson. 2002. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu. Rev. Immunol. 20:101–123. [DOI] [PubMed] [Google Scholar]
- 19.Jahng, A.W., I. Maricic, B. Pedersen, N. Burdin, O. Naidenko, M. Kronenberg, Y. Koezuka, and V. Kumar. 2001. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J. Exp. Med. 194:1789–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar, V., V. Bhardwaj, L. Soares, J. Alexander, A. Sette, and E. Sercarz. 1995. Major histocompatibility complex binding affinity of an antigenic determinant is crucial for the differential secretion of interleukin 4/5 or interferon gamma by T cells. Proc. Natl. Acad. Sci. USA. 92:9510–9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuda, J.L., O.V. Naidenko, L. Gapin, T. Nakayama, M. Taniguchi, C.R. Wang, Y. Koezuka, and M. Kronenberg. 2000. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sidobre, S., O.V. Naidenko, B.C. Sim, N.R. Gascoigne, K.C. Garcia, and M. Kronenberg. 2002. The V alpha 14 NKT cell TCR exhibits high-affinity binding to a glycolipid/CD1d complex. J. Immunol. 169:1340–1348. [DOI] [PubMed] [Google Scholar]
- 23.Cui, J., N. Watanabe, T. Kawano, M. Yamashita, T. Kamata, C. Shimizu, M. Kimura, E. Shimizu, J. Koike, H. Koseki, et al. 1999. Inhibition of T helper cell type 2 cell differentiation and immunoglobulin E response by ligand-activated Valpha14 natural killer T cells. J. Exp. Med. 190:783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park, S.H., A. Weiss, K. Benlagha, T. Kyin, L. Teyton, and A. Bendelac. 2001. The mouse CD1d-restricted repertoire is dominated by a few autoreactive T cell receptor families. J. Exp. Med. 193:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cardell, S., S. Tangri, S. Chan, M. Kronenberg, C. Benoist, and D. Mathis. 1995. CD1-restricted CD4+ T cells in major histocompatibility complex class II–deficient mice. J. Exp. Med. 182:993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacDonald, H.R. 2002. Development and selection of NKT cells. Curr. Opin. Immunol. 14:250–254. [DOI] [PubMed] [Google Scholar]
- 27.Coetzee, T., N. Fujita, J. Dupree, R. Shi, A. Blight, K. Suzuki, and B. Popko. 1996. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 86:209–219. [DOI] [PubMed] [Google Scholar]
- 28.Chun, T., M.J. Page, L. Gapin, J.L. Matsuda, H. Xu, H. Nguyen, H.S. Kang, A.K. Stanic, S. Joyce, W.A. Koltun, et al. 2003. CD1d-expressing dendritic cells but not thymic epithelial cells can mediate negative selection of NKT cells. J. Exp. Med. 197:907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pellicci, D.G., A.P. Uldrich, K. Kyparissoudis, N.Y. Crowe, A.G. Brooks, K.J. Hammond, S. Sidobre, M. Kronenberg, M.J. Smyth, and D.I. Godfrey. 2003. Intrathymic NKT cell development is blocked by the presence of alpha-galactosylceramide. Eur. J. Immunol. 33:1816–1823. [DOI] [PubMed] [Google Scholar]
- 30.Forsthuber, T., H.C. Yip, and P.V. Lehmann. 1996. Induction of TH1 and TH2 immunity in neonatal mice. Science. 271:1728–1730. [DOI] [PubMed] [Google Scholar]
- 31.Singh, A.K., M.T. Wilson, S. Hong, D. Olivares-Villagomez, C. Du, A.K. Stanic, S. Joyce, S. Sriram, Y. Koezuka, and L. Van Kaer. 2001. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J. Exp. Med. 194:1801–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharif, S., G.A. Arreaza, P. Zucker, Q.S. Mi, J. Sondhi, O.V. Naidenko, M. Kronenberg, Y. Koezuka, T.L. Delovitch, J.M. Gombert, et al. 2001. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune type 1 diabetes. Nat. Med. 7:1057–1062. [DOI] [PubMed] [Google Scholar]
- 33.Hong, S., M.T. Wilson, I. Serizawa, L. Wu, N. Singh, O.V. Naidenko, T. Miura, T. Haba, D.C. Scherer, J. Wei, et al. 2001. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat. Med. 7:1052–1056. [DOI] [PubMed] [Google Scholar]
- 34.Wilson, S.B., S.C. Kent, K.T. Patton, T. Orban, R.A. Jackson, M. Exley, S. Porcelli, D.A. Schatz, M.A. Atkinson, S.P. Balk, et al. 1998. Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature. 391:177–181. [DOI] [PubMed] [Google Scholar]
- 35.Beaun, L., V. Laloux, J. Novak, B. Lucas, and A. Lehuen. 2002. NKT cells inhibit the onset of diabetes by impairing the development of pathogenic T cells specific for pancreatic beta cells. Immunity. 17:725–736. [DOI] [PubMed] [Google Scholar]
- 36.Shamshiev, A., A. Donda, T.I. Prigozy, L. Mori, V. Chigorno, C.A. Benedict, L. Kappos, S. Sonnino, M. Kronenberg, and G. De Libero. 2000. The alphabeta T cell response to self-glycolipids shows a novel mechanism of CD1b loading and a requirement for complex oligosaccharides. Immunity. 13:255–264. [DOI] [PubMed] [Google Scholar]
- 37.Cipriani, B., L. Chen, K. Hiromatsu, H. Knowles, C.S. Raine, L. Battistini, S.A. Porcelli, and C.F. Brosnan. 2003. Upregulation of group 1 CD1 antigen presenting molecules in guinea pigs with experimental autoimmune encephalomyelitis: an immunohistochemical study. Brain Pathol. 13:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Battistini, L., F.R. Fischer, C.S. Raine, and C.F. Brosnan. 1996. CD1b is expressed in multiple sclerosis lesions. J. Neuroimmunol. 67:145–151. [DOI] [PubMed] [Google Scholar]
- 39.Katz-Levy, Y., K.L. Neville, J. Padilla, S. Rahbe, W.S. Begolka, A.M. Girvin, J.K. Olson, C.L. Vanderlugt, and S.D. Miller. 2000. Temporal development of autoreactive Th1 responses and endogenous presentation of self myelin epitopes by central nervous system-resident APCs in Theiler's virus-infected mice. J. Immunol. 165:5304–5314. [DOI] [PubMed] [Google Scholar]
- 40.Vanderlugt, C.L., and S.D. Miller. 2002. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2:85–95. [DOI] [PubMed] [Google Scholar]
- 41.Moody, D.B., T. Ulrichs, W. Muhlecker, D.C. Young, S.S. Gurcha, E. Grant, J.P. Rosat, M.B. Brenner, C.E. Costello, G.S. Besra, and S.A. Porcelli. 2000. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature. 404:884–888. [DOI] [PubMed] [Google Scholar]
- 42.Heller, F., I.J. Fuss, E.E. Nieuwenhuis, R.S. Blumberg, and W. Strober. 2002. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 17:629–638. [DOI] [PubMed] [Google Scholar]
- 43.Baron, J.L., L. Gardiner, S. Nishimura, K. Shinkai, R. Locksley, and D. Ganem. 2002. Activation of a nonclassical NKT cell subset in a transgenic mouse model of hepatitis B virus infection. Immunity. 16:583–594. [DOI] [PubMed] [Google Scholar]
- 44.Anderson, A.C., L.B. Nicholson, K.L. Legge, V. Turchin, H. Zaghouani, and V.K. Kuchroo. 2000. High frequency of autoreactive myelin proteolipid protein-specific T cells in the periphery of naive mice: mechanisms of selection of the self-reactive repertoire. J. Exp. Med. 191:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong, F.S., J. Karttunen, C. Dumont, L. Wen, I. Visintin, I.M. Pilip, N. Shastri, E.G. Pamer, and C.A. Janeway, Jr. 1999. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat. Med. 5:1026–1031. [DOI] [PubMed] [Google Scholar]
- 46.Miyamoto, K., S. Miyake, and T. Yamamura. 2001. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature. 413:531–534. [DOI] [PubMed] [Google Scholar]
- 47.Brossay, L., S. Tangri, M. Bix, S. Cardell, R. Locksley, and M. Kronenberg. 1998. Mouse CD1-autoreactive T cells have diverse patterns of reactivity to CD1+ targets. J. Immunol. 160:3681–3688. [PubMed] [Google Scholar]