

Abstract
Cell surface proteins major histocompatibility complex (MHC) class I–related chain A (MICA) and UL16-binding proteins (ULBP) 1, 2, and 3 are up-regulated upon infection or tumor transformation and can activate human natural killer (NK) cells. Patches of cross-linked raft resident ganglioside GM1 colocalized with ULBP1, 2, 3, or MICA, but not CD45. Thus, ULBPs and MICA are expressed in lipid rafts at the cell surface. Western blotting revealed that glycosylphosphatidylinositol (GPI)-anchored ULBP3 but not transmembrane MICA, MHC class I protein, or transferrin receptor, accumulated in detergent-resistant membranes containing GM1. Thus, MICA may have a weaker association with lipid rafts than ULBP3, yet both proteins accumulate at an activating human NK cell immune synapse. Target cell lipid rafts marked by green fluorescent protein–tagged GPI also accumulate with ULBP3 at some synapses. Electron microscopy reveals constitutive clusters of ULBP at the cell surface. Regarding a specific molecular basis for the organization of these proteins, ULBP1, 2, and 3 and MICA are lipid modified. ULBP1, 2, and 3 are GPI anchored, and we demonstrate here that MICA is S-acylated. Finally, expression of a truncated form of MICA that lacks the putative site for S-acylation and the cytoplasmic tail can be expressed at the cell surface, but is unable to activate NK cells.
Keywords: immunological synapse, natural killer cell, fluorescence imaging, lipid rafts, intercellular communication
Introduction
NK cell activation can be triggered by an absence of inhibitory receptor ligation (i.e., by detection of missing self) and also by ligation of specific activating receptors. NKG2D is a C-type lectin–activating receptor that recognizes cell surface MHC class I–like proteins up-regulated upon cell stress, including viral or bacterial infection and tumor transformation (for review see references 1, 2). Human ligands for NKG2D, UL16-binding protein (ULBP) 1, 2, and 3, and MHC class I–related chain A (MICA; references 3–5) have Ig domains similar to the α1/α2 and the α1/α2/α3 domains, respectively, of MHC class I protein, but do not associate with β2-microglobulin nor present peptide (for review of crystal structures see reference 6).
NKG2D is expressed in various immune cells including NK, NKT, CD8+ αβ T cells, γδ T cells, and macrophages. Its function can be regulated by noncovalent association with either DNAX-activating protein (DAP) 10 or DAP12 (7, 8). In NK cells, NKG2D-mediated cytotoxicity can be triggered by signaling from immunoreceptor tyrosine-based activation motifs in DAP12 or via a Syk-independent pathway activated by DAP10 (9, 10). Activation of NK cells via NKG2D can overcome inhibitory signaling from self recognition (5, 11, 12). In T cells, NKG2D is thought to associate principally with DAP10 to mediate costimulatory functions (8, 13, 14).
Lipid rafts are critical in facilitating numerous cellular processes, including immune cell signaling (for review see reference 15). Clearly, NK cell lipid rafts play an important role in NK cell activation and cytotoxicity (16), for example, by facilitating the function of 2B4 (17). However, the role of target cell lipid rafts in controlling NK cell activation has not yet been addressed. Here, we investigate the organization of human NKG2D ligands at target cell surfaces and the activating NK cell immune synapse.
Materials and Methods
Cells and Antibodies.
Daudi, a Burkitt's lymphoma B cell line that lacks expression of β2-microglobulin, was transduced with amphotropic retroviruses encoding β2-microglobulin (Daudi/Class I+) and further transduced to express ULBP1, 2, or 3 (Daudi/Class I+/ULBP1, 2, or 3) or MICA (Daudi/Class I+/MICA) as described previously (5). Transfectants of Namalwa (American Type Culture Collection), expressing GPI–green fluorescent protein (GFP) were created by electroporation of pcDNA3.1/Hygro (Invitrogen) encoding the Acrosin signal sequence–enhanced GFP-Thy1GPI (a gift from G. Kondoh, Osaka University, Osaka, Japan; reference 18) as a KpnI/NotI fragment and selection by flow cytometry. Human peripheral blood NK cell lines and clones were cultured as described previously (19).
For imaging, 15–30 μg/ml mouse anti–human ULBP1 (M295), ULBP2 (M311), ULBP3 (M551), and MICA (M673) mAb that have been described previously (5) were used. 5 μg/ml mouse anti–human CD45 mAb (HI30; BD Biosciences) and 10 μg/ml mouse anti–human transferrin receptor (TfR) mAb (H68.4; Zymed Laboratories) were used. Alexa Fluor 488 or 633 goat anti–mouse IgG (Molecular Probes) were used at 4 μg/ml.
Site-directed Mutagenesis.
The MICA gene (GenBank/EMBL/DDBJ accession no. AAD52060 with the exception of one substitution, Val 256 Ile; reference 5) was subcloned as a BamHI/NotI fragment into pcDNA3.1/Neo (Invitrogen). In vitro site-directed mutagenesis was performed (QuickChange Kit; Stratagene) according to the manufacturer's instructions using the following primers: forward, 5′-CTATGTCCGTTGATGTAAGAAGA-3′ and reverse, 5′-TCTTCTTACAACTACGGACATAG-3′. Constructs encoding the correct mutation, verified by sequencing the full insert, were transfected into Daudi/Class I+ cells by electroporation. Cells were selected for MICA expression by flow cytometry.
Patching of GM1 Ganglioside with Cholera Toxin β Subunit (CT-B).
For cross-linking of GM1, 106 cells were first incubated with 10 μg/ml CT-B (Sigma-Aldrich) on ice for 30 min and washed in 1% BSA/5% horse serum/PBS before incubation with anti–CT-B rabbit antiserum (Sigma-Aldrich) in 1% BSA/5% horse serum/PBS for 30 min on ice. Patching was induced by incubation with Alexa Fluor 488 goat anti–rabbit IgG or Alexa Fluor 350 goat anti–rabbit IgG (4 μg/ml; Molecular Probes) for 20 min at 37°C. Immediately after patching, cells were fixed in 2% paraformaldehyde for 15 min at room temperature (RT). After washing, cells were incubated with the primary mAb for 45 min at 4°C, washed in PBS, and incubated for 45 min at 4°C with 3 μg/ml Cy5 goat anti–mouse IgG (Jackson ImmunoResearch Laboratories) or 4 μg/ml Alexa Fluor 488 goat anti–mouse IgG. As a positive control, cells that had been GM1 patched were stained with an additional secondary mAb, 4 μg/ml Alexa Fluor 568 goat anti–rabbit IgG for 45 min at 4°C.
Laser Scanning Confocal Microscopy and Image Analysis.
Cells were imaged (TCS SP2; Leica) as described previously (19), except that here, for imaging immune synapses, cells were fixed in 4% paraformaldehyde for 15 min at RT and cells (106 of each) were coincubated in 50 μl of prewarmed culture medium to facilitate rapid conjugation.
To quantify colocalization between GM1 and proteins, images were first corrected for background; a binary mask, outlining the plasma membrane, was applied to both images; and the correlation coefficient was calculated on a pixel-by-pixel basis according to
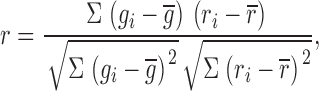 |
where gi and ri are the intensities of the green and red channels in pixel i, and 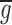 and
and 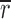 are the average intensities of the green and red channels within the region of interest. Appropriate code was written in Matlab (The Mathworks Inc.). The correlation coefficient theoretically ranges from −1 to 1, where 1 indicates perfect overlap, 0 indicates random distribution, and −1 indicates avoidance.
are the average intensities of the green and red channels within the region of interest. Appropriate code was written in Matlab (The Mathworks Inc.). The correlation coefficient theoretically ranges from −1 to 1, where 1 indicates perfect overlap, 0 indicates random distribution, and −1 indicates avoidance.
Preparation of Detergent-resistant Membranes (DRMs) and Western Blotting.
To prepare DRMs, 5 × 107 cells were washed in cold PBS and lysed for 30 min on ice in 1 ml MNE (25 mM Mes, pH 6.5, 150 mM NaCl, and 1 mM EDTA) containing 1% Triton X-100, phosphatase inhibitors (5 mM NaF and 0.1 mM Na3VO4), and protease inhibitors. Lysates were sheared with a cold 23-gauge needle and mixed with an equal volume of 80% (wt/vol) sucrose/MNE. This was overlaid with 2 ml 30% (wt/vol) sucrose/MNE followed by 1 ml 5% (wt/vol) sucrose/MNE. Equilibrium ultracentrifugation at 200,000 g was performed in a precooled SW50.1 swing-out rotor (Beckman Coulter) for 18 h at 4°C.
For Western blotting, equal volumes from 10 fractions were mixed with 5× Laemmli sample buffer and separated by 10 or 15% (for the detection of GM1 ganglioside) of reducing SDS-PAGE. Proteins were transferred onto 0.2 μm nitrocellulose (Optitran; Schleicher and Schuell) or polyvinyldifluoride (Hybond P; Amersham Biosciences) and probed with the indicated mAb followed by 16 ng/ml horseradish peroxidase (HRP) rabbit anti–mouse IgG (H + L) (Immunopure® antibody; Pierce Chemical Co.) where needed. For Western blotting, HRP-conjugated CT-B (Sigma-Aldrich) was used at 84 ng/ml, mouse anti–human TfR was used at 1 μg/ml, mouse anti–human ULBP3 was used at 0.66 μg/ml, mouse anti–human MICA was used at 1.5 μg/ml, and mouse anti–human MHC class I (HC10; American Type Culture Collection) was used at 1.5 μg/ml. Blots were developed with chemiluminescent substrate (SuperSignal®; Pierce Chemical Co.).
Analysis of Protein Acylation.
2 × 107 Daudi/Class I+/MICA (per IP) were resuspended in 2 ml RPMI 1640 culture medium supplemented with 5% dialysed FBS and 10 mM sodium pyruvate and subsequently labeled with 400 μCi (9,10-[3H]N)palmitic acid (40–60 Ci/mmol; Amersham Biosciences) and 100 μCi [35S]methionine (60 Ci/mmol; ICN Biomedicals) for 5 h at 37°C. Cells were lysed in 1 ml RIPA buffer containing 0.1 mM PMSF and protease inhibitors as described previously (20). DNA was sheared by passage through a narrow-gauge syringe needle, debris was pelleted in a microfuge at 4°C, and 2 μg of each of the primary mAb was added to the supernatant. After overnight incubation at 4°C immunocomplexes were recovered on protein G-Sepharose (Amersham Biosciences), washed, released from the beads with 25 μl Laemmli gel sample buffer, and heated to 90°C for 3 min. Radiolabeled proteins were analyzed by SDS-PAGE on 10% acrylamide minigels under nonreducing conditions and visualized by fluorography using Kodak X-Omat AR films as described previously (20).
NK Cell Cytotoxicity Assays.
The susceptibility of various target cells NK cytotoxicity was assessed in 5-h [35S]Met release assays performed in triplicate as described previously (19). Spontaneous release of 35S was <20% of the maximal release.
Electron Microscopy.
Daudi transfectants were fixed with 2% paraformaldehyde/PBS for 30 min at RT. Cells were washed in PBS, and excess aldehyde was quenched using 50 mM glycine/PBS. Cells were embedded in 12% gelatin in PBS and incubated in 2.3 M sucrose at 4°C overnight. Blocks were mounted and frozen in liquid nitrogen. Ultrathin sections were cut (Ultracut FCS; Leica) and picked up in a 1:1 mixture of 2% methylcellulose and 2.3 M sucrose. Sections were labeled with mAb in 1% BSA/0.5% BSA-C (Aurion) as a blocking agent. This was followed by rabbit anti–mouse bridging antibody (DakoCytomation), which was detected using 10-nm gold particles conjugated to protein A (obtained from J.W. Slot, Utrecht University, Utrecht, Netherlands). Sections were contrast stained and supported in a mixture of methylcellulose and uranyl acetate before being imaged (EM400; Phillips).
Results and Discussion
Colocalization of ULBP1, 2, and 3 and MICA with Cross-linked Lipid Rafts.
To study the distribution of ULBP1, 2, and 3 and MICA with respect to lipid rafts on Daudi/Class I+ transfectants, we cross-linked the lipid raft resident GM1 ganglioside with CT-B to create distinct patches visible by fluorescence microscopy (Fig. 1 a). After patching, the cells were fixed and stained to show the distribution of NKG2D ligands. As negative controls, we looked at the distribution of CD45 and TfR that do not accumulate in GM1 patches. Strikingly, the majority of ULBP and MICA staining (Fig. 1 a, red) accumulated at the same areas on the cell surface as the GM1 patches (Fig. 1 a, green), whereas CD45 remained even around the plasma membrane.
Figure 1.
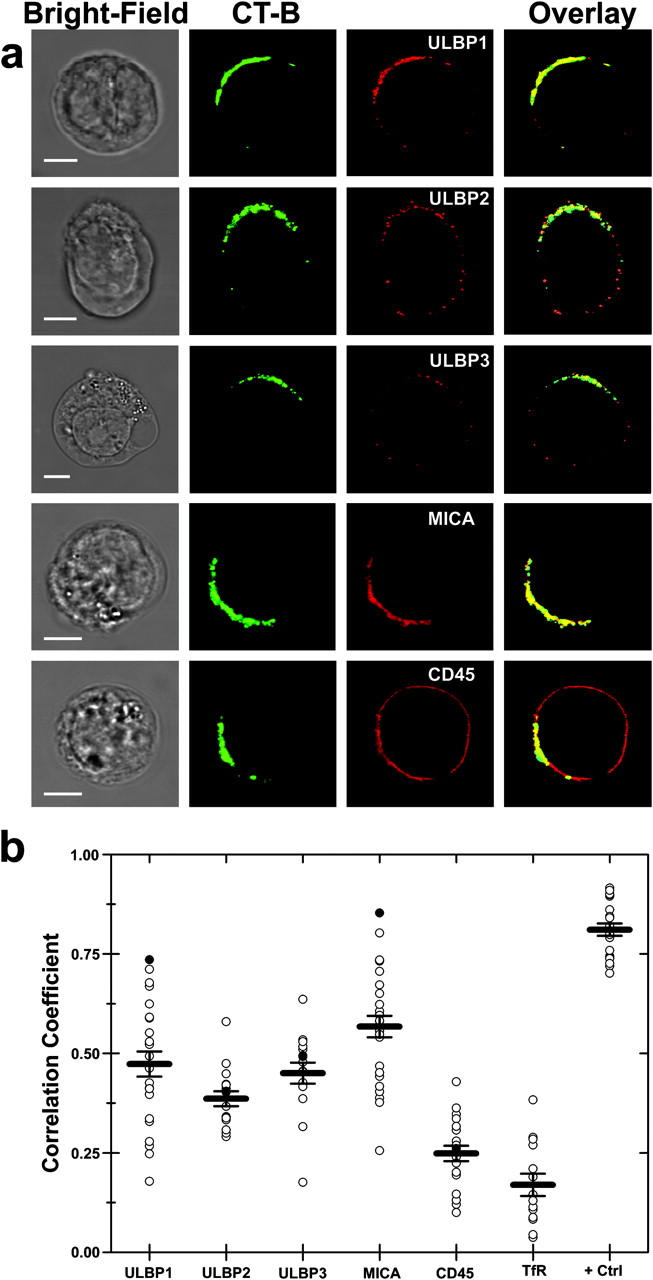
NKG2D ligands colocalize with cross-linked patches of CT-B in Daudi transfectants. (a) After cross-linking GM1, Daudi/Class I+ cells transfected with ULBP1, 2, 3, or MICA were fixed and stained for CT-B (second column) and the appropriate NKG2D ligand (third column). As a control, the distribution of CD45 was compared (last row). Images show single confocal planes. Bars, 5 μm. (b) Correlation coefficients calculated for colocalization of CT-B and NKG2D ligands in individual GM1-patched cells are shown (open circles). Filled circles mark the specific cell shown in panel a. Between 14 and 27 cells of each type that had clearly visible patching of GM1 were selected for quantitative analysis. The mean for each cell type is shown with standard errors. The positive control (+Ctrl) corresponds to labeling of CT-B with two different secondary antibodies.
We developed software to calculate the correlation coefficient between the intensity corresponding to CT-B and each of the NKG2D ligands, similar to a method described previously (21). A practical upper limit of the calculated correlation coefficient was obtained by targeting anti–CT-B with two different secondary antibodies. The distribution of calculated correlation coefficients was quite broad. Nevertheless, our analysis shows clearly that ULBP and MICA colocalize within the GM1 patches (Fig. 1 b). Thus, ULBP and MICA are preferentially concentrated in glycosphingolipid-rich lipid rafts.
ULBP3 But Not MICA Associates with DRMs.
GPI-linked proteins have a propensity for robust association with biochemically isolated DRMs as a result of their Triton X-100 insolubility at 4°C and their low density (22). At the same time, other membrane proteins, such as TfR, do not copurify with DRMs (23, 24). Thus, we set out to investigate whether ULBP3 and MICA associate with DRMs; the mAbs for ULBP1 and ULBP2 were found to be unsuitable for Western blotting.
DRMs were isolated by fractionating cell lysates and analyzed by Western blotting. HRP-conjugated CT-B demonstrated that GM1 ganglioside accumulated in DRMs as expected (Fig. 2, a and b). Western blotting revealed that ULBP3 accumulated in the DRM fractions from both Daudi/Class I+/ULBP3 transfectants and Namalwa, a B cell line endogenously expressing ULBP3 (Fig. 2 a). Thus, ULBP3 is constitutively associated with DRMs.
Figure 2.

Probing the cell surface organization of NKG2D ligands in DRMs and by electron microscopy. DRMs were prepared from (a) Daudi/Class I+/ULBP3 and Namalwa or (b) Daudi/Class I+/MICA cells. 10 fractions were collected from the top of the gradient (fraction 1 denotes the top of the gradient and fraction 10 denotes the bottom), and equal volumes of each were resolved by reducing electrophoresis and probed by Western blotting for each molecule indicated. DRM and detergent-soluble (Det. Sol.) fractions are marked. Molecular masses are indicated in kilodaltons. For TfR, a fainter second band corresponding to the size of TfR dimers was also evident (not depicted). ULPB3, but not MICA, associates with DRMs. Ultrathin cryosections of Daudi/Class I+/ULBP1 were immunolabeled for (c) CD45 or (d) ULBP1. Primary antibodies were detected with 10 nm of gold particles conjugated to protein A. ULBP1 is expressed in clusters at the cell surface (arrowheads), as highlighted by the enlarged view of a small region of cell surface (inset, bottom right). Bar, 0.2 μm.
MICA was absent from the DRM fractions as were TfR and MHC class I proteins (Fig. 2 b). Thus, although MICA clearly is expressed in particular membrane microdomains (Fig. 1), it may have a weaker association to lipid rafts that is not detected by the stringent technique used to isolate DRMs. This is reminiscent of T cell receptor colocalizing with patches of cell surface GM1, yet not routinely in biochemically isolated DRMs (25).
It may be important to note that Western blotting of gels in reducing conditions identified MICA with an apparent molecular weight approximately twice that predicted by its amino acid sequence. By cleaving N-linked oligosaccharides in lysates of cells expressing MICA, a band corresponding to a molecular mass of ∼45 kD can be detected by Western blotting (unpublished data). Thus the high apparent molecular weight of MICA in Fig. 2 b is likely due to extensive glycosylation.
Electron Microscopy Reveals Clusters of ULBP at the Cell Surface.
For immunoelectron microscopy, cells were fixed rapidly at 5°C, cryoprotected, and snap frozen in liquid nitrogen; therefore, movement and redistribution during subsequent labeling procedures should not occur. CD45 was distributed evenly over the cell surface (Fig. 2 c). However, immunogold labels for ULBP1 tended to concentrate in distinct clusters of 2–5 particles (Fig. 2 d). The cell surface between these clusters was free of label, suggesting that ULBP1 is expressed in distinct “islands” within the plasma membrane. ULBP-rich membrane domains spanned ∼30–100 nm, which is consistent with estimates of the size of lipid rafts. Similar observations were seen for ULBP3 (unpublished data). This suggests that ULBPs are constitutively expressed in lipid rafts, and not just upon cross-linking GM1 as shown in Fig. 1.
Accumulation of ULBP3, MICA, and Target Cell Lipid Rafts at the Activating NK Cell Immune Synapse.
To assess the distribution of NKG2D ligands at human NK cell immune synapses (26), NK cells were coincubated with Daudi/Class I+/MICA for 15 min before conjugates were fixed and stained. The peripheral blood NK cell line efficiently lysed Daudi/Class I+/MICA (unpublished data). MICA was found to accumulate at 50% of intercellular contacts (n = 301; Fig. 3 a) and was even seen to accumulate at two immune synapses formed by a single NK cell (Fig. 3 a, first row). Thus, MICA accumulates at activating NK cell immune synapses.
Figure 3.
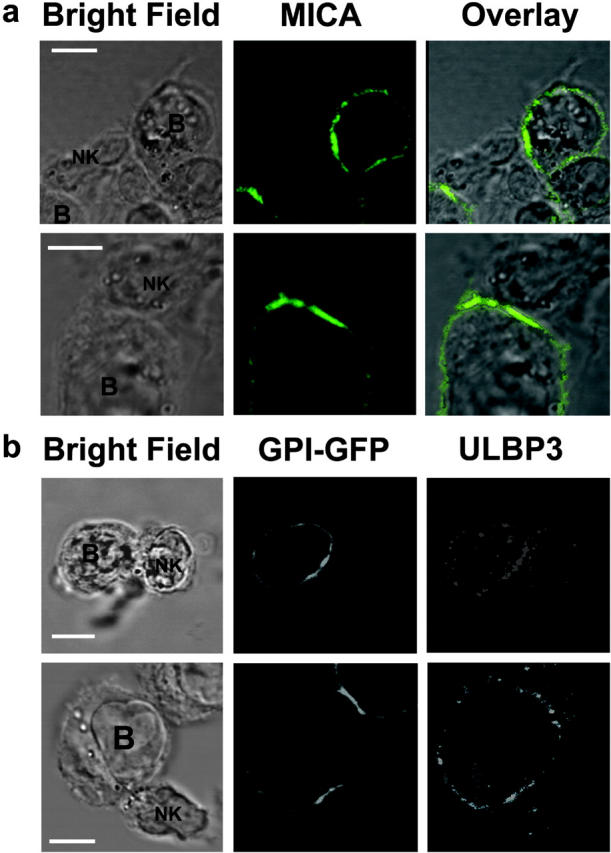
Recruitment of ULBP3 and MICA to the NK cell immune synapse. (a) CD56+ CD3− human peripheral blood NK cells were coincubated with Daudi/Class I+/MICA cells for 15 min, fixed, and stained with anti-MICA mAb, and the distribution at the immunological synapse was determined. The first row shows a single NK cell forming two activating synapses with two different Daudi/Class I+/MICA cells. Data shown are representative of three experiments in which 301 NK-Daudi/Class I+/MICA conjugates were assessed. (b) A CD56+ CD3− human peripheral blood NK clone was coincubated with GPI-GFP–expressing Namalwa transfectants for 15 min, fixed, and stained for ULBP3. Images are representative of 13 out of 14 conjugates where ULBP3 was clearly seen to accumulate at the synapse. Bars, 5 μm.
To assess the role of target cell lipid rafts at the activating NK cell immune synapse, we transfected Namalwa with a construct encoding GPI-anchored GFP (GPI-GFP). Western blotting of DRMs confirmed that this transfectant expressed GFP in the GM1-rich membrane compartment (unpublished data). We imaged conjugates formed between this transfectant and an NK clone that efficiently lysed Namalwa. Fig. 3 b clearly demonstrates the simultaneous accumulation of ULBP3 and GPI-GFP (i.e., target cell lipid rafts) at the activating NK cell immune synapse.
Due to the lower level of expression of ULBP3 in this cell line compared with the Daudi transfectants, it was difficult to image ULBP3 at these synapses. Nevertheless, even though we could clearly see accumulation of ULBP3 at the immune synapse in only 25% of conjugates, in nearly all of these cases (13 out of 14), GPI-GFP simultaneously accumulated at the synapse. Thus, these data provide the first evidence that target cell lipid rafts containing NKG2D ligands are important in NK cell activation.
MICA Is S-acylated.
Many proteins that associate with lipid rafts are attached to saturated lipid groups likely to prefer the ordered environment of rafts. S-acylated proteins are often targeted to lipid rafts; however, one exception is TfR. Thus, we set out to determine if MICA is lipid modified. The fact that MICA contains a dicysteine motif at positions 331 and 332 juxtaposed to its putative transmembrane sequence suggested that the protein might be S-acylated. To test this, MICA was immunoprecipitated from lysates of cells preincubated with radiolabeled palmitic acid. Immunoprecipitated MICA was radiolabeled, demonstrating that MICA is indeed S-acylated (Fig. 4 a).
Figure 4.

MICA is S-acylated, and a truncated form of MICA is unable to activate NK cells. (a) Daudi/Class I+/MICA cells were labeled for 5 h with either [35S]methionine (35S MET) or with [3H]palmitate (3H PAL), lysed, immunoprecipitated with anti-MICA or anti-TfR mAb, and analyzed by SDS-PAGE and fluorography. Molecular weight markers (MW) are also shown. The two bands seen for TfR correspond to the size of TfR monomers and dimers. The mobility of MICA was higher in this gel than in that used in Fig. 2, probably due to the nonreducing conditions. Fluorograms were exposed for 81 d (3H PAL) or 7 d (35S MET). (b and c) Cell surface staining by anti-MICA mAb of (b) Daudi/Class I+/MICA and (c) Daudi/Class I+/MICA/C331*. Plots show staining with an isotype-matched control mAb (shaded) and staining with anti-MICA (line). (d) Susceptibility of Daudi (□), Daudi/Class I+ (♦), Daudi/Class I+/MICA (•), and Daudi/Class I+/MICA/C331* (▴) by a peripheral blood NK cell line, assessed in a 5-h [35S]Met release assay. Data are representative of three independent experiments, each performed in triplicate.
A Truncated Form of MICA Is Unable to Activate NK Cells.
Toward demonstrating functional significance for the transmembrane and/or cytoplasmic portions of MICA, we generated a transfectant of Daudi expressing a truncated form of MICA in which the cysteine at position 331 was replaced with a stop codon (Daudi/Class I+/MICA/C331*). The level of cell surface staining of this transfectant with a MICA mAb was similar to that of wild-type MICA in Daudi/Class I+/MICA (Fig. 4, b and c). We tested the susceptibility of these transfectants to NK cell cytotoxicity (Fig. 4 d). A peripheral blood NK cell line was efficiently able to kill untransfected Daudi, which lacks endogenous expression of MHC class I protein, but was inhibited from killing Daudi-expressing MHC class I protein (Daudi/Class I+). In agreement with previous data (5), Daudi-expressing MHC class I protein and MICA was efficiently killed, demonstrating how activation via NKG2D recognition can overcome inhibitory signaling. However, Daudi transfectants expressing MHC class I protein and the truncated form of MICA were not efficiently killed. Thus, removal of the putative site of S-acylation and the cytoplasmic tail of MICA abrogates cytolysis by human NK cells. From this first study, it is possible that either S-acylation or another, currently unknown, function of the cytoplasmic portion of MICA could account for the loss of function of the truncated MICA. Thus, we are currently establishing and testing the effect of numerous point mutations in MICA to determine precisely which amino acid residues control the localization and function of MICA.
In summary, we demonstrated by fluorescence imaging, Western blotting of DRMs, and electron microscopy that the stress-inducible ligands ULBP1, 2, 3, and MICA are expressed in specific membrane microdomains (Figs. 1 and 2). In addition, we show that these proteins accumulate at activating NK cell immune synapses (Fig. 3). The molecular mechanism by which these proteins are organized at the cell surface likely involves attachment to specific lipids, which may be important in triggering NK immune responses (Fig. 4). It has been demonstrated previously that viruses can prevent cell surface expression of NKG2D ligands to evade NK cell activation. In light of our analysis, it might be interesting to investigate if some viruses or tumors have evolved mechanisms to subvert NK activation by disrupting the cell surface organization of NKG2D ligands.
Acknowledgments
We thank A. Rae for cell sorting. We thank K. Yanagi for help in subcloning GPI-GFP.
We acknowledge support from the Medical Research Council, the Biotechnology and Biological Sciences Research Council, the Human Frontier Science Program, and a Wenner-Gren Foundations Fellowship (to B. Önfelt).
K. Eleme, S.B. Taner, and B. Önfelt contributed equally to this work.
References
- 1.Gleimer, M., and P. Parham. 2003. Stress management: MHC class I and class I-like molecules as reporters of cellular stress. Immunity. 19:469–477. [DOI] [PubMed] [Google Scholar]
- 2.Raulet, D.H. 2003. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3:781–790. [DOI] [PubMed] [Google Scholar]
- 3.Bauer, S., V. Groh, J. Wu, A. Steinle, J.H. Phillips, L.L. Lanier, and T. Spies. 1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729. [DOI] [PubMed] [Google Scholar]
- 4.Wu, J., Y. Song, A.B. Bakker, S. Bauer, T. Spies, L.L. Lanier, and J.H. Phillips. 1999. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 285:730–732. [DOI] [PubMed] [Google Scholar]
- 5.Cosman, D., J. Mullberg, C.L. Sutherland, W. Chin, R. Armitage, W. Fanslow, M. Kubin, and N.J. Chalupny. 2001. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 14:123–133. [DOI] [PubMed] [Google Scholar]
- 6.Radaev, S., and P.D. Sun. 2003. Structure and function of natural killer cell surface receptors. Annu. Rev. Biophys. Biomol. Struct. 32:93–114. [DOI] [PubMed] [Google Scholar]
- 7.Gilfillan, S., E.L. Ho, M. Cella, W.M. Yokoyama, and M. Colonna. 2002. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 3:1150–1155. [DOI] [PubMed] [Google Scholar]
- 8.Diefenbach, A., E. Tomasello, M. Lucas, A.M. Jamieson, J.K. Hsia, E. Vivier, and D.H. Raulet. 2002. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 3:1142–1149. [DOI] [PubMed] [Google Scholar]
- 9.Zompi, S., J.A. Hamerman, K. Ogasawara, E. Schweighoffer, V.L. Tybulewicz, J.P. Di Santo, L.L. Lanier, and F. Colucci. 2003. NKG2D triggers cytotoxicity in mouse NK cells lacking DAP12 or Syk family kinases. Nat. Immunol. 4:565–572. [DOI] [PubMed] [Google Scholar]
- 10.Billadeau, D.D., J.L. Upshaw, R.A. Schoon, C.J. Dick, and P.J. Leibson. 2003. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 4:557–564. [DOI] [PubMed] [Google Scholar]
- 11.Lanier, L.L. 2003. Natural killer cell receptor signaling. Curr. Opin. Immunol. 15:308–314. [DOI] [PubMed] [Google Scholar]
- 12.Cerwenka, A., J.L. Baron, and L.L. Lanier. 2001. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA. 98:11521–11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groh, V., R. Rhinehart, J. Randolph-Habecker, M.S. Topp, S.R. Riddell, and T. Spies. 2001. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2:255–260. [DOI] [PubMed] [Google Scholar]
- 14.Jamieson, A.M., A. Diefenbach, C.W. McMahon, N. Xiong, J.R. Carlyle, and D.H. Raulet. 2002. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 17:19–29. [DOI] [PubMed] [Google Scholar]
- 15.Dykstra, M., A. Cherukuri, H.W. Sohn, S.J. Tzeng, and S.K. Pierce. 2003. Location is everything: lipid rafts and immune cell signaling. Annu. Rev. Immunol. 21:457–481. [DOI] [PubMed] [Google Scholar]
- 16.Lou, Z., D. Jevremovic, D.D. Billadeau, and P.J. Leibson. 2000. A balance between positive and negative signals in cytotoxic lymphocytes regulates the polarization of lipid rafts during the development of cell-mediated killing. J. Exp. Med. 191:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watzl, C., and E.O. Long. 2003. Natural killer cell inhibitory receptors block actin cytoskeleton-dependent recruitment of 2B4 (CD244) to lipid rafts. J. Exp. Med. 197:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondoh, G., X.H. Gao, Y. Nakano, H. Koike, S. Yamada, M. Okabe, and J. Takeda. 1999. Tissue-inherent fate of GPI revealed by GPI-anchored GFP transgenesis. FEBS Lett. 458:299–303. [DOI] [PubMed] [Google Scholar]
- 19.McCann, F.E., B. Vanherberghen, K. Eleme, L.M. Carlin, R.J. Newsam, D. Goulding, and D.M. Davis. 2003. The size of the synaptic cleft and distinct distributions of filamentous actin, ezrin, CD43, and CD45 at activating and inhibitory human NK cell immune synapses. J. Immunol. 170:2862–2870. [DOI] [PubMed] [Google Scholar]
- 20.Magee, A.I., J. Wootton, and J. de Bony. 1995. Detecting radiolabeled lipid-modified proteins in polyacrylamide gels. Methods Enzymol. 250:330–336. [DOI] [PubMed] [Google Scholar]
- 21.Parmryd, I., J. Adler, R. Patel, and A.I. Magee. 2003. Imaging metabolism of phosphatidylinositol 4,5-bisphosphate in T-cell GM1-enriched domains containing Ras proteins. Exp. Cell Res. 285:27–38. [DOI] [PubMed] [Google Scholar]
- 22.Brown, D.A., and J.K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 68:533–544. [DOI] [PubMed] [Google Scholar]
- 23.Montixi, C., C. Langlet, A.M. Bernard, J. Thimonier, C. Dubois, M.A. Wurbel, J.P. Chauvin, M. Pierres, and H.T. He. 1998. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 17:5334–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodgers, W., and J.K. Rose. 1996. Exclusion of CD45 inhibits activity of p56lck associated with glycolipid-enriched membrane domains. J. Cell Biol. 135:1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janes, P.W., S.C. Ley, A.I. Magee, and P.S. Kabouridis. 2000. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin. Immunol. 12:23–34. [DOI] [PubMed] [Google Scholar]
- 26.Davis, D.M. 2002. Assembly of the immunological synapse for T cells and NK cells. Trends Immunol. 23:356–363. [DOI] [PubMed] [Google Scholar]