

Abstract
Neural precursor cells from adults have exceptional proliferative and differentiative capability in vitro yet respond minimally to in vivo brain injury due to constraining mechanisms that are poorly defined. We assessed whether cell cycle inhibitors that restrict stem cell populations in other tissues may participate in limiting neural stem cell reactivity in vivo. The cyclin-dependent kinase inhibitor, p21cip1/waf1 (p21), maintains hematopoietic stem cell quiescence, and we evaluated its role in the regenerative response of neural tissue after ischemic injury using the mice deficient in p21. Although steady-state conditions revealed no increase in primitive cell proliferation in p21-null mice, a significantly larger fraction of quiescent neural precursors was activated in the hippocampus and subventricular zone after brain ischemia. The hippocampal precursors migrated and differentiated into a higher number of neurons after injury. Therefore, p21 is an intrinsic suppressor to neural regeneration after brain injury and may serve as a common molecular regulator restricting proliferation among stem cell pools from distinct tissue types.
Keywords: neural stem cells, cell cycle, p21, neural regeneration, brain ischemia
Introduction
Therapeutic effects of stem cell expansion in vitro and regeneration in vivo in part depend upon stem cell proliferation. Understanding the molecular basis for the constraint of stem cell cycling is critical for rationally determined strategies to manipulate stem cells therapeutically. As largely modeled in hematopoiesis, maintenance of mature cell production requires a cytokine-responsive progenitor cell pool with prodigious proliferative capacity and a smaller population of stem cells intermittently feeding daughter cells into a proliferative compartment. In contrast to progenitor cells, the stem cell pool itself is relatively quiescent and cytokine resistant, a state which appears particularly critical in adults (1). In the central nervous system (CNS), evidence suggests that the proliferative pools of adult neural progenitor cell (NPC) are derived from a quiescent multipotent precursor or neural stem cell (NSC) (2–6). Given a much lower turnover rate in CNS (7), the mitotic quiescence may also involve NPC. Perhaps due to the extraordinary quiescence that cannot be sufficiently stimulated by the signals from damaged tissue, endogenous NSCs or NPCs do not produce substantial recovery in cases of severe injury (8). For example, recent studies demonstrated that the precursors from subventricular zone (SVZ) in adult brain can migrate into the injured areas and participate in the neural repair after stroke (9, 10), but the replacement appeared to be very limited (11). This suggests a dominant role of intrinsic inhibitors in suppressing the proliferation of NSC and NPC after injury. Therefore, understanding the molecular mediators in cell cycle arrest underlying neural quiescence may ultimately provide molecular targets for enhancing endogenous repair mechanisms after acute and chronic damages.
The cell cycle machinery is negatively controlled by several cyclin-dependent kinase inhibitors (CKIs) (12). We demonstrated previously that the founding member of CKI families, p21cip1/waf1 (p21), maintains relative quiescence of the hematopoietic stem cell (HSC) (13, 14). p21 was found to be expressed at a higher level in individual quiescent human HSCs than in differentiating progenitor cells, and its absence resulted in a higher fraction of cycling cells in HSC-enriched populations. Hypothesizing that p21-mediated quiescence may also be present in neural precursors (we use “precursors” in our text since the cells cannot be clearly defined as NSC or NPC in vivo), we examined the role of p21 in a model of adult neural precursors within the hippocampus and the lateral ventricle using genetically engineered mice deficient in p21 gene expression before and after ischemic brain injury.
Materials and Methods
Generation of Homozygous Mice
We obtained heterozygous 129/SV p21+/− mice (15) from the laboratory of Tyler Jacks (Massachusetts Institute of Technology, Cambridge, MA) and homozygous 129/SV p53+/+ or 129/SV p53−/− mice from Jackson Laboratory under the permission of the Subcommittee on Research Animal Care of the Massachusetts General Hospital. The inbred 129/SV heterozygous p21+/− mice were bred to yield homozygous and wild-type offsprings. Mice were genotyped by PCR using primers specific for the wild-type and knockout alleles. The littermates at 8–12 wk of age from the same +/− parents were used in further experiments. Animal care and experimental protocols complied with NIH guidelines set forth in The Principles of Laboratory Animal Care. Before all the operations, mice were anesthetized with 1.5% isoflurane in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical).
Middle Cerebral Artery Occlusion (MCAO)
Cerebral ischemia was performed in spontaneously ventilating mice as described previously (16). Body temperature was maintained at 37 ± 0.5°C with a heating blanket. (FHC). Regional cerebral blood flow was measured by laser-Doppler flowmetry (PF2B; Perimed) (16). The left middle cerebral artery was occluded with an 8-0 nylon monofilament (Ethicon) coated with a mixture of silicone resin (Xantopren; Bayer Dental) and a hardener (Elastomer Activator; Bayer Dental) as described (16). 20 min later, the filament was withdrawn and cerebral blood flow reperfusion was confirmed by laser-Doppler flowmetry. After these procedures, the mice were kept at 32°C for 2 h. Infarct area was quantified on sections stained by an image analysis system (M4; Imaging Research) and calculated by summing the volumes of each section.
Physiology Parameters.
In randomly selected animals (n = 3–4/group), mean artery blood pressure were monitored as described (16). Arterial blood samples were analyzed for oxygen (PaO2) and carbon dioxide (PaCO2) before and during ischemia using a blood gas/pH analyzer (Corning 178; Ciba Corning Diagnostics). Physiological parameters were as follows: 111 ± 8 and 111 ± 12 mmHg for the mean artery blood pressure; 7.44 ± 0.03 and 7.41 ± 0.03 for pH; 30 ± 5 and 28 ± 3 for PaCO2 in p21+/+ and p21−/− animals, respectively. Regional cerebral blood flow during middle cerebral artery occlusion (MCAO) and reperfusion were as follows: 9.7 ± 4.0% and 8.8 ± 4.3 during ischemia in p21+/+ and p21−/−, respectively, and 2.2 ± 24.3% and 76.9 ± 20.1% after ischemia in p21+/+ and p21−/−, respectively (n = 6/group). Infarct volumes were 11.8 ± 4.6 mm3 and 11.4 ± 2.7 mm3 in p21+/+ and p21−/−, respectively (n = 5/group).
RT-PCR for p21 and GAPDH
Total RNA was isolated from hippocampus or SVZ with TRIzol method (Life Technologies). 1 μg RNA was treated with 1 U of amplification-grade DNase I (Life Technologies) to eliminate residual genomic DNA and reverse transcribed into first-strand cDNA with oligo-dT primers and 200 U of Superscript II reverse transcriptase (Life Technologies). Then the specific primer pair for mouse p21 (5′-AGCCTGAAGACTGTGATGGG-3′ and 5′-AAAGTTCCACCGTTCTCGG-3′) (17) or mouse GAPDH (5′-TAAAGGGCATCCTGAGCTACACT-3′ and 5′-TTACTCCTTCGAGGCCATGTAGG-3′) was used to amplify the first cDNA template. The PCR cycles and temperature conditions were optimized to reflect a linear relationship between the input template and the final PCR product (p21: 45 s at 94°C, 45 s at 60°, and 1 min at 72°C for 40 cycles; GAPDH: 30 s at 95°C, 30 s at 55°C, and 1 min at 72°C for 25 cycles). PCR was performed in Peltier Thermal Cycler (PTC-200; MJ Research). 1% agarose gel was used to display the PCR product, and the relative density ratio between p21 and GAPDH was analyzed with the NIH imaging system.
5-Bromodeoxyuridine Incorporation In Vivo and Stereological Analysis
Animals received i.p. injections of 5-bromodeoxyuridine (BrdU; 50 mg/kg, dissolved at 5 mg/ml in PBS [Sigma-Aldrich]). Two daily injections (two times/day) were given to naive animals or to operated animals on day 7 and 8 or day 14 and 15 after MCAO. The animals were killed on day 1 or day 28 after the last BrdU injection (i.e., on day 9, 16, or 35 after MCAO). For histological evaluation, the animals were perfused transcardially with 4% paraformaldehyde in PBS under deep anesthesia, and a stereological estimation (18) was used to quantify the positive cells. BrdU-positive cells were counted in the dentate gyrus in four sections per animal (one of every fifteenth serial, 40-μm sections) throughout the rostro caudal extent of the granule cell layer (GCL) using a 100× objective. The GCL area (mm2) was measured on adjacent sections stained with cresyl violet. The total granule cell volume (mm3) was estimated by summing the traced areas for each section multiplied by the distance between sampled sections. The number of BrdU-labeled cells per mm3 was then calculated from the sectional and total volumes of the GCL. The identical stereological approach was applied to examine BrdU incorporation in the SVZ (19, 20).
Immunohistochemistry Staining
Immunohistochemistry was performed on 40-μm floating coronal sections pretreated by denaturing DNA as reported previously (21). We used mouse anti-BrdU (Becton Dickinson) 1:400 for DAB staining or rat anti-BrdU ascites fluid (Harlan Sera-Lab), 1:100 for double fluorescence labeling; 1:1,000 for rabbit GFAP antibody cy3 conjugated (Sigma-Aldrich); 1:1,000 mouse anti-NeuN (Chemicon); 1:100 for goat anti-doublecortin (DCX) (Santa Cruz Biotechnology, Inc.); and 1:500 for mouse anticalbindin (Swant). To determine the number of BrdU-labeled cells, we stained for BrdU by using the peroxidase method (ABC system, with biotinylated horse anti–mouse IgG antibodies and diamino-benzidine as chromogen [Vector Laboratories]). The fluorescent secondary antibodies used were appropriate cy2- and cy3-labeled IgG (Jackson ImmunoResearch Laboratories), 1:200 dilution. The same stereological estimation was used to quantify the double-positive cells as described in 5-Bromodeoxyuridine Incorporation In Vivo and Stereological Analysis. For the TUNEL assay, a well established protocol in our laboratory was used in this study (22).
Enzyme Immunoassay for Fibroblast Growth Factor-2
For enzyme immunoassay (EIA), the brains were removed without transcardial perfusion, and the hippocampi were dissected and frozen immediately at −80°C. Stored hippocampi were homogenized and centrifuged at 14,000 g for 30 min at 4°C. Protein concentration of each supernatant was determined by a protein assay kit (Bio-Rad Laboratories). EIA for fibroblast growth factor (FGF)-2 was performed by using an assay kit (Quantikine HS; R&D Systems) according to the manufacturer's instruction and our previous work (19).
Neurosphere Culture
Culture of neurospheres was based on the previously published method (23, 24) with minor modifications. Details are provided in Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20031385/DC1.
Online Supplemental Material
Photographic images along with culture conditions for neurospheres (Fig. S1) and experimental design for the in vivo assessments used in the brain ischemic model (Fig. S2) are available at http://www.jem.org/cgi/content/full/jem.20031385/DC1.
Results
Cell Proliferation in the Neurogenic Regions Under Steady-state (Noninjured) Conditions.
We assessed cell proliferation within the subgranular zone (SGZ) of hippocampal dentate gyrus and the SVZ of the lateral ventricle. Both regions contain well-defined neural precursors that maintain ongoing low-level neurogenesis in adults (25, 26). To directly determine the cell cycling status of the neural precursors in vivo, we examined by immunohistochemistry the incorporation of the thymidine analogue, BrdU, into brain cells in 8–12-wk-old mice. It has been shown previously that cycling cells in this model do not express mature neuron markers according to a defined experimental protocol (19, 27), and therefore, BrdU incorporation in the regions is thought to mainly reflect proliferative activity of precursor cells. BrdU uptake over 48 h was analyzed to assess cell proliferative activity. Under steady-state conditions, a low level of BrdU labeling (black cells) was detected in the noninjured mice (Fig. 1). Labeled cells were found mainly in the SGZ and SVZ as reported previously (19). However, the numbers of BrdU-positive cells in p21−/− and p21+/+ mice did not differ significantly in either the SGZ (Fig. 1 A) or SVZ (Fig. 1 B), unexpectedly demonstrating no impact of p21 deletion on the proliferation of neural precursors under steady-state or noninjured conditions. To further confirm this unexpected finding, the neurosphere culture system (23) was used to enumerate NSCs from hippocampus and lateral ventricle tissue. Consistent with the above in vivo data, the number of neurospheres from p21−/− mice was not significantly different from p21+/+ mice (SGZ: 121 ± 150 and 179 ± 203 in p21−/− and p21+/+ mice, respectively; SVZ: 496 ± 163 and 501 ± 249 in p21+/+ and p21−/− mice, respectively; mean ± SD, n = 4/each). In addition, we did not observe significant differences in the neural or glial differentiation potential of the cells regardless of the integrity of their p21 gene locus (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20031385/DC1).
Figure 1.


Increased cell proliferation in p21−/− brain tissues after MCAO. BrdU was injected into the littermate or age-matched p21−/− or p21+/+ mice on day 7 and 8 or day 14 and 15; and animals were killed 9 d or 16 d after MCAO for 20 min. Immunohistochemistry was performed on free-floating 40-μm coronal sections pretreated by denaturing DNA, and a specific stereological analysis (18) was applied to enumerate the labeled cells per volume as detailed in Materials and Methods. A few BrdU-labeled cells were detected in the noninjured p21−/− and p21+/+ mice. There was an increased BrdU labeling (black cells) in the region corresponding to the SGZ of dentate gyrus in hippocampus (A) or the SVZ in lateral ventricle (B) after MCAO. Mean values from multiple experiments are summarized in the graphs under each histological picture. The noninjured samples are marked as control in the graph. Black and white bars indicate mean values ± SD from p21−/− and p21+/+ genotype, respectively. A significant increase of BrdU-labeled cells in both genotypes and a larger fraction of BrdU-positive cells in p21−/− genotype was observed 9 d after MCAO (P < 0.01, n = 5, assessed by ANOVA with Bonferroni's post hoc analysis). To further confirm the increased cell proliferation, the injured brain tissue sections were incubated with anti-PCNA antibody, and second antibody was labeled with Rhodamine X (red). The PCNA-expressing cells were significantly more abundant in p21−/− SVZ than in p21+/+ SVZ (color pictures in B). The color images were taken with confocal microscopy.
Cell Proliferation in SGZ and SVZ After Brain Ischemia.
Reasoning that the potential biological consequences of the absence of p21 might be revealed under conditions of damage stress, we exposed the animals to ischemic injury by MCAO (16, 19; Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20031385/DC1). MCAO has been demonstrated as an unique model in which robust neurogenesis can be achieved in weeks (9, 19, 28), and in this study, we chose a mild condition during the procedure (MCAO for 20 min) as a first attempt to assess the direct impact of p21 absence on the proliferation of neural precursors. 1 wk after MCAO, there was an increased BrdU incorporation in both p21−/− and p21+/+ injured mice (Fig. 1). Moreover, the increase of BrdU incorporation in the injured animals was significantly higher in p21−/− mice than in p21+/+ mice (SGZ: 3.0- and 1.5-fold increase in p21−/− and p21+/+ mice, respectively, P = 0.0003; SVZ: 3.1- and 1.4-fold increase in p21−/− and p21+/+ mice, respectively, P = 0.0029). These differences were found at 1 wk and then interestingly reverted to the baseline 2 wk after injury in both p21−/− and p21+/+ animals (Fig. 1, graphs), indicating a specific association with the acute injury. To confirm that the BrdU incorporation indicated the cell proliferation activity in the regions, we also examined the proliferating cell nuclear antigen (PCNA) and found that the PCNA-positive cells were more abundant in p21−/− SVZ than in p21+/+ SVZ after injury (Fig. 1 B, top right panels). Therefore, cell proliferation in SGZ or SVZ is responsive to ischemic injury despite the absence of overt injury to those sites (see the data in the next paragraph), and p21 appears to limit the proliferative response to the injury. The physiologic relevance of p21 in that context was further supported by the up-regulation of p21 mRNA 4 d after brain ischemia in the SGZ and SVZ regions of wild-type animals (Fig. 2), which was consistent with the elevated levels of the p21 protein reported by others (29).
Figure 2.

Up-regulation of p21 expression in brain tissues after MCAO. RT-PCR for p21 or GAPDH was performed on the noninjured and injured (MCAO) tissues from hippocampus (Hipp) or subventricular zone (SVZ) 4 d after MCAO. An enhanced intensity of p21 expression was noted on the RT-PCR gels, and there was a significant increase of p21 mRNA levels if normalized to the transcript of a housekeeping gene (GAPDH) after MCAO (black bars) in both anatomic sites (two experiments, two pairs of animals in each experiment) as shown in the graph. Numbers above the gels indicate samples from different individual animals. The noninjured samples are marked as control in the graph. Detailed RT-PCR conditions are described in the methods.
Minimal Cell Death in SGZ and SVZ After MCAO.
Because SGZ and SVZ lie outside the core territory supplied by the middle cerebral artery, under the mild injury condition applied in this study, significant cell death was not anticipated in these specific regions. To further assess this, we then examined the tissue sections using terminal deoxynucleotidyl transferase–mediated deoxyuridinetriphosphate nick end labeling (TUNEL) within the hippocampus and adjacent structures after MCAO. Unlike our previous demonstration of TUNEL-positive staining in striatum after MCAO (30), BrdU-stained cells in the SVZ and hippocampus exhibited normal neuronal morphology without morphological features of apoptosis. Further, there were no TUNEL-positive cells observed on day 1, 2, or 9 after MCAO in these two regions (not depicted). Response to brain injury in an area remote from injured cells and tissue has advantages over models in which cells themselves are injured possibly irreparably. Relatively mild injury and distal site evaluation allowed us to more accurately quantify cell proliferation without the confounding effects of DNA damage, cell apoptosis, or infiltrating inflammatory cells.
Migration and Differentiation Potential of the Neural Precursor Cells from SGZ.
To explore the potential of migration and differentiation of the precursors detected by BrdU incorporation, the migration marker for neurons, DCX (31, 32), and the specific marker for mature neurons, NeuN, were used to costain the BrdU-positive cells in dentate gyrus of hippocampus 9 and 35 d after MCAO (Fig. S2). Doublecortin is known to be microtubule-associated protein widely expressed in migrating and differentiating neurons (32, 33). We found that the DCX-positive cells were mainly located in SGZ. The majority (96%) of BrdU-positive cells in the region acquired the expression of DCX and had the morphological characteristic of the neuroblast cell 1 d after the 2-d BrdU injection procedure (Fig. 3, A–C). The increase of DCX/BrdU double-positive cells correlated with the increase of BrdU incorporation after injury, indicating that these neuroblast cells were immediate progenies of the activated precursors and, moreover, that there was a higher abundance of the neural precursors generated in p21−/− brain after injury (Fig. 3, D and E). The precursor nature of the BrdU-incorporated cells shortly after injury was further confirmed by the absence of NeuN in the BrdU-positive cells in SGZ 9 d after MCAO (1 d after the 2-d BrdU injection procedure) (Fig. 4 A, first column). Subsequently, to reveal a further differentiating phenotype, separate groups of animals injected with BrdU at day 7 and 8 after injury or no injury were maintained for an additional 4 wk. Brain sections were stained for BrdU (cy2, green) plus neuronal or glial markers (cy3, red) and examined with confocal microscopy. Colocalization of the glial marker GFAP with BrdU was rarely observed in steady-state or ischemic conditions in the regions, but a majority (82–93%) of the survived BrdU-marked cells on the first week after MCAO had migrated from SGZ to GCL and were positive for NeuN (Fig. 4 A, second and third columns) during the additional 4 wk. There was an increase of NeuN/BrdU-costained cells at this later time point after injury in all groups. Strikingly, after injury the number of BrdU/NeuN-costained cells was significantly higher in p21−/− mice than in the p21+/+ littermates (5.6-fold increase in p21−/− mice and 1.9-fold increase in p21+/+ mice, P = 0.0001) (Fig. 4 B). In addition, the expression of a distinct marker for mature neurons, calbindin (21), further confirmed the maturation of the neuron-like cells as the coexpression of calbindin with BrdU was similar to that of NeuN in the cells. Notably, unlike NeuN/BrdU coexpression, DCX+BrdU+ cells appearing earlier after MCAO were not present in GCL at this later time point, which is consistent with the recent study by others (34). Together, these data strongly demonstrate a significant impact of p21 on the regeneration of the neuron-like cells from their precursors after brain ischemia.
Figure 3.


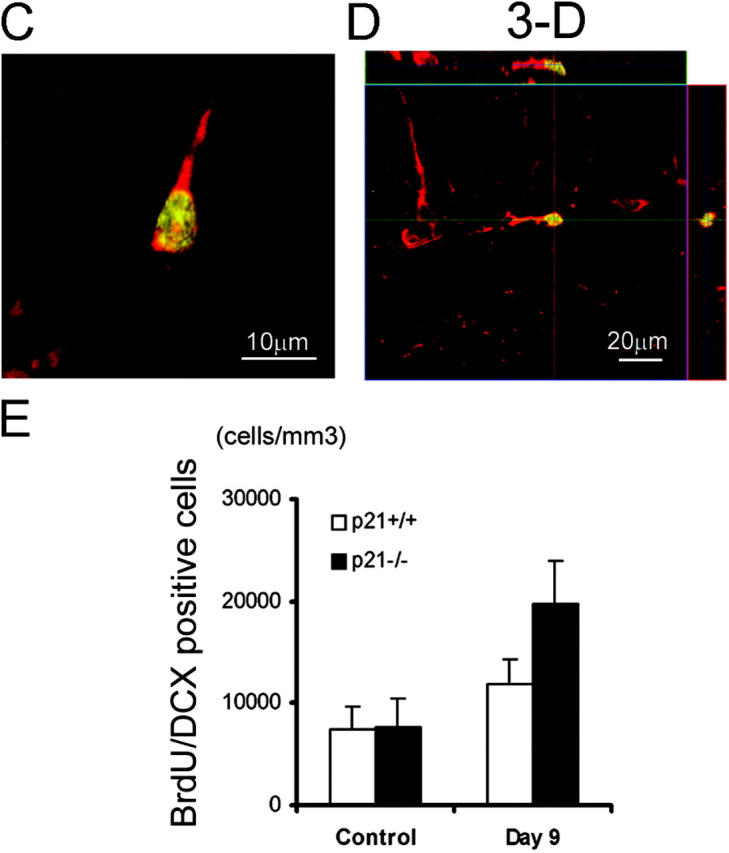
Expression of the neuronal migration marker (DCX) by the proliferating cells in SGZ 1 wk after MCAO. BrdU was injected on day 7 and 8, and animals were killed on day 9 after MCAO. Immunohistochemistry was performed on floating 40-μm coronal sections pretreated by denaturing DNA, and a specific stereological analysis (18) was applied to enumerate the labeled cells per volume as detailed in Materials and Methods. Brain sections were stained for BrdU immunoreactivity (cy2, green) with the neuronal migration indicator, DCX (cy3, red) and examined in SGZ of dentate gyrus with confocal microscopy for BrdU/DCX-coexpressing cells (merged, yellow) (A). A significant increase of BrdU/DCX double-positive cells in p21−/− SGZ especially after MCAO was visualized among the different samples (B). The morphology of the neuroblast cell expressing both BrdU and DCX is shown by a single cell in two-dimension (C) and three-dimension (D). Based on the data from multiple experiments, these differentiating cells positive for DCX/BrdU are significantly more in p21−/− SGZ than in p21+/+ SGZ (P < 0.01, n = 4, assessed by ANOVA with Bonferroni's post hoc analysis) (E). The black and white bars in the graphs indicate mean values ± SD from the p21−/− and p21+/+ genotype, respectively. The noninjured samples are marked as control and Day 9 indicates 9 d after MCAO in the graph (E).
Figure 4.

Expression of the neuronal maturation markers (NeuN and Calbindin) by the proliferated cells in GCL 5 wk after MCAO. BrdU was injected on day 7 and 8, and animals were killed on day 9 or 35 after MCAO. Brain sections were stained for BrdU immunoreactivity (cy2, green) and the neuronal differentiation marker (cy3, red) NeuN (A) or calbindin (C) and examined with confocal microscopy. Immunohistochemistry was performed on floating 40-μm coronal sections pretreated by denaturing DNA and a specific stereological analysis (18) was applied to enumerate the labeled cells per volume as detailed in Materials and Methods. NeuN was extensively examined on day 9 and 35 after MCAO. 9 d after MCAO, most BrdU-positive cells in SGZ did not express NeuN (A, first column). Strikingly, 35 d after MCAO a significant increase of BrdU/NeuN double-positive cells in both genotypes and a larger fraction of BrdU/NeuN-positive cells in p21−/− brain was observed and relocated from SGZ to GCL after MCAO (P < 0.01, n = 5, assessed by ANOVA with Bonferroni's post hoc analysis) (A and B). The black and white bars in the graphs indicate mean values ± SD from p21+/+ and p21−/− genotype, respectively. The noninjured samples are marked as control and MCAO indicates 35 d after MCAO in the graph. Calbindin was used as an independent differentiation marker showing its coexpression in with BrdU in the cells located in GCL 35 d after injury (C, merged yellow color a representative image from the p21−/− brain).
Independent Effect of p21 from the Known Upstream Regulators.
p21 has been demonstrated to be transcriptionally up-regulated by p53 upon DNA damage (35), and we sought to functionally determine whether the restriction of p21 on neural regeneration after brain ischemia is dependent on the presence of p53. Thus, we examined BrdU incorporation before and after MCAO in p53−/− mice compared with p53+/+ controls (Fig. 5). In contrast to p21−/− and p21+/+ animals, there was no difference in BrdU incorporation between p53−/− and p53+/+, although an increase in BrdU incorporation after ischemia was observed (n = 5/group). Therefore, the inhibitory role of p21 in neural proliferation in vivo appears to be independent of p53. Further, to rule out that the increased proliferation in the absence of p21 after ischemia may be due to elevated levels of some key neural growth factors in p21-null animals, we measured the concentrations of FGF-2 and epidermal growth factor (EGF) in hippocampus 1 wk after injury (19). FGF-2 and EGF have been shown to be potent mitogens stimulating the proliferation of NSC and NPC from adult brain. FGF-2 was able to stimulate in vivo neurogenesis through a systemic injection (36), and in particular we demonstrated recently that FGF-2 delivery by viral vector promotes neurogenesis in the SGZ after brain ischemia (19). We in this study that the levels of FGF-2 were increased after injury, as we reported previously (19), but there was no difference between p21−/− and p21+/+ animals (Fig. 6). In agreement with a previous report that EGF has no proliferative effect in the SGZ in vivo (7), EGF was not detectable at the protein level in SGZ after MCAO (unpublished data), likely due to location or development stage–dependent expression of EGF (37). Admittedly, our data cannot exclude influences from other potential neural growth factors yet to be defined. Nevertheless, these data suggest that p21 promotes proliferation of NSC and NPC in adult brain independent of raised levels of the essential growth factors of EGF and FGF-2.
Figure 5.

No difference of the cell proliferation in SGZ between p53−/− and p53+/+ mice. Identical methods were used as described in the study on BrdU incorporation in p21−/− or p21+/+ mice (Fig. 1), and there was no difference of BrdU incorporation between p53−/− and p53+/+ in SGZ either before or after MCAO (P > 0.05, n = 5, assessed by ANOVA with Bonferroni's post hoc analysis). Mean values from multiple experiments are summarized in the graph under the histological picture. White and black bars in the graphs indicate mean values ± SD from p53−/− and p53+/+ genotype, respectively. The noninjured samples are marked as control in the graph.
Figure 6.

No difference in the levels of FGF-2 growth factor in hippocampus between p21−/− and p21+/+ mice. FGF-2 concentration in hippocampus before and 1 wk after MCAO was measured by EIA as described in Materials and Methods. Black and white bars in the graph indicate mean values ± SD from p21−/− and p21+/+ samples, respectively. Five animals in each group were used in this experiment.
Discussion
This study provides the first evidence that cell cycle activation of neural precursors in vivo is intrinsically inhibited by p21 after brain injury, through a p53-independent pathway. Similar responses in both hippocampus and lateral ventricle indicate that the effect of p21 is not highly location specific. In the mild and remote injury model we used, hippocampal cells were stressed as a result of focal ischemia and were stimulated to divide, similar to periventricular cells. It is anticipated that these cells may not only participate in the recovery process after focal ischemia but also provide a source of cells potentially renewable for repairing injured striatal tissue, especially in the light of the recent studies demonstrating the migration of the neural precursors from SVZ to the injured sites after severe brain ischemia (9, 10). Therefore, this study provides a rationale for future strategies to suppress p21 within injured brain after severe ischemia.
It is unlikely that the BrdU-positive cells were the result of DNA repair rather than proliferation based on the following reasons: (a) the documented intensity and location of the BrdU staining specific for neural cell proliferation was consistent with the previous validations by others (7, 19, 28); (b) the “off-on” coexpression of NeuN with BrdU suggested neuronal differentiation; (c) there was a minimal degree of cell apoptosis or inflammatory response in the regions; and (d) there was a correlated appearance of the independent indicator for cell proliferation, PCNA, with BrdU incorporation. Arguably, it might be possible that the increased BrdU incorporation in the p21−/− injured brain may reflect the reentry into cell cycle of some postmitotic differentiated cells as demonstrated in the p27−/− or p19−/− mice (38). However, the fact that BrdU-incorporated cells exhibited little coexpression of NeuN 1 wk and acquired NeuN later on after injury opposes the possibility of “dedifferentiation” of the postmitotic differentiated cells. In addition, it might be also possible that abnormal alterations in cell cycle kinetics, as mediated by the absence of p21, may alter the neural fate choices or downstream differentiation potential in the way that would contribute to the composition of cell subtypes in the regenerated tissue as a result of the experimental ischemic insult. Although this issue perhaps needs formal exploration within CNS in the future, we did not observe the altered lineage distribution in the neurosphere culture system in the absence of p21 (Fig. S1), arguing against the possibility.
Our study provides intriguing parallels with the role of p21 in stem cell regulation in hematopoietic tissue (14). A functional parallelism between neurogenesis and hematopoiesis has been proposed to map the hierarchy of neural precursor cells. This has been partially substantiated by the studies suggesting transdifferentiation (39–41) and shared gene transcripts between these two stem cell populations (42–45). Together with our previous work in both mouse and human HSCs (14, 46), we present a functional approach, defining a common cell cycle mediator restricting cell cycle entry in primitive quiescent cells from different tissues. Of note, we did not observe an increase of NSC/NPC proliferation in p21−/− mice under homeostatic conditions, which contrasts with the increased stem cell number and more active cell cycling status we observed in hematopoiesis (14). Several reasons might account for this difference. First, there are fundamental differences between neurogenesis and hematopoiesis in the turnover rate and longevity of newly born cells. The demand for steady-state proliferation of neural precursors in CNS is much lower than in the hematopoietic system. Second, possible compensation from other CKIs or other molecules in p21−/− mice might contribute to the invisible change of NSC proliferation under noninjured conditions. However, data indicating that p21-null and p27- or p18-null have very different phenotypes in ischemic brain (unpublished data), similar to what we found in the hematopoietic system (13), does not seem to support the possibility. Third, it is possible that altered BrdU incorporation cannot be specifically detected in NSC due to the lack of our ability to phenotypically distinguish NSC from NPC in vivo. Notably, a distinct temporal pattern of increasing NeuN/BrdU double-positive cells was observed in the p21-null setting in the current study (Fig. 4 B). After MCAO, there were 3.0-fold increases of single BrdU-positive cells at 1 wk that increased to a 5.6-fold increase of BrdU/NeuN double-positive cells at 1 mo. This magnitude of rise over time was smaller in the p21+/+ animals where the increases were 1.5–1.9, respectively, thereby suggesting that the absence of p21 may have engaged a more primitive, self-expanding population of stem cells. This speculation is supported by a recent study indicating a possible role of p21 in regulating NSC self-renewal caused by the Bmi-1 protein (47), whereas further proof of this possibility awaits a clear phenotype of NSC in vivo equivalent to that defined for HSC (48).
In conclusion, p21 appears to specifically restrict entry of quiescent precursors into cell cycle in both adult neural and hematopoietic tissue types despite the distinct proliferative kinetics of the two systems. Functionally similar HSC, NSC/NPC, and possibly other stem cell type (49) may share the molecular commonality of p21-restricted cell proliferation. As a corollary, altering p21 expression may be viewed as a possible means of enhancing stem cell response to tissue injury in general.
Acknowledgments
This work was supported by National Institutes of Health grants P50 NS10828 (to M.A. Moskowitz), DK02761 and HL70561 (to T. Cheng), and HL65909 and DK50234 (to D.T. Scadden); a Doris Duke Charitable Foundation grant (to D.T. Scadden); a scholar award from the American Society of Hematology (to T. Cheng); and American Heart Association grant N0335154 (to J. Qiu).
J. Qiu and Y. Takagi contributed equally to this work.
The online version of this article contains supplemental material.
Abbreviations used in this paper: BrdU, 5-bromodeoxyuridine; CKI, cyclin-dependent kinase inhibitor; CNS, central nervous system; DCX, doublecortin; EGF, epidermal growth factor; EIA, enzyme immunoassay; FGF, fibroblast growth factor; GCL, granule cell layer; HSC, hematopoietic stem cell; MCAO, middle cerebral artery occlusion; NPC, neural progenitor cell; NSC, neural stem cell; p21, p21cip1/waf1; PCNA, proliferating cell nuclear antigen; SGZ, subgranular zone; SVZ, subventricular zone; TUNEL, terminal deoxynucleotidyl transferase–mediated deoxyuridinetriphosphate nick end labeling.
References
- 1.Wilpshaar, J., M. Bhatia, H.H. Kanhai, R. Breese, D.K. Heilman, C.S. Johnson, J.H. Falkenburg, and E.F. Srour. 2002. Engraftment potential of human fetal hematopoietic cells in NOD/SCID mice is not restricted to mitotically quiescent cells. Blood. 100:120–127. [DOI] [PubMed] [Google Scholar]
- 2.Bonfanti, L.G.A., R. Galli, and A.L. Vescovi. 2001. Multipotent stem cells in the adult central nervous system. Stem Cells and CNS Development. M.S. Rao, editor. Humana Press, Totowa, NJ. 31–48.
- 3.Palmer, T.D. S. Colamarino, and F.H. Gage. 2001. Mobilizing endogenous stem cells. Stem Cells and CNS Development. M.S. Rao, editor. Humana Press, Inc., Totowa, NJ. 263–290.
- 4.Morrison, S.J. 2002. Stem cells of the nervous system. Mouse Development. J. Rossant and P.P.L. Tam, editors. Academic Press, Toronto, Canada. 235–252.
- 5.Doetsch, F., I. Caille, D.A. Lim, J.M. Garcia-Verdugo, and A. Alvarez-Buylla. 1999. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 97:703–716. [DOI] [PubMed] [Google Scholar]
- 6.Morshead, C.M., B.A. Reynolds, C.G. Craig, M.W. McBurney, W.A. Staines, D. Morassutti, S. Weiss, and D. van der Kooy. 1994. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 13:1071–1082. [DOI] [PubMed] [Google Scholar]
- 7.Gage, F.H. 2000. Mammalian neural stem cells. Science. 287:1433–1438. [DOI] [PubMed] [Google Scholar]
- 8.Horner, P.J., and F.H. Gage. 2000. Regenerating the damaged central nervous system. Nature. 407:963–970. [DOI] [PubMed] [Google Scholar]
- 9.Arvidsson, A., T. Collin, D. Kirik, Z. Kokaia, and O. Lindvall. 2002. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 8:963–970. [DOI] [PubMed] [Google Scholar]
- 10.Nakatomi, H., T. Kuriu, S. Okabe, S. Yamamoto, O. Hatano, N. Kawahara, A. Tamura, T. Kirino, and M. Nakafuku. 2002. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 110:429–441. [DOI] [PubMed] [Google Scholar]
- 11.Snyder, E.Y., and K.I. Park. 2002. Limitations in brain repair. Nat. Med. 8:928–930. [DOI] [PubMed] [Google Scholar]
- 12.Sherr, C.J., and J.M. Roberts. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13:1501–1512. [DOI] [PubMed] [Google Scholar]
- 13.Cheng, T., N. Rodrigues, D. Dombkowski, S. Stier, and D. Scadden. 2000. Stem cell repopulation efficiency but not pool size is governed by p27. Nat. Med. 6:1235–1240. [DOI] [PubMed] [Google Scholar]
- 14.Cheng, T., N. Rodrigues, H. Shen, Y. Yang, D. Dombkowski, M. Sykes, and D.T. Scadden. 2000. Hematopoietic stem cell quiescence maintained by p21(cip1/waf1). Science. 287:1804–1808. [DOI] [PubMed] [Google Scholar]
- 15.Brugarolas, J., C. Chandrasekaran, J.I. Gordon, D. Beach, T. Jacks, and G.J. Hannon. 1995. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 377:552–557. [DOI] [PubMed] [Google Scholar]
- 16.Huang, Z., P.L. Huang, N. Panahian, T. Dalkara, M.C. Fishman, and M.A. Moskowitz. 1994. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 265:1883–1885. [DOI] [PubMed] [Google Scholar]
- 17.Gartel, A.L., X. Ye, E. Goufman, P. Shianov, N. Hay, F. Najmabadi, and A.L. Tyner. 2001. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA. 98:4510–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long, J.M., A.N. Kalehua, N.J. Muth, J.M. Hengemihle, M. Jucker, M.E. Calhoun, D.K. Ingram, and P.R. Mouton. 1998. Stereological estimation of total microglia number in mouse hippocampus. J. Neurosci. Methods. 84:101–108. [DOI] [PubMed] [Google Scholar]
- 19.Yoshimura, S., Y. Takagi, J. Harada, T. Teramoto, S.S. Thomas, C. Waeber, J.C. Bakowska, X.O. Breakefield, and M.A. Moskowitz. 2001. FGF-2 regulation of neurogenesis in adult hippocampus after brain injury. Proc. Natl. Acad. Sci. USA. 98:5874–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teramoto, T., J. Qiu, J.C. Plumier, and M.A. Moskowitz. 2003. EGF amplifies the replacement of parvalbumin-expressing striatal interneurons after ischemia. J. Clin. Invest. 111:1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn, H.G., H. Dickinson-Anson, and F.H. Gage. 1996. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16:2027–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Namura, S., J. Zhu, K. Fink, M. Endres, A. Srinivasan, K.J. Tomaselli, J. Yuan, and M.A. Moskowitz. 1998. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci. 18:3659–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynolds, B.A., and S. Weiss. 1992. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 255:1707–1710. [DOI] [PubMed] [Google Scholar]
- 24.Johansson, C.B., S. Momma, D.L. Clarke, M. Risling, U. Lendahl, and J. Frisen. 1999. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 96:25–34. [DOI] [PubMed] [Google Scholar]
- 25.Gage, F.H., G. Kempermann, T.D. Palmer, D.A. Peterson, and J. Ray. 1998. Multipotent progenitor cells in the adult dentate gyrus. J. Neurobiol. 36:249–266. [DOI] [PubMed] [Google Scholar]
- 26.Zhang, R.L., Z.G. Zhang, L. Zhang, and M. Chopp. 2001. Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience. 105:33–41. [DOI] [PubMed] [Google Scholar]
- 27.Venkatraman, G., and M.B. Luskin. 2001. Neuronal restricted precursors. Stem Cells and CNS Development. M. Rao, editor. Humana Press, Inc., Totowa, NJ. 93–122.
- 28.Arvidsson, A., Z. Kokaia, and O. Lindvall. 2001. N-methyl-d-aspartate receptor-mediated increase of neurogenesis in adult rat dentate gyrus following stroke. Eur. J. Neurosci. 14:10–18. [DOI] [PubMed] [Google Scholar]
- 29.van Lookeren Campagne, M., and R. Gill. 1998. Cell cycle-related gene expression in the adult rat brain: selective induction of cyclin G1 and p21WAF1/CIP1 in neurons following focal cerebral ischemia. Neuroscience. 84:1097–1112. [DOI] [PubMed] [Google Scholar]
- 30.Endres, M., S. Namura, M. Shimizu-Sasamata, C. Waeber, L. Zhang, T. Gomez-Isla, B.T. Hyman, and M.A. Moskowitz. 1998. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J. Cereb. Blood Flow Metab. 18:238–247. [DOI] [PubMed] [Google Scholar]
- 31.Jin, K., M. Minami, J.Q. Lan, X.O. Mao, S. Batteur, R.P. Simon, and D.A. Greenberg. 2001. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. USA. 98:4710–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magavi, S.S., B.R. Leavitt, and J.D. Macklis. 2000. Induction of neurogenesis in the neocortex of adult mice. Nature. 405:951–955. [DOI] [PubMed] [Google Scholar]
- 33.Gleeson, J.G., P.T. Lin, L.A. Flanagan, and C.A. Walsh. 1999. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 23:257–271. [DOI] [PubMed] [Google Scholar]
- 34.Brown, J.P., S. Couillard-Despres, C.M. Cooper-Kuhn, J. Winkler, L. Aigner, and H.G. Kuhn. 2003. Transient expression of doublecortin during adult neurogenesis. J. Comp. Neurol. 467:1–10. [DOI] [PubMed] [Google Scholar]
- 35.Vogelstein, B., D. Lane, and A.J. Levine. 2000. Surfing the p53 network. Nature. 408:307–310. [DOI] [PubMed] [Google Scholar]
- 36.Tao, Y., I.B. Black, and E. DiCicco-Bloom. 1996. Neurogenesis in neonatal rat brain is regulated by peripheral injection of basic fibroblast growth factor (bFGF). J. Comp. Neurol. 376:653–663. [DOI] [PubMed] [Google Scholar]
- 37.Lazar, L.M., and M. Blum. 1992. Regional distribution and developmental expression of epidermal growth factor and transforming growth factor-alpha mRNA in mouse brain by a quantitative nuclease protection assay. J. Neurosci. 12:1688–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zindy, F., J.J. Cunningham, C.J. Sherr, S. Jogal, R.J. Smeyne, and M.F. Roussel. 1999. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA. 96:13462–13467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brazelton, T.R., F.M. Rossi, G.I. Keshet, and H.M. Blau. 2000. From marrow to brain: expression of neuronal phenotypes in adult mice. Science. 290:1775–1779. [DOI] [PubMed] [Google Scholar]
- 40.Bjornson, C.R., R.L. Rietze, B.A. Reynolds, M.C. Magli, and A.L. Vescovi. 1999. Turning brain into blood: a hematopoietic fate adopted by adult neural stem cells in vivo. Science. 283:534–537. [DOI] [PubMed] [Google Scholar]
- 41.Mezey, E., K.J. Chandross, G. Harta, R.A. Maki, and S.R. McKercher. 2000. Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science. 290:1779–1782. [DOI] [PubMed] [Google Scholar]
- 42.Hackney, J.A., P. Charbord, B.P. Brunk, C.J. Stoeckert, I.R. Lemischka, and K.A. Moore. 2002. A molecular profile of a hematopoietic stem cell niche. Proc. Natl. Acad. Sci. USA. 99:13061–13066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramalho-Santos, M., S. Yoon, Y. Matsuzaki, R.C. Mulligan, and D.A. Melton. 2002. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 298:597–600. [DOI] [PubMed] [Google Scholar]
- 44.Ivanova, N.B., J.T. Dimos, C. Schaniel, J.A. Hackney, K.A. Moore, and I.R. Lemischka. 2002. A stem cell molecular signature. Science. 298:601–604. [DOI] [PubMed] [Google Scholar]
- 45.Tsai, R.Y., and R.D. McKay. 2002. A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev. 16:2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stier, S., T. Cheng, R. Forkert, C. Lutz, D.M. Dombkowski, J.L. Zhang, and D.T. Scadden. 2003. Ex vivo targeting of p21Cip1/Waf1 permits relative expansion of human hematopoietic stem cells. Blood. 102:1260–1266. [DOI] [PubMed] [Google Scholar]
- 47.Molofsky, A.V., R. Pardal, T. Iwashita, I.K. Park, M.F. Clarke, and S.J. Morrison. 2003. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 425:962–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morrison, S.J., N. Uchida, and I.L. Weissman. 1995. The biology of hematopoietic stem cells. Annu. Rev. Cell Dev. Biol. 11:35–71. [DOI] [PubMed] [Google Scholar]
- 49.Topley, G.I., R. Okuyama, J.G. Gonzales, C. Conti, and G.P. Dotto. 1999. p21(WAF1/Cip1) functions as a suppressor of malignant skin tumor formation and a determinant of keratinocyte stem-cell potential. Proc. Natl. Acad. Sci. USA. 96:9089–9094. [DOI] [PMC free article] [PubMed] [Google Scholar]