

Abstract
PML–RARA was proposed to initiate acute promyelocytic leukemia (APL) through PML–RARA homodimer–triggered repression. Here, we examined the nature of the PML–RARA protein complex and of its DNA targets in APL cells. Using a selection/amplification approach, we demonstrate that PML–RARA targets consist of two AGGTCA elements in an astonishing variety of orientations and spacings, pointing to highly relaxed structural constrains for DNA binding and identifying a major gain of function of this oncogene. PML–RARA-specific response elements were identified, which all conveyed a major transcriptional response to RA only in APL cells. In these cells, we demonstrate that PML–RARA oligomers are complexed to RXR. Directly probing PML–RARA function in APL cells, we found that the differentiation enhancer cyclic AMP (cAMP) boosted transcriptional activation by RA. cAMP also reversed the normal silencing (subordination) of the transactivating function of RXR when bound to RARA or PML–RARA, demonstrating that the alternate rexinoid/cAMP-triggered APL differentiation pathway also activates PML–RARA targets. Finally, cAMP restored both RA-triggered differentiation and PML–RARA transcriptional activation in mutant RA-resistant APL cells. Collectively, our findings directly demonstrate that APL cell differentiation parallels transcriptional activation through PML–RARA-RXR oligomers and that those are functionally targeted by cAMP, identifying this agent as another oncogene-targeted therapy.
Keywords: therapy, leukemia, selex, transcription, oncogene
Introduction
Acute promyelocytic leukemia (APL) is triggered by a chromosomal translocation that yields expression of the PML–RARA oncogene, a fusion protein between a transcription factor, the retinoic receptor (RARA), and an organizer of nuclear domains involved in apoptosis, PML. RARA belongs to the nuclear receptor family and binds DNA as a heterodimer with the nuclear receptor RXR. Response elements consist of direct repeats (DRs) of a (A/G)G(G/T)TCA consensus with spacings of 2 or 5, whereas RXR homodimers bind DR1 and DR6 elements (1). RXR-specific agonists (rexinoids) fail to activate transcription from RAR–RXR complexes, a process referred to as RXR subordination (2). This results from a steric hindrance of RARA-bound corepressors that impede the binding of coactivators onto RXR (3). Numerous observations indicate that PML–RARA is a potent repressor, which could turn down the expression of RA target genes critical to myeloid differentiation (4). PML harbors a strong dimerization domain that allows formation of PML–RARA homodimers, which bind DNA independently of RXR (5, 6). PML–RARA homodimers bind corepressors more tightly than RAR–RXR heterodimers, which could account for enhanced repression (7–9). It was also proposed that the binding of two corepressor molecules, rather than one, may contribute to PML–RARA-triggered repression (10, 11).
APL is a target for differentiation therapy because several agents can induce in vivo differentiation of the leukemic cells, yielding clinical remissions (12). At least two of these agents, retinoic acid or arsenic directly targets PML–RARA through each of its constitutive moieties (13–15). Targeting by these two agents results in the degradation of the PML–RARA oncogene, yet, the role of degradation versus transcriptional activation has been unclear, in part because RA can simultaneously activate RAR and PML–RARA. Similarly, cAMP is well known to synergize with RA to trigger differentiation in several cell types including APL cells (16, 17). In the case of F9 embryonal carcinoma cells, the molecular basis of the RA/cAMP cross-talk for parietal endoderm differentiation was shown to be cAMP-triggered RARA phosphorylation, although cAMP did not enhance expression of the RA target genes tested (18). Finally, previous studies have suggested the existence, in APL cells, of an alternate RXR/cAMP differentiation pathway, independent from PML–RARA (19, 20). This pathway could be active in non-APL leukemias and extend the concept of differentiation therapy to other hematological malignancies (21).
In this study, we have performed an unbiased selection for DNA sequences that bind to PML–RARA homodimers and identified binding sites consisting of two copies of an AGGTCA core in a wide variety of spatial arrangements, demonstrating that PML–RARA deregulates a much wider network of target genes than previously thought. All of these sequences could confer retinoic acid response, some of them exclusively, in PML–RARA-expressing cells. We show that in APL cells PML–RARA homodimers are RXR bound. A dramatic synergy between RA, RAR-specific agonists, as well as RXR-specific agonists and cAMP was identified at the transcription activation level, which results, at least in part, from cAMP-triggered RXR desubordination. Hence, in APL, a PML–RARA-RXR oligomer binds an extended repertoire of response elements and is functionally targeted by the differentiation inducer cAMP.
Materials and Methods
Preparation of the Cell Extracts.
Cos-7 cells transiently transfected with pSG5-PML–RARA were lysed in ice for 30 min in the lysis buffer (50 mM Tris, pH 8.0, 0.6% [vol/vol] Igepal CA-630 [Sigma-Aldrich], 480 mM NaCl, 10% [vol/vol] glycerol, 0.1 mM EDTA, 1 mM DTT, 1.5 mM sodium vanadate, 0.4 mM PMSF, 3 μg/ml aprotinin, 1 μg/ml pepstatin, 1 μg/ml leupeptin). The centrifugation supernatant was stored at −80°C.
Selection and Amplification of Binding Sequences.
The amplification and binding selection method was modified as previously described (22), using an oligonucleotide pool of randomized single-stranded oligonucleotide template 5′-CACATTCCTTCGGCGGATCCAGTCG-(N)25-TAGGCTCTAGAATTCCATGCGCAGC. Double-stranded oligonucleotides were generated by annealing the 5′ primer (5′-CACATTCCTTCGGCGGATCCAGTCG) to the single-stranded template and extending with Klenow fragment of DNA polymerase I. The double-stranded templates were end labeled with γP32-ATP. End-labeled templates were mixed with extract of PML–RARA-transfected cells in the binding buffer (25 mM Tris, pH 8.0, 0.15 mM DTT, 140 mM NaCl, 0.075% [vol/vol] Igepal CA-630 [Sigma-Aldrich], 10% [vol/vol] glycerol, 0.2 mg/ml BSA). DNA–protein complexes were separated on a 3.75% polyacrylamide gel buffered with 45 mM Tris, 45 mM borate, and 0.1 mM EDTA at 4°C for 2 h. An oligonucleotide probe of the same length as a randomized matrix (75 bp) containing the RAREB (23) served as a marker for migration of the two PML–RARA-DNA complexes. The gel slices corresponding to the position of the positive controls DIM and OLI (see Fig. 1 A) were excised. The eluted selected pool was PCR amplified with 5′ (see above) and 3′ primer (5′-GCTGCGCATGGAATTCTAGAGCCTA), gel purified, and labeled as described above for subsequent rounds of selection. For each subsequent round, we decreased a quantity of template twice or thrice, starting from 400 pmol DNA. The concentration of the cell extract was chosen from a pilot experiment where 1 pmol of the nonlabeled template and labeled RAREB were mixed with various amounts of the extract. The concentration of the extract able to shift 7–14% of the RAREB was used for the preparative selection step. After five (for band OLI) and six (for band DIM) rounds, the selected oligonucleotides were subcloned into the pGEM-7Zf(+) cloning vector and 174 of them were sequenced.
Figure 1.

Selection of high affinity binding sites for PML-RARA homodimers. (A) Band shift assays representing the different steps of the selection/amplification procedure with Cos cell–expressed PML–RARA protein. The positive control (β) is the RAREB response element. V represents the initial pool of random oligonucleotides and OLI and DIM point to the two complexes containing PML–RARA oligomers and dimers, respectively. The fifth (5) and sixth (6) round of purification derived from the OLI complex and the sixth (6) round of purification from the DIM complex are shown. Note that the selected sequences are specific for PML–RARA, as they can be supershifted with anti-PML antibodies (PML-Ab). (B) Optimized binding site in a DR5 architecture (DR5) bind PML–RARA and RARA/RXR with a higher affinity than RAREB (β).
Oligonucleotides.
Oligonucleotides representing the DRs were 5′-AATCGTCGACACCAAAGGTCA -spacer- AGGTCACGACTCGAGCATT, where the spacers were as follows: Dr1 A, Dr2 AA, Dr3 AAA, Dr4 ACAA, Dr5 ACGAA, Dr6 ACGGAA, Dr8 ACGGCGAA, Dr10 ACGGAACGAA, Dr12 ACGGATTACGAA, Dr14 ACGGATCTTACGAA, Dr16 ACGGATCGATTACGAA, Dr20 ACGGATCGTAGAATTACGAA. The palindromes were prepared from the sequences IR0 5′-AATCGTCGACCCAAAGGTCATGACCTTTGTCTCGAGCATT and ER8 5′-AATCGTCGACCACGTGACCTTTACCCAAAGGTCACGATCTCGAGCATT. As negative controls, we used the sequence DR5 carrying base substitutions in one or both hexamers.
Electrophoretic Mobility Shift Assay (EMSA).
For EMSAs, the whole cell extract of the Cos cells transiently transfected with pSG5 expression vector(s) containing PML-RAR. RXR and RAR genes were presented to the variety of the consensus PML-RARA–binding sequences. PCR-constructed DNA probes were gel purified and labeled as described above and mixed with cellular extract in the binding buffer as above with 0.02 μg/μl poly(dI-dC) at room temperature. DNA–protein complexes and free DNA were separated on a 3.75% polyacrylamide gel.
Cell Culture, Transient Transfections, and Reporter Gene Assays.
Double-stranded oligonucleotides, described above for EMSA, were digested with XhoI/SalI and ligated into SalI-digested pTK-Luc (23). All plasmids were selected in which a single copy of the binding site was oriented in the same direction. For pTK-Dr constructs, the binding sites were all oriented such that the TGACCT sequence was followed by the promoter.
Cos cells were transiently transfected with 0.24 μg pTK-Luc reporter plasmids, 0.04 μg pRLtk (Promega), and 0.03 μg expression vector per 4 cm2 wells. Cells were grown in medium containing 10% FBS for 2 h and then the media was changed to 5% charcoal resin-stripped (super-stripped) FBS and either 1 μM RA (Sigma-Aldrich) or ethanol carrier alone, or others as indicated in the figures. After 24 h, cells were lysed and normalized luciferase activities were determined.
U-937, NB4, NB4LR2, and NB4-MR4 cells were electroporated in 250 μl Optimem 1 (GIBCO BRL) with 6 μg pTK-Luc reporter plasmids, 0.8 μg pRLtk (Promega) per 20 million cells in a 4-mm thick cuvette by an impulse of 250 V, 950 F. After transfection, cells were grown in medium containing 10% FBS for 3 h and then either RA (Sigma-Aldrich) or cAMP (Sigma-Aldrich), or other reagents as indicated. After 18 h, cells were lysed and reporter gene activities were determined. Differentiation of the APL cell lines NB4, NB4LR2, and NB4MR4 (24) were assessed using NBT reduction, CD11b expression, and MGG stains as previously described (17, 25).
Quantitative PCR.
CD37 gene expression was monitored with the Light Cycler system (Roche Diagnosis) using SBYR green fluorescent probe. TBP gene expression was quantified in the same run as an internal reference. The quantification data were analyzed using Light Cycler software.
Online Supplemental Material.
Identity of RAR, PML–RARA, and RXR DNA–protein complexes is shown in Fig. S1. Extracts from Cos cells transfected with RAR, RXR, or PML–RARA were analyzed by band shifting using an optimized DR5 probe. Complexes were super shifted using anti-RARA (A), anti-PML (P), or anti-RXRA (X) antibodies. The different complexes are indicated by letters. Fig. S1 is available at http://www.jem.org/cgi/content/full/jem.20032226/DC1.
Results
Selection of High Affinity Binding DNA Sites for PML–RARA.
The wild-type nuclear receptor RARA and the PML–RARA fusion protein recognize the same response element from the RARB promoter (6), noted here as RAREB. Extracts from PML–RARA-transfected Cos cells generate two distinct DNA–protein complexes with the RAREB probe, which were not observed using the extracts from untransfected cells and were supershifted by either anti-RAR or anti-PML antibodies (6). The lowest band (DIM) corresponds to the PML–RARA homodimer (5, 6, 10) and the upper (OLI) corresponds to a PML–RARA oligomer (Fig. 1 A; references 6 and 11). The presence of PML–RARA-DNA complex near the top of the gel has been reported by others and is due to the formation of an oligomeric PML–RARA complex greater than a dimer in size (26 and references therein). To identify all possible high affinity binding sites for PML–RARA, we used the selection amplification technique starting from a pool of DNA sequences containing 25 random base pairs. PML–RARA-bound DNA was separated from free DNA by gel migration. Initially, PML–RARA-DNA complexes are undetectable. Therefore, we used RAREB as a marker for migration of the DIM and OLI PML–RARA complexes (Fig. 1 A). After three rounds of the selection/amplification, the specific DNA–protein complexes became visible. As expected, PML–RARA binding of oligonucleotide increased with additional selection rounds and the resulting complexes were all supershifted by PML antibodies (Fig. 1 A). DNA selected by PML–RARA binding was subcloned and 172 colonies were sequenced.
Most of the selected sequences contain an AGGTCA consensus (Fig. 2 A), which is the common target of type II nuclear receptors, including RARA. As expected, most selected sequences (109) contain the consensus repeated twice. The most spectacular feature of this family of PML–RARA binding sites is their architecture. Indeed, we found all possible orientations of the two AGGTCA consensus: direct, inverted, and everted repeat. Moreover, among these three orientations, a wide variety of spacing was observed (Fig. 2 B). Clearly, this is very different from known binding sites for RAR and RXR heterodimers or homodimers and highly unusual for nuclear receptors. Although tolerant to the binding site architecture, PML–RARA nevertheless has preferred spacing for each repeat configuration. The number of selected sequences decreases as follows: DR2 > DR4 > IR0 = DR3 = DR5 > ER8 > DR6, but importantly, spacers up to 12 and 13 were also selected (Fig. 2). PML-RAR is more selective for inverted repeat. The number of IR0 sequences is higher than for all other spacers. Among everted repeats, ER8 is clearly preferential for PML–RARA binding.
Figure 2.

Summary of the binding elements identified by random selection. (A) Nucleotide frequency within the identified core motif. (B) Summary of the various architectures and spacings identified among the 109 sequences with two AGGTCA core motifs. (C) Preference for specific nucleotides adjacent to the AGGTCA core in the various architectures. Comparison between the natural RAREB response element from the RARB promoter and the optimized DR5 site identified here. Nonconserved residues are indicated by x.
Our selection approach, in addition to defining a core consensus and spacer lengths, allows the identification of preferred neighboring bases. We found a strong pressure for an AA doublet before the AGGTCA core, whatever the architecture of the PML–RARA binding site. Similarly, the favored first doublet downstream to the core is CG. The bases neighboring the sites of contact with zinc fingers often interact with other segments of the protein to orient it along the DNA (27, 28). In the context of a DR, this extended consensus influences the composition of the spacer, allowing us to determine an optimal and extended PML–RARA binding consensus that defines the internal bases of the various DR motifs (Fig. 2 C). Interestingly, in the DR5 architecture, this consensus is strikingly similar to the natural response element from the RARB gene, the most potent RARE known to date, with the notable exception of the T within the cores (Fig. 2; reference 23). Our optimized binding site, in the DR5 architecture, binds PML/RAR dimers with a 10-fold higher affinity than the RAREB, and RARA/RXR heterodimers with a fivefold higher affinity (Fig. 1 B), providing a new tool to probe endogenous RAR–RXR complexes or to monitor RA response in vivo.
Analysis of PML–RARA Binding to DRs of Optimized Half Sites.
The effect of mutations within the AGGTCA core motif was previously studied (26). Therefore, we evaluated the binding affinity of PML–RARA to a family of sequences derived from our optimized consensus in which DRs of the consensus have spacings varying from 0 to 20. Similarly to RAREB, two PML–RARA-DNA complexes were detected in EMSA for all binding sites tested, which were specifically supershifted using anti-RARA or anti-PML antibodies and not by anti-RXR antibodies (Fig. 3 A and unpublished data). Among these sites, the highest affinity was found for DR2 and DR4, consistent with the greater prevalence of these spacer lengths among selected sequences (Fig. 2 B). Strikingly, PML–RARA oligomers bound DRs with wide spacing (DR > 6; Fig. 3 A), which were not efficiently recognized by RAR–RXR (unpublished data). Sequences in which one or both of the core motifs were destroyed bound very weakly (one core motif mutated) or not at all (two mutations). Conversely, when PML–RARA harbored mutations that abolish either dimerization or DNA binding, no binding was observed (unpublished data).
Figure 3.

Binding of PML-RAR and PML-RAR–RXR to direct, inverted, or everted optimized repeats. (A) Extracts of Cos cells transfected with PML–RARA were analyzed by band shifting using as probes DRs separated with the indicated number of bases, as well as palindromes (inverted or everted repeats, IR0 and ER8). On the right, controls were the RAREB probe (β), untransfected cells (Ø), or a DR5 where one hexamer was mutated (−). Ratio of bound/nonbound probe is shown above. Bold line, OLI complex; thin line, DIM complex. (B) Same as A, using extracts of Cos cells transfected with PML–RARA and RXRA. Note that a single complex corresponding to PML–RARA-RXRA oligomers (arrow) is observed with most probes. A rapidly migrating complex (arrowhead) is observed with DR1 and DR6 corresponds to RXRA homodimers.
PML–RARA Is Complexed to RXR in NB4 Cells and Binds Widely Spaced DRs.
PML–RARA may bind RXR (6) and, in the presence of RXR, the major form of PML–RARA-containing complex migrated slower than the DIM complex and can be interpreted as an oligomer containing both PML–RARA and RXR, most likely a tetramer (PML–RARA-RXR)x2 bound to DNA (5, 6, 10, 11), whereas an excess of RXR over PML–RARA generated a fast migrating RXR homodimer complex with DNA. Supershift experiments with anti-RAR, anti-RXR, and anti-PML, as well as use of PML–RARA mutants (DNA binding, dimerization, RXR binding), were all consistent with this view (not depicted). The (PML–RARA-RXR)x1 heterodimer, which was previously detected using the RAREB probe (6), was only detected here as the transitory product in RXR titration experiments (not depicted). The binding selectivity of RXR homodimer obtained with DR1/DR20 elements confirmed its preferential binding to DR1 and DR6 (1). In contrast, PML–RARA-RXR oligomers bound DNA with much less restrictions. DRs with spacing varying from 1 to 20, as well as both IR or ER palindromes, were all efficiently bound (Fig. 3 B). The binding preferences were somehow distinct from those of PML–RARA homodimers. As expected, for DRs with small spacing (DR < 6), the affinity profile of PML–RARA-RXR oligomers is close to that of the normal RAR–RXR heterodimers, with a greater affinity for DR1 and DR5 than for DR4 (Fig. 3). Yet, in sharp contrast to RAR–RXR, PML–RARA-RXR oligomers bound DR with very long spacing (DR6–20).
The availability of very high affinity binding sites for PML–RARA allowed us to detect endogenous PML–RARA in leukemic cells by EMSA. Using several methods to prepare nuclear extracts, we constantly found PML–RARA as PML–RARA-RXR oligomers and, importantly, we could never detect PML–RARA homodimers although the probes used here (DR8, DR2, and ER8) were optimized for homodimer binding (Fig. 4 A). This specific DNA–protein complex involves the protein moieties containing PML, RAR, and RXR as demonstrated by supershifting (Fig. 4 B). We constantly found that the PML–RARA-RXR oligomers derived from NB4 cells had a slightly retarded mobility when compared with the ones derived from transfected Cos cells. This likely reflects the poor efficiency of posttranslational modifications such as phosphorylation or sumolation in transfected cells (5, 29). This experiment very strongly suggests that in APL, the predominant form of the fusion protein is a PML–RARA-RXR oligomer.
Figure 4.

Identification of PML–RARA-RXR tetramers in APL cells. Binding sites ER8, DR8, and DR2 were presented to increasing amounts of the NB4 nuclear extracts. The gel mobility of the complexes was compared with extracts of Cos cells transfected with RXRA (RXR), PML–RARA (P/R), or both (P/R+RXR). Identity of DNA–protein complexes was tested by supershifting with anti-RARA (α-A), anti-PML (α-P), and anti-RXR (α-X) antibodies. The arrow points to the PML–RARA-RXR oligomers, OLI and DIM, and to the PML–RARA-only complexes.
Identification of PML–RARA-specific Transcriptional Response Elements.
To test if these selected DNA sequences are not only effective binding sites, but also functional response elements, we inserted a single copy of the DR with a 1–16-bp spacer upstream to a TK minimal promoter fused with a luciferase reporter cDNA (pTK luciferase vector). In the absence of PML–RARA expression in the Cos cells, no significant change in reporter gene activity was found upon treatment with RA, except with DR-5 and DR-2 inserts that are well-known to bind endogenous RAR–RXR heterodimers (Fig. 5 A). When a PML–RARA expression vector was cotransfected with the reporters in the absence of RA, a significant transcription repression was observed from all reporters that contained bindings sites (Fig. 5 B). In contrast, when PML–RARA-expressing cells were treated with RA, an induction of luciferase transcription was observed for many repeats, including DR8, DR12, IR0, and ER8 (Fig. 5 A). Comparing the level of reporter expression in the absence of PML–RARA, we found that in most cases, RA derepressed, rather than activated (DR5 and DR6), but for DR8 elements, some PML–RARA-dependent activation was observed (Fig. 5 B). Similar results were obtained when PML–RARA was cotransfected along with an expression vector for RXR (unpublished data).
Figure 5.
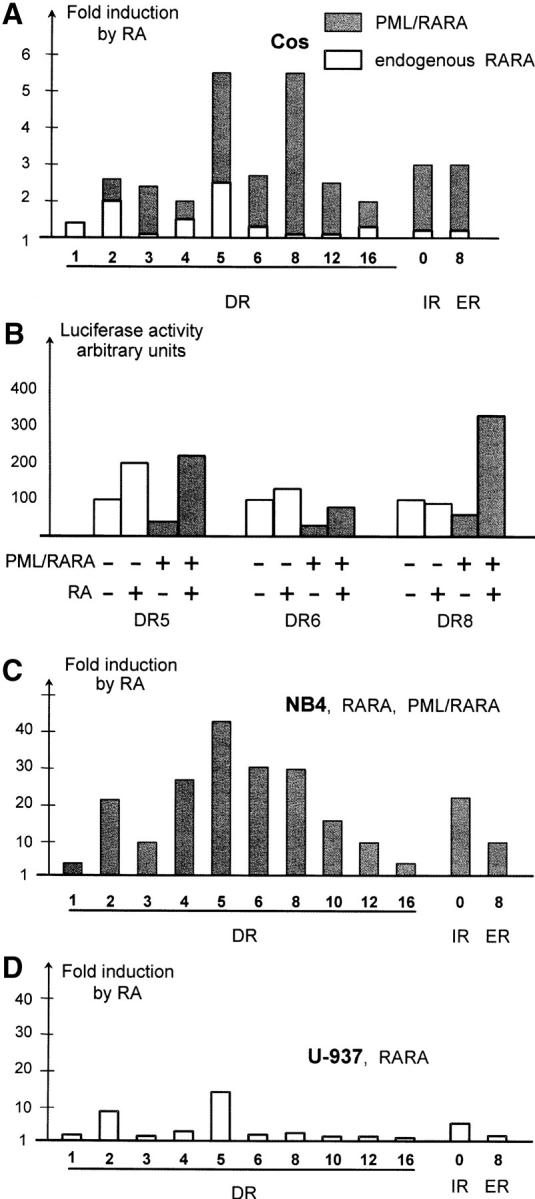
Identification of PML–RARA-specific response elements. (A) DRs separated with spacers of the indicated length, as well as ER8 and IR0, were inserted in a TK-Luc vector and tested for induction in the presence of RA. Values reported are averaged from three independent experiments in Cos cells. Shaded bars denote the induction in the presence of cotransfected PML–RARA expression vector, striped bars represent induction by endogenous RARA–RXR complexes. (B) PML–RARA induces repression on several PML–RARA binding sites. Note that RA only relieves repression on DR5 and DR6, but activates on DR8. (C) Reporter constructs were electroporated in NB4 APL cells that express endogenous RARA and PML–RARA proteins. (D) As in C, except that non-APL U937 cells that express RARA, but not PML–RARA, were used.
We then turned to a system that more closely reflects the situation of leukemic cells by electroporating these reporters in NB4 cells, which express levels of PML–RARA, RAR, and RXR similar to those present in APL blasts. A very strong transcriptional activation/derepression was observed after RA treatment for all reporters tested, except DR1 and DR16. In particular, DR4, DR6, and DR8 motifs conferred a major response to RA (>30-fold; Fig. 5 C). To address the specificity of response in a cell line that does not express PML–RARA, we studied hematopoietic U937 cells. As expected, we only found a significant RA response with the RAR–RXR targets DR2 and DR5 (Fig. 5 D). We have thus identified a subset of response elements that confer response to PML–RARA, but not RARA. Among these, some are specific for other nuclear receptors (DR4, thyroid hormone receptor), whereas others (DR8, DR10, etc.) have never been identified as nuclear receptor binding sites.
To demonstrate in an in vivo setting that these elements are specific for PML–RARA, we performed a search for such elements in the promoters of known human genes in the Eukaryotic Promoter Database (http://www.epd.isb-sib.ch). 13 genes contain DR7-DR16 elements within their proximal promoter and do not contain DR < 6. Expression of four of these genes containing large DRs was monitored by quantitative PCR during a time course of RA response in NB4 or HL60 cells (CD37, COX8, RAD51C, and PDHA1). CD37 (DR11) was induced fivefold by RA after 12 h in APL, but not in HL60 acute myeloid leukemia cells that differentiate in response to RA, but do not express PML–RARA (Fig. 6). Strikingly, comparing the baseline CD37 gene expression in NB4 and HL60 cells, we found that it was expressed at much higher levels in HL60 cells (20-fold). Hence, this gene that harbors a PML–RARA-specific binding site within its promoters is repressed in NB4 compared with HL60 cells and partially derepressed upon RA exposure (Fig. 6). The three others genes were not induced by RA. Hence, the presence of the PML–RARA-specific response elements is not sufficient for RA induction in APL cells, possibly reflecting low accessibility of these binding sites in vivo due to chromatin structure.
Figure 6.

Identification of natural PML–RARA-specific RA targets in APL cells. CD37 mRNA expression relative to TBP mRNA in NB4 cells (solid lines) or HL60 (dotted line) were monitored using quantitative RT-PCR after exposure to 1 μM RA for the indicated time.
cAMP Desubordinates RXR Transactivation in Either PML–RARA/RXR or RAR–RXR Complexes.
cAMP greatly enhances arsenic- or RA-triggered differentiation ex vivo or in vivo (17, 30–32), and triggers APL cell differentiation in vivo (17). In NB4 cells, cAMP administration modestly activated the DR5 reporter (1.5–2-fold), but did not affect DR8 or DR10 (unpublished data). Although additive effects were noted on DR5 reporters, a sharp synergy (up to sixfold) between cAMP and RA was noted on PML–RARA-specific DR8 and DR10 (Fig. 7 A), strongly suggesting that cAMP potentiation of RA-induced differentiation results from a direct enhancement of the transcription of primary PML–RARA target genes. A dose response analysis of RA-triggered PML–RARA-dependent activation demonstrated that activation plateaued above 10−8 M (Fig. 7 B) and that cAMP greatly enhanced the maximal level of activation and was sensitized to the effects of RA. Similarly, combining forskolin and phosphodiesterase inhibitors also enhanced RA-triggered transcription (unpublished data).
Figure 7.

Synergistic transcriptional activation by RA and cAMP. (A) NB4 cells were electroporated with the indicated reporters and treated with the following retinoids, as indicated: RA, BMS753 (RARA+), or BMS649 (RXR+), in the presence or absence of 160 μM cAMP for 18 h. (B) Dose response of RA-triggered activation on PML–RARA-specific reporter DR8 in NB4 cells, in the presence or absence of cAMP. (C) Same as in A, except that NB4LR2 cells were tested. (D) Same as in A and C, except that U937 cells were tested.
Our demonstration that in APL cells RXR is bound to PML–RARA and that cAMP enhances transcriptional activation by this complex led us to reevaluate the existence of the PML–RARA-independent RXR/cAMP differentiation pathway by using RARA- and RXR-specific ligands (19). As expected, RARA-specific agonist strongly activated PML–RARA-specific reporters and response was increased by cAMP. Although neither RXR-specific agonists nor cAMP induced significant activation on DR5-, DR6-, or DR8-containing reporters, their association induced a robust sevenfold increase in luciferase activity (Fig. 7 A), suggesting that cAMP desubordinates RXR and allows it to become a transcriptional activator on the DR8 reporter, which binds PML–RARA. Similar results were obtained with BMS 749, a compound with both RXR agonist and RARA antagonist activities (unpublished data). As a control, we used NB4LR2 APL cells, which harbor a frameshift mutation in the RARA moiety of PML–RARA that deletes the RA binding pocket (33). PML–RARA-dependent activation was observed exclusively by combining a rexinoid and cAMP (Fig. 7 C), but not with rexinoids alone nor RARA agonists in the presence or absence of cAMP. Altogether, these experiments demonstrate that cAMP allows RXR to enhance PML–RARA-dependent transcription, resulting in an induction of differentiation.
To verify that this is also the case in the context of the wild-type RAR–RXR heterodimers, we tested the ability of RARA agonists or rexinoids to activate transcription from a DR5-containing reporter in U937 cells. As expected, RARA agonists strongly activated transcription and cAMP further enhanced response, whereas cAMP was absolutely required for rexinoid-mediated transactivation (Fig. 7 D). Hence, cAMP also desubordonnates RXR in the context of the endogenous RXR/RAR heterodimers.
cAMP Can Restore RA Sensitivity and PML–RARA Transcription in an RA-resistant NB4 Clone.
In NB4MR4 APL cells, a point mutation in the E domain of PML–RARA impairs RA binding, resulting in RA resistance (34). Yet, in studying an animal model of RA-resistant APL in which the PML–RARA transgene harbors this same mutation (35), we found that the cAMP/RA association triggered terminal differentiation in vivo (17). Testing the hypothesis that cAMP may also circumvent RA resistance in the NB4MR4 cell line, we found that cAMP indeed restored RA-triggered differentiation (Fig. 8). When we probed PML–RARA signaling in these cells using DR5 or DR8, neither RA nor cAMP induced luciferase expression, but the combination of these two drugs triggered a major transcriptional activation (Fig. 8 and not depicted). Hence, RA/cAMP transcriptional activation through PML–RARA strictly parallels APL cell differentiation ex vivo and in vivo.
Figure 8.

cAMP restores RA-triggered differentiation in NB4MR4 cells. NBT reduction (A), CD11b expression (B), or May Grunwald Giemsa staining (C) were measured after a 4-d treatment with the indicated drug (10−6 M) and/or 200 μM cAMP in NB4 or NB4MR4 cells, as indicated. (D) Transcriptional activation on a DR8 reporter upon electroporation in NB4MR4 cells and treatment with the indicated drugs. Note that activation of PML–RARA-dependent transcription mirrors differentiation induction.
That cAMP restored RA response in NB4MR4 cells was surprising. RA might be isomerized into 9-cis-RA, a compound that efficiently binds and activates RXR. Thus, we used RARA- and RXR-specific ligands and assessed both differentiation and transcriptional activation. As expected, neither differentiation nor transactivation was found with either compound alone (Fig. 8). In contrast, in the presence of cAMP, either RARA- or RXR-specific agonists induced both transcriptional activation of PML/RAR-specific reporters and differentiation. Although this was expected for the rexinoid, reporter activation and differentiation induction by the RARA agonist/cAMP association was surprising and clearly distinguishes NB4LR2 from NB4MR4 cells. RA-triggered NB4MR4 differentiation was impaired by the RARA antagonists BMS 493 and BMS 009, or the protein kinase A inhibitor H89 (unpublished data). These observations imply that transcriptional activation and differentiation induction in these cells ex vivo (Fig. 8) or in vivo (17) are mediated by the RARA moiety of the fusion, likely through a change in its RA binding properties triggered by cAMP signaling.
Discussion
High affinity PML–RARA binding sites consist of a highly conserved consensus for RARA arranged in a variety of spatial arrangements, extending previous reports that PML–RARA may bind DR1–DR5 response elements (5, 6, 10). This extended consensus (A-AGGTCA-C) is likely the best binding site, not only for PML–RARA, but also for RARA, especially as DR2 and DR5. Our observations imply that PML-induced dimerization, remote from the DNA binding domains, allows the two RARA moieties of PML–RARA to bind very distant monomeric DNA sites, even if contacts between them are spatially impossible. The spectrum of response elements for PML–RARA and PML–RARA-RXR (DR1–DR16) is much broader than one for the wild-type RAR–RXR (DR1, DR2, and DR5). Hence, the fusion protein is likely to repress a very large number of genes including, but not limited to, all those controlled by all type II nuclear receptors. Accordingly in APL cells, RA activates a much larger number of genes than it does in non-APL leukemias (13). This broader spectrum of genes regulated by PML–RARA (or RA) identifies a major gain of function of this oncogene.
In vitro DNA binding assays show that PML–RARA may interact with RXR, yielding PML–RARA-RXR oligomers. The presence of RXR changes the DNA binding preferences of PML–RARA. Oligomers, but not homodimers, recognizes DR1 well, binds DR5 more avidly than DR4, and binds IR0 stronger than ER8. In NB4 cells, RA stimulation of DR1 in NB4 is modest, likely reflecting the purely repressive function of RAR–RXR on DR1 elements (36), and RA stimulation of DR5 reporters is stronger than DR4, all suggestive for the predominance of PML–RARA-RXR oligomers. The DNA binding experiments with NB4 nuclear extracts directly show that with the stoichiometry of PML–RARA and RXR within these cells, PML–RARA is entirely complexed with RXR. Yet, the most demonstrative experiment for the presence of endogenous PML–RARA-RXR oligomers is the strong transcriptional response of PML–RARA-specific reporters (DR8 and DR10) to the rexinoid/cAMP treatment. PML–RARA homodimers and PML–RARA-RXR oligomers have very different mechanisms of protein–DNA interactions. Although for homodimers, each DNA recognition surfaces binds a single hexamer, oligomers harbor at least four DNA recognition surfaces of which two only interact with DNA. Two adjacent or distant RARA or RXR moieties become tethered onto DNA without directly binding it. On the targets on the RAR–RXR heterodimer (DR1, DR2, and DR5), binding is likely to be dependent on RARA and its bound RXR. On other small DRs (DR3 and DR4) as well as large ones (>6) binding is likely to involve the two RARAs from each of the two fusions or the bound RXRs. DNA binding has been implicated in RARs degradation (15, 37), possibly through a shift in RAR–RXR conformation, which may not occur in the context of oligomers. In any case, the existence of PML–RARA-RXR oligomers in APL cells does not favor the previously proposed model in which transformation is only accounted by an enhanced binding affinity of corepressors on PML–RARA homodimers (8, 9), as RXRs are most likely to shift the RA-triggered corepressor dissociation curve toward that of the normal RARA–RXR complexes.
Some authors found that like RARA, PML–RARA is an RA-dependent activator (38–40), whereas others show that even in the presence of RA, PML–RARA remains a repressor (10, 41). Both our transient transfection data using PML–RARA-specific reporters and repression of the natural PML–RARA-specific target CD37 in APL compared with HL60 cells, strongly suggest that the fusion remains a potent repressor even in the presence of RA. Similarly, recent studies in Xenopus oocytes (42) have led support to the observations with the RARB gene that the fusion is a pure repressor, even in the presence of RA (41). cAMP dramatically enhances transcriptional response to RA or RARA-specific agonists on both RARA- and PML–RARA-specific elements, providing a plausible mechanism for cAMP-facilitated RA-triggered APL cell differentiation (30). Here we also show that cAMP unravels the ability of rexinoid to activate either RARA or PML–RARA-specific reporters, identifying the first physiological signal that mediates RXR desubordination. Previously, RXR desubordination had only been achieved in artificial settings such as a combination of RARA antagonist and RXR agonists (43) or overexpression of coactivators (3). cAMP greatly enhances expression of the PGC-1 coactivator (44, 45) and recent reports have pointed to dynamic equilibrium between corepressor, coactivators, and RA-bound conformations of RARA (46), which may explain our findings. Alternatively, as in F9 cells (18), cAMP might also facilitate RA- and rexinoid-triggered differentiation through direct phosphorylation of the RARA moiety of PML–RARA, which may directly or indirectly alter the binding efficiency of corepressors and coactivators. This last hypothesis is favored by the paradoxical restoration of RA and RARA agonist response by cAMP in NB4MR4 cells, which suggests that cAMP-triggered RARA phosphorylation induces a conformation change that restores its ability to bind RA. Whatever the mechanism(s) involved, that cAMP strongly activates PML–RARA targets, identifies this clinically relevant agent as yet another oncogene-targeted therapy.
Using a variety of drugs, mutant cell lines, and PML–RARA-specific reporters, we directly show a very strict parallelism between transcriptional activation of PML–RARA targets and NB4 differentiation. The complete reversal of RA resistance by cAMP in NB4MR4 cells coupled to restoration of PML–RARA-dependent transactivation, also stresses the central role of the fusion over the normal RARA receptor in differentiation induction. Similarly, that the rexinoid/cAMP combination activates transcription from PML–RARA-specific reporters, strongly suggests that the previously described PML–RARA-independent cAMP/rexinoid differentiation pathway (19) in fact also triggers differentiation through PML–RARA targets, with cAMP desubordinating PML–RARA-bound RXR. Previous studies had outlined distinct gene activation profiles in RA- or cAMP/rexinoid-treated NB4 cells (19), and experiments based on differentiation kinetics of variant NB4 cells had suggested that cAMP acts downstream of RA-induced differentiation priming (32). Yet, our data unambiguously demonstrates an upstream effect of cAMP on PML–RARA signaling, which does not exclude a later cooperation with cAMP downstream targets.
Monitoring PML–RARA targets through transient transfections or quantitative PCR demonstrates that they are activated/derepressed by a number of differentiating agents. Yet, arsenic trioxide rapidly degrades PML–RARA and a sharp RA/arsenic synergism was observed in both mice (47, 48) and patients, which is inconsistent with an absolute dependence of differentiation for PML–RARA-triggered gene activation. In contrast, this last observation favors a model in which differentiation is only dependent on derepression (13). Altogether, our data show that PML–RARA-RXR oligomers silence a wide range of nuclear receptor target genes and that all known relevant differentiating agents RA, arsenic, cAMP, HDAC inhibitors, and rexinoids relieve this repression through various molecular mechanisms that nevertheless, all target the PML–RARA-RXR–HDAC complexes, allowing spontaneous differentiation of the leukemic blasts.
Acknowledgments
We gratefully acknowledge the sequencing facility of CEPH and particularly H. Buy, P. Chambon (IGBMC) for antibodies, and all members of the APL group for advice and discussion. We thank L. Delva for decisive help with NB4 electroporation and H. Gronemeyer for receptor-specific ligands. We are grateful to W. Miller for providing NB4-MR4 cells and to Marjorie Carrere for dedicated technical help. We thank M. Lanotte, L. Delva, A. Bazarbachi, A. Saib, C. Lavau, J. Zhu, and V. Lallemand-Breitenbach for comments on the manuscript.
This work was supported by the E. Lilly Foundation, Assistance Publique-Hôpitaux de Paris (to D. Vitoux), and ARECA.
The online version of this article contains supplemental material.
Abbreviations used in this paper: APL, acute promyelocytic leukemia; DR, direct repeat; EMSA, electrophoretic mobility shift assay.
References
- 1.Castelein, H., A. Janssen, P.E. Declercq, and M. Baes. 1996. Sequence requirements for high affinity retinoid X receptor-alpha homodimer binding. Mol. Cell. Endocrinol. 119:11–20. [DOI] [PubMed] [Google Scholar]
- 2.Forman, B.M., K. Umesono, J. Chen, and R.M. Evans. 1995. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell. 81:541–550. [DOI] [PubMed] [Google Scholar]
- 3.Germain, P., J. Iyer, C. Zechel, and H. Gronemeyer. 2002. Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature. 415:187–192. [DOI] [PubMed] [Google Scholar]
- 4.Minucci, S., C. Nervi, F. Lo Coco, and P.G. Pelicci. 2001. Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene. 20:3110–3115. [DOI] [PubMed] [Google Scholar]
- 5.Jansen, J.H., A. Mahfoudi, S. Rambaud, C. Lavau, W. Wahli, and A. Dejean. 1995. Multimeric complexes of the PML-retinoic acid receptor alpha fusion protein in acute promyelocytic leukemia cells and interference with retinoid and peroxisome-proliferator signaling pathways. Proc. Natl. Acad. Sci. USA. 92:7401–7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez, A., P. Kastner, S. Sethi, Y. Lutz, C. Reibel, and P. Chambon. 1993. PML/RAR homodimers: distinct binding properties and heteromeric interactions with RXR. EMBO J. 12:3171–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He, L.-Z., F. Guidez, C. Tribioli, D. Peruzzi, M. Ruthardt, A. Zelent, and P.P. Pandolfi. 1998. Distinct interactions of PML-RARalpha and PLZF-RARalpha with co-repressors determine differential responses to RA in APL. Nat. Genet. 18:126–135. [DOI] [PubMed] [Google Scholar]
- 8.Grignani, F., S. de Matteis, C. Nervi, L. Tomassoni, V. Gelmetti, M. Cioce, M. Fanelli, M. Ruthardt, F.F. Ferrara, I. Zamir, et al. 1998. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 391:815–818. [DOI] [PubMed] [Google Scholar]
- 9.Lin, R.J., L. Nagy, S. Inoue, W.L. Shao, W.H. Miller, and R.M. Evans. 1998. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 391:811–814. [DOI] [PubMed] [Google Scholar]
- 10.Lin, R., and R. Evans. 2000. Acquisition of oncogenic potential by RAR chimeras in acute promyelocytic leukemia through formation of homodimers. Mol. Cell. 5:821–830. [DOI] [PubMed] [Google Scholar]
- 11.Minucci, S., M. Maccarana, M. Cioce, P. De Luca, V. Gelmetti, S. Segalla, L. Di Croce, S. Giavara, C. Matteucci, A. Gobbi, et al. 2000. Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol. Cell. 5:811–820. [DOI] [PubMed] [Google Scholar]
- 12.Degos, L., and Z.Y. Wang. 2001. All trans retinoic acid in acute promyelocytic leukemia. Oncogene. 20:7140–7145. [DOI] [PubMed] [Google Scholar]
- 13.Zhu, J., Z. Chen, V. Lallemand-Breitenbach, and H. de Thé. 2002. How acute promyelocytic leukemia revived arsenic. Nat. Rev. Cancer. 2:705–713. [DOI] [PubMed] [Google Scholar]
- 14.Zhu, J., M.H.M. Koken, F. Quignon, M.K. Chelbi-Alix, L. Degos, Z.Y. Wang, Z. Chen, and H. de Thé. 1997. Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 94:3978–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu, J., M. Gianni, E. Kopf, N. Honore, M. Chelbi-Alix, M. Koken, F. Quignon, C. Rochette-Egly, and H. de Thé. 1999. Retinoic acid induces proteasome-dependent degradation of retinoic acid receptor alpha (RAR alpha) and oncogenic RAR alpha fusion proteins. Proc. Natl. Acad. Sci. USA. 96:14807–14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benoit, G., M. Roussel, F. Pendino, E. Segal-Bendirdjian, and M. Lanotte. 2001. Orchestration of multiple arrays of signal cross-talk and combinatorial interactions for maturation and cell death: another vision of t(15;17) preleukemic blast and APL-cell maturation. Oncogene. 20:7161–7177. [DOI] [PubMed] [Google Scholar]
- 17.Guillemin, M.C., E. Raffoux, D. Vitoux, S. Kogan, H. Soilihi, V. Lallemand-Breitenbach, J. Zhu, A. Janin, M.T. Daniel, B. Gourmel, et al. 2002. In vivo activation of cAMP signaling induces growth arrest and differentiation in acute promyelocytic leukemia. J. Exp. Med. 196:1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taneja, R., C. Rochette-Egly, J.-L. Plassat, L. Penna, M.-P. Gaub, and P. Chambon. 1997. Phosphorylation of activation functions AF-1 and AF-2 of RAR alpha and RAR gamma is indispensable for differentiation of F9 cells upon retinoic acid and cAMP treatment. EMBO J. 16:6452–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benoit, G., L. Altucci, M. Flexor, S. Ruchaud, J. Lillehaug, W. Raffelsberger, H. Gronemeyer, and M. Lanotte. 1999. RAR-independent RXR signaling induces t(15;17) leukemia cell maturation. EMBO J. 18:7011–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benoit, G.R., M. Flexor, F. Besancon, L. Altucci, A. Rossin, J. Hillion, Z. Balajthy, L. Legres, E. Segal-Bendirdjian, H. Gronemeyer, et al. 2001. Autonomous rexinoid death signaling is suppressed by converging signaling pathways in immature leukemia cells. Mol. Endocrinol. 15:1154–1169. [DOI] [PubMed] [Google Scholar]
- 21.Altucci, L., and H. Gronemeyer. 2001. The promise of retinoids to fight against cancer. Nat. Rev. Cancer. 1:181–193. [DOI] [PubMed] [Google Scholar]
- 22.Subauste, J.S., and R.J. Koenig. 1998. Characterization of the DNA-binding and dominant negative activity of V-erbA homodimers. Mol. Endocrinol. 12:1380–1392. [DOI] [PubMed] [Google Scholar]
- 23.de Thé, H., M.d.M. Vivanco-Ruiz, P. Tiollais, H. Stunnenberg, and A. Dejean. 1990. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature. 343:177–180. [DOI] [PubMed] [Google Scholar]
- 24.Roussel, M.J., and M. Lanotte. 2001. Maturation sensitive and resistant t(15;17) NB4 cell lines as tools for APL physiopathology: nomenclature of cells and repertory of their known genetic alterations and phenotypes. Oncogene. 20:7287–7291. [DOI] [PubMed] [Google Scholar]
- 25.Gianni, M., M.H.M. Koken, M.K. Chelbi-Alix, G. Benoit, M. Lanotte, Z. Chen, and H. de Thé. 1998. Combined arsenic and retinoic acid treatment enhances differentiation and apoptosis in arsenic-resistant NB4 cells. Blood. 91:4300–4310. [PubMed] [Google Scholar]
- 26.Hauksdottir, H., and M.L. Privalsky. 2001. DNA recognition by the aberrant retinoic acid receptors implicated in human acute promyelocytic leukemia. Cell Growth Differ. 12:85–98. [PMC free article] [PubMed] [Google Scholar]
- 27.Jamieson, A.C., J.C. Miller, and C.O. Pabo. 2003. Drug discovery with engineered zinc-finger proteins. Nat. Rev. Drug Discov. 2:361–368. [DOI] [PubMed] [Google Scholar]
- 28.Kamashev, D.E., A.V. Balandina, and V.L. Karpov. 2000. Tramtrack protein-DNA interactions. A cross-linking study. J. Biol. Chem. 275:36056–36061. [DOI] [PubMed] [Google Scholar]
- 29.Lallemand-Breitenbach, V., J. Zhu, F. Puvion, M. Koken, N. Honore, A. Doubeikovsky, E. Duprez, P.P. Pandolfi, E. Puvion, P. Freemont, et al. 2001. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As(2)O(3)-induced PML or PML/retinoic acid receptor α degradation. J. Exp. Med. 193:1361–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quenech'Du, N., S. Ruchaud, N. Khelef, N. Guiso, and M. Lanotte. 1998. A sustained increase in the endogenous level of cAMP reduces the retinoid concentration required for APL cell maturation to near physiological levels. Leukemia 12:1829–1833. [DOI] [PubMed] [Google Scholar]
- 31.Zhu, Q., J.W. Zhang, H.Q. Zhu, Y.L. Shen, M. Flexor, P.M. Jia, Y. Yu, X. Cai, S. Waxman, M. Lanotte, et al. 2002. Synergic effects of arsenic trioxide and cAMP during acute promyelocytic leukemia cell maturation subtends a novel signaling cross-talk. Blood. 99:1014–1022. [PubMed] [Google Scholar]
- 32.Ruchaud, S., E. Duprez, M.C. Gendron, G. Houge, H.G. Genieser, B. Jastorff, S.O. Doskeland, and M. Lanotte. 1994. Two distinctly regulated events, priming and triggering, during retinoid-induced maturation and resistance of NB4 promyelocytic leukemia cell line. Proc. Natl. Acad. Sci. USA. 91:8428–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duprez, E., G. Benoit, M. Flexor, J.R. Lillehaug, and M. Lanotte. 2000. A mutated PML/RARA found in the retinoid maturation resistant NB4 subclone, NB4-R2, blocks RARA and wild-type PML/RARA transcriptional activities. Leukemia. 14:255–261. [DOI] [PubMed] [Google Scholar]
- 34.Shao, W., L. Benedetti, W.W. Lamph, C. Nervi, and W.H.J. Miller. 1997. A retinoid-resistant acute promyelocytic leukemia subclone expresses a dominant negative PML-RAR alpha mutation. Blood. 89:4282–4289. [PubMed] [Google Scholar]
- 35.Kogan, S.C., S.H. Hong, D.B. Shultz, M.L. Privalsky, and J.M. Bishop. 2000. Leukemia initiated by PMLRARalpha: the PML domain plays a critical role while retinoic acid-mediated transactivation is dispensable. Blood. 95:1541–1550. [PubMed] [Google Scholar]
- 36.Kurokawa, R., M. Söderström, A. Hörlein, S. Halachmi, M. Brown, M.G. Rosenfeld, and C.K. Glass. 1995. Polarity-specific activities of retinoic acid receptors determined by a co-repressor. Nature. 377:451–454. [DOI] [PubMed] [Google Scholar]
- 37.Gianni, M., A. Bauer, E. Garratini, P. Chambon, and C. Rochette-Egly. 2002. Phosphorylation by p38MAPK and recruitment of SUG-1 are required for RA-induced RAR gamma degradation and transactivation. EMBO J. 15:3760–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grignani, F., P. Ferrucci, U. Testa, G. Talamo, M. Fagioli, M. Alcalay, A. Mencarelli, F. Grignani, C. Peschle, I. Nicoletti, et al. 1993. The acute promyelocytic leukemia specific PML/RARα fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell. 74:423–431. [DOI] [PubMed] [Google Scholar]
- 39.Kastner, P., A. Perez, Y. Lutz, C. Rochette-Egly, M.-P. Gaub, B. Durand, M. Lanotte, R. Berger, and P. Chambon. 1992. Structure, localization and transcriptional properties of two classes of retinoic acid receptor alpha fusion proteins in acute promyelocytic leukemia (APL): structural similarities with a new family of oncoproteins. EMBO J. 11:629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandolfi, P.P., F. Grignani, M. Alcalay, A. Mencarelli, A. Biondi, F. LoCoco, F. Grignani, and P.G. Pelicci. 1991. Structure and origin of the acute promyelocytic leukemia myl/RARα cDNA and characterization of its retinol-binding and transactivation properties. Oncogene. 6:1285–1292. [PubMed] [Google Scholar]
- 41.de Thé, H., C. Lavau, A. Marchio, C. Chomienne, L. Degos, and A. Dejean. 1991. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 66:675–684. [DOI] [PubMed] [Google Scholar]
- 42.Segalla, S., L. Rinaldi, C. Kilstrup-Nielsen, G. Badaracco, S. Minucci, P.G. Pelicci, and N. Landsberger. 2003. Retinoic acid receptor alpha fusion to PML affects its transcriptional and chromatin-remodeling properties. Mol. Cell. Biol. 23:8795–8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen, J.-Y., J. Clifford, C. Zusi, J. Starrett, D. Tortolani, J. Ostrowski, P.R. Reczek, P. Chambon, and H. Gronemeyer. 1996. Two distinct actions of retinoid-receptor ligands. Nature. 382:819–822. [DOI] [PubMed] [Google Scholar]
- 44.Herzig, S., F. Long, U.S. Jhala, S. Hedrick, R. Quinn, A. Bauer, D. Rudolph, G. Schutz, C. Yoon, P. Puigserver, et al. 2001. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 413:179–183. [DOI] [PubMed] [Google Scholar]
- 45.Yoon, J.C., P. Puigserver, G. Chen, J. Donovan, Z. Wu, J. Rhee, G. Adelmant, J. Stafford, C.R. Kahn, D.K. Granner, et al. 2001. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 413:131–138. [DOI] [PubMed] [Google Scholar]
- 46.Sohn, Y.C., S.W. Kim, S. Lee, Y.Y. Kong, D.S. Na, S.K. Lee, and J.W. Lee. 2003. Dynamic inhibition of nuclear receptor activation by corepressor binding. Mol. Endocrinol. 17:366–372. [DOI] [PubMed] [Google Scholar]
- 47.Lallemand-Breitenbach, V., M.-C. Guillemin, A. Janin, M.-T. Daniel, L. Degos, S.C. Kogan, J.M. Bishop, and H. de Thé. 1999. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J. Exp. Med. 189:1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rego, E.M., L.Z. He, R.P. Warrell, Jr., Z.G. Wang, and P.P. Pandolfi. 2000. Retinoic acid (RA) and As2O3 treatment in transgenic models of acute promyelocytic leukemia (APL) unravel the distinct nature of the leukemogenic process induced by the PML-RARalpha and PLZF-RARalpha oncoproteins. Proc. Natl. Acad. Sci. USA. 97:10173–10178. [DOI] [PMC free article] [PubMed] [Google Scholar]