

Abstract
The T-box transcription factor T-bet is known to control lineage commitment and interferon-γ production by T helper 1 (Th1) CD4 lymphocytes. We report here that T-bet is essential for development of CD8 lymphocyte-dependent autoimmune diabetes (type 1 diabetes [T1D]) in the rat insulin promoter–lymphocytic choriomeningitis virus (LCMV) transgenic model for virally induced T1D. In the absence of T-bet, autoaggressive (anti-LCMV) CD8 lymphocytes were reduced in number and produced less IFN-γ, but increased IL-2 compared with controls. Further analysis showed that T-bet intrinsically controls the generation, but not apoptosis, maintenance, or secondary expansion of antiviral effector/memory CD8 lymphocytes. This observation points toward a therapeutic opportunity for the treatment of T1D and other autoimmune disorders.
Keywords: autoimmunity, T cell memory, cytokines, lymphocytic choriomeningitis virus, transcription factors
Introduction
Modulating the cytokine balance of autoreactive T lymphocytes can be of great value for preventing development of autoimmune disorders. In type 1 diabetes (T1D), autoaggression is supported by IFN-γ, IL-12, and cytotoxic activity, whereas counter-regulation is achieved by Th2 cytokines such as IL-4, IL-5, and possibly IL-10. Therefore, we wished to explore the role of the recently discovered (1) transcription factor T-bet in T1D because it was known that T-bet controls lineage commitment and IFN-γ production by CD4 T cells (1, 2). The significant Th2 shift in T-bet–deficient mice can lead to an asthmalike syndrome (3). This type of skewing of the cytokine equilibrium occurs on a systemic level and also extends to the mucosal compartment (4, 5). Thus, T-bet appears to be a master controller of the Th1 cytokine pathway and might constitute a mechanism for “cytokine memory” in T helper cells, as much as GATA-3 controls Th2 lineage commitment (6). Interestingly, although Th1 cell function is markedly impaired in the absence of T-bet, IFN-γ production by CD8 T cells after polyclonal stimulation is unaffected (2).
A suitable diabetes model to test the role of T-bet on CD8 responses in vivo during T1D is the rat insulin promoter (RIP)–lymphocytic choriomeningitis virus (LCMV) mouse developed by Oldstone and Zinkernagel (7, 8). These transgenic mice express viral antigens derived from LCMV under control of the RIP in pancreatic β cells (9). Viral infection with LCMV induces rapid CD8-dependent diabetes in RIP-LCMV-glycoprotein (GP) transgenic lines. Importantly, the infection is first cleared in these mice before the autoimmune process fully develops. Therefore, islet autoimmunity is governed by different immunological rules compared with clearance of the systemic LCMV infection, and treatments that abrogate the diabetogenic process need not necessarily impair viral clearance (10, 11). Lymphocytes infiltrating the islets are of the experienced (effector/memory) phenotype, which is characterized functionally by concomitant production of TNF-α and IFN-γ, but not IFN-γ alone (12, 13). Islet destruction depends on production of inflammatory cytokines such as IL-12 and IFN-γ in addition to cytolytic effector function by autoaggressive (anti-LCMV) CD8 lymphocytes (10, 14).
Based on these arguments, the RIP-LCMV model appeared to be an ideal tool for evaluating the role of T-bet in CD8 responses in autoimmunity. The suitability of RIP-LCMV mice as a valid diabetes model for our analysis is further supported by the fact that more recently a strong role for CD8 cells as the final effectors in β cell destruction has been found (14–17). In addition, antigenic spreading occurs in our RIP-LCMV-GP lines as evidenced by autoantibodies to insB and GAD preceding the onset of T1D (18). Finally, diabetes in RIP-GP mice occurs in a CD4-independent fashion (9, 19), and the effect of T-bet on CD8 responses can be segregated from its known effect on CD4 T cells. In the studies reported here, we found a dramatic reduction of disease. This is associated with a decrease of islet infiltration and significant reduction of LCMV-specific (“anti-self”) memory CD8 responses. Blocking the activity of T-bet might be of unique therapeutic value in preventing T cell–mediated autoimmune disorders.
Materials and Methods
Mice.
Mice deficient in T-bet (T-bet−/−) were generated on a C57BL/6 × 129 background as described previously (2). T-bet−/− mice were backcrossed three times to C57BL/6 before being crossed to RIP-GP (7, 9) to obtain T-bet+/− × RIP-GP mice. These mice were intercrossed to obtain T-bet+/+, T-bet+/−, and T-bet−/− × RIP-GP. In some experiments, nontransgenic T-bet−/− or +/− and +/+ littermate control mice were used. Mice were housed under specific pathogen-free conditions at La Jolla Institute for Allergy and Immunology.
Viruses.
LCMV Armstrong clone 53b (20) plaque-purified three times on Vero cells and stocks prepared by a single passage on BHK-21 cells were used throughout experiments. 6–10-wk-old mice were infected with a single dose of 105 PFU i.p.
Blood Glucose Values.
Blood glucose was monitored with OneTouch Ultra at weekly intervals. Diabetes was defined as blood glucose values (BGVs) over 300 mg/dl (21).
Peptides.
Peptides used for viral studies were the dominant Db-restricted LCMV epitopes GP33–41 and NP396–404 (The Scripps Research Institute Peptide Core Facility; reference 22) as well as I-Ab–restricted epitope GP61–80 (Chiron Corp.; reference 23).
Immunohistochemistry.
Tissues were immersed in Tissue-Tek OCT (Bayer) and quick frozen on dry ice. Using cryomicrotome and sialin-coated Superfrost Plus slides (Fisher Scientific), 6–10-μm tissue sections were cut. Sections were fixed with 90% ethanol at −20°C, and, after washing in PBS, an avidin/biotin-blocking step was included (Vector Laboratories). Primary and biotinylated secondary antibodies (Vector Laboratories) were reacted with the sections for 60 min each, and color reaction was obtained by sequential incubation with avidin–peroxidase conjugate (Vector Laboratories) and diaminobenzidine–hydrogen peroxide. Primary antibodies used were rat anti–mouse CD8a (Ly2) and rat anti–mouse CD4 (L3T4; BD Biosciences).
Flow Cytometry.
For intracellular stains, single cell suspensions were restimulated for 5 h with 1 μg/ml MHC class I–restricted viral peptides, 2 μg/ml MHC class II–restricted viral peptides, or 2 μg/ml anti-CD3 and anti-CD28 in the presence of brefeldin A (Sigma-Aldrich). Cells were stained for surface expression of CD4 and CD8, fixed, permeabilized, and stained for intracellular IFN-γ, TNF-α, or IL-2. CCR7 staining was performed by incubation with ELC-Ig followed by goat anti–human Fc-FITC (24). DbGP33 and DbNP396 MHC class I tetramers were produced as described previously (25). Samples were acquired using a FACSCalibur™ (Becton Dickinson).
ELISPOT Assay.
Membrane-bottom 96-well plates were coated overnight with IL-4 or IFN-γ capture antibodies. Splenocytes were plated at various dilutions and cultured at 37°C, 5% CO2 with GP61 MHC II–restricted viral peptide. After 24 h (IFN-γ) or 48 h (IL-4), the plates were washed, the cells were removed, and an IFN-γ or IL-4 detection antibody was added at 4°C overnight. A streptavidin/angiotensin-converting enzyme color reaction was used to identify cell cytokine secretion patterns, and the spots were counted with the aid of a dissection microscope.
In Vitro Restimulations.
For LCMV-specific responses, memory splenocytes were cultured for 6 d on LCMV-infected irradiated peritoneal exudate cells. After culture, viable cells were collected, and cells were incubated for 5 h with peptide in the presence of brefeldin A before staining for intracellular IFN-γ. The fold increase of specific CD8+ cells was calculated by dividing the absolute number of IFN-γ+ CD8+ cells after in vitro culture by the absolute number of IFN-γ+ CD8+ cells at the start of the culture.
ELISA.
Quantitation of cytokines in cell culture supernatants after in vitro stimulation was performed in a sandwich ELISA. For IL-4, IL-10, and IFN-γ assays, 96-well plates were precoated overnight at 4°C with purified capture antibodies (BD Biosciences). After washing and blocking, serial dilution of supernatants and recombinant cytokine standards (BD Biosciences) were incubated overnight at 4°C. Plates were further incubated with matched biotinylated detection antibodies and a streptavidin–peroxidase conjugate. For color reaction, H2O2-activated 2,2'-Azino-di-[3-ethylbenzthiazolin-6-sulfonic acid] (ABTS; Sigma-Aldrich) solution was added. Plates were read at 405 nm. For TGFβ quantitation, the TGFβ1 Emax ImmunoAssay kit (Promega) was used.
Adoptive Transfers.
CD8+ T cells were purified from GP33 TCR-tg (26) or T-bet+/+, +/−, or −/− donors by negative selection (Dynal). 104 GP33 TCR-Tg cells were transferred i.v. into T-bet+/+, +/−, or −/− recipients.
Generation of Mixed Bone Marrow Chimeras.
Bone marrow was isolated from tibias and femurs of donor T-bet−/−, T-bet+/−, or C57Bl/6 Thy1.1 congenic mice. Bone marrow was depleted of CD4 and CD8 T cells, and a 1:1 mixture containing a total of 107 cells was injected i.v. into lethally irradiated (1,100 rad) T-bet+/− or T-bet−/− recipients. Mice were allowed to reconstitute over a period of 6 wk, which was confirmed by FACS® analysis of peripheral blood lymphocytes before infection with LCMV.
Results
Drastically Reduced T1D in T-bet–deficient Mice.
To examine a role for T-bet in autoimmune diabetes (T1D), T-bet−/− mice were crossed to RIP-GP transgenic mice. T-bet+/+, T-bet+/− or T-bet−/− × RIP-GP littermates were infected with 105 PFU LCMV Armstrong to induce diabetes. BGVs were measured weekly, and by 2 wk after infection, 80–100% of T-bet+/+ or T-bet+/− × RIP-GP mice were diabetic (Fig. 1 A), with average BGVs of ∼500 mg/dl (Fig. 1 B). In these experiments, a small percentage of the diabetic mice reverted to normal BGVs at >3 wk after infection. This has been described previously in mice of a mixed 129 × B6 background (27). However, by 35 d after infection, 75–80% of T-bet+/+ and T-bet+/− × RIP-GP mice remained diabetic. In contrast, most of the T-bet−/− × RIP-GP had normal BGVs on day 14 (Fig. 1 B), averaging ∼200 mg/dl. In addition, diabetes incidence in T-bet−/− × RIP-GP remained dramatically reduced with only 10% diabetic (Fig. 1 A) 35 d after infection. Thus, elimination of T-bet results in a highly significant reduction (P < 0.0001 by Student's t test) of BGVs and autoimmune diabetes.
Figure 1.

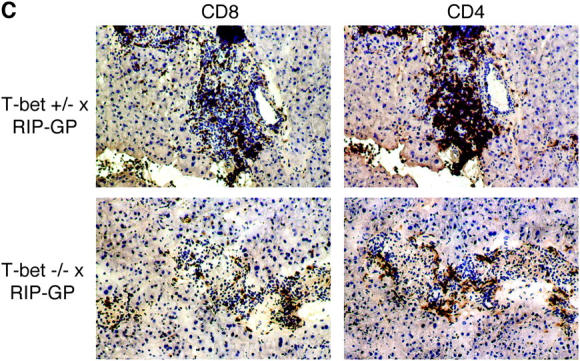
T-bet−/− mice are resistant to diabetes. T-bet+/+, T-bet+/−, or T-bet−/− × RIP-GP mice were infected i.p. with 105 PFU LCMV Armstrong. Incidence of diabetes (A) or BGVs of individual mice on day 14 after infection (B) are shown. Mice were considered diabetic when BGVs were >300 mg/dl. Tissue sections were cut and analyzed for islet infiltration (C) using monoclonal antibodies specific for CD8 and CD4. Sections were counterstained with hematoxylin. Staining shown is from the same islets present on consecutive sections, and represents the average degree of infiltration present. Photographs were shot at a magnification of 20, and some photos were additionally enlarged using photo-processing software.
Reduced Islet Infiltration in T-bet–deficient Mice.
Pancreata from T-bet × RIP-GP mice were examined by immunohistochemistry for the presence of CD4 and CD8 T cells. Islets from diabetic T-bet+/− × RIP-GP mice were heavily infiltrated with both CD4 and CD8 T cells. In contrast, islets from T-bet−/− × RIP-GP had less severe infiltration, with CD4 and CD8 T cell present mostly around the periphery of the islets (Fig. 1 C).
Defect in Effector/Memory CD8s in T-bet–deficient Mice.
The CD8 immune response to LCMV is characterized by an initial primary expansion of LCMV-specific, IFN-γ single positive T cells, peaking ∼7–8 d after infection. This is followed by a contraction phase leaving resting LCMV memory CTLs that can turn into IFN-γ/TNF-α double positive effector/memory CD8 T cells upon antigenic restimulation. Subsequently, memory CD8 T cells are maintained at a similar frequency for life. The frequency of LCMV-specific CD8 lymphocytes was evaluated over time. Primary CD8 T cell responses examined on day 8 after LCMV infection were not impaired in T-bet–deficient mice as evidenced by unimpaired IFN-γ production (Fig. 2 A) and cytotoxicity toward LCMV-infected targets (not depicted). Accordingly, viral clearance was not affected (unpublished data). This was not unexpected because this particular primary antiviral response occurs independently of many immunological parameters, including B7, IFN-γ, CD4 help, CD28, and CD40L (28–35). In addition, previous studies have demonstrated that the primary immune response on day 8 does not correlate with disease, whereas effector/memory cells do (36, 37).
Figure 2.

Drastic reduction of autoaggressive memory effector CD8 lymphocytes in the absence of T-bet. T-bet+/−, T-bet+/−, and T-bet−/− mice were infected with 105 PFU LCMV and killed at the indicated days after infection. Spleen cells were cultured with GP33 peptide for 5 h for intracellular IFN-γ staining or for 72 h for ELISA (insets). Data for the intracellular cytokine staining are graphed as the percent IFN-γ+ of CD8 gated populations and are an average of at least three mice per group per time point (A). Diabetic T-bet+/− or protected T-bet−/− × RIP-GP mice were killed 4 wk after infection, and spleen (B) or pancreatic LNs (C) were pooled. Single cell suspensions were incubated with GP33 or NP396 MHC-I peptides, or anti-CD3/CD28 and stained for intracellular IFN-γ and TNF-α. Plots are gated on CD8+ T cells with percentages shown in relative quadrants.
In contrast, and consistent with the reduction of autoimmune diabetes, antigen-specific effector/memory CD8 T cells were dramatically reduced by day 14 after LCMV infection, with 2% IFN-γ positive cells in T-bet−/− mice after culture with GP33, compared with 10% in T-bet+/+ mice (Fig. 2 A). The reduction seen at this time point is likely to be highly relevant in terms of diabetes induction because it corresponds exactly to the peak of clinical disease in control mice. This lower number of LCMV-specific CD8 T cells after day 14 is maintained in T-bet−/− mice over time (Fig. 2 A). In agreement with the reduced number of IFN-γ positive effector/memory CD8 lymphocytes after 5 h stimulation with peptide is the reduced amount of IFN-γ in tissue culture supernatants assessed by ELISA after a 3-d stimulation period (Fig. 2 A). Despite the significantly reduced IFN-γ production, there was no compensatory increase in the production of Tc2 cytokines, such as IL-4, IL-10, and TGF-β. LCMV-specific production of IL-4, IL-10, or TGF-β by CD8 T cells could not be detected by ELISA or ELISPOT in T-bet−/− or T-bet+/− mice (see Fig. 6 D and not depicted). Thus, antigen-specific CD8 effector/memory responses in T-bet–deficient mice are markedly impaired.
Figure 6.
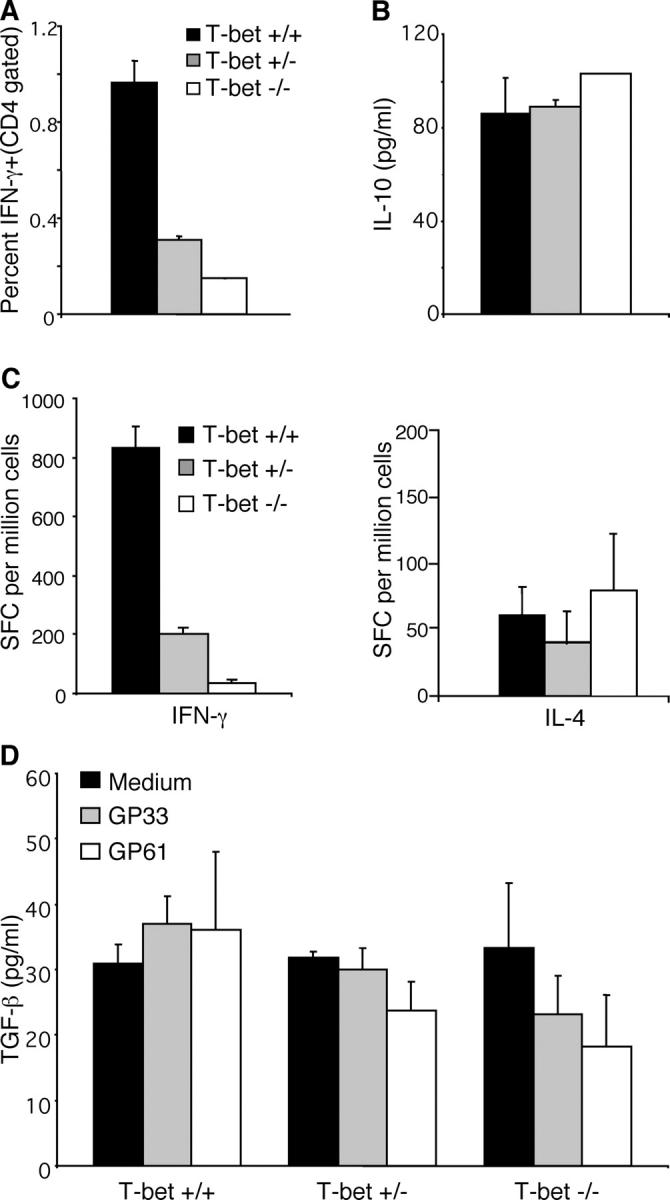
Reduced LCMV-specific Th1 without increased Th2 cells in T-bet−/− mice. Spleen cells from mice infected with LCMV 30 d previously were cultured with LCMV MHC-II peptide GP61, and intracellular IFN-γ production by CD4 T cells was determined. Data are an average of three mice per group (A). ELISA for IL-10 (B) or ELISPOT assays for IFN-γ and IL-4 (C) were performed using day 8 spleen cells cultured with GP61 MHC class-II peptide. For IFN-γ responses by CD4 cells, statistically significant results (P < 0.005 by Student's t test) were obtained when comparing IFN-γ production by T-bet+/+, T-bet+/−, and T-bet−/−. For TGF-β ELISA, spleen cells from mice infected with LCMV 30 d previously were cultured with medium alone, MHC-I peptide GP33, or MHC-II peptide GP61 as indicated (D).
Phenotypically, effector/memory CD8 cells are predominantly present in islets of diabetic RIP-GP mice and secrete TNF-α in addition to IFN-γ (12). To assess the activity of autoaggressive (LCMV specific) CD8 lymphocytes in proximity to the target organ, antigen-specific production of IFN-γ and TNF-α by CD8 T cells was examined in T-bet × RIP-GP mice 1 mo after infection. Cells were pooled from the spleen or pancreatic draining LNs of T-bet+/− and T-bet−/− × RIP GP mice and were stimulated in vitro with the MHC class I LCMV peptides GP33 or NP396, or with anti-CD3 and anti-CD28. Intracellular cytokine staining was performed to determine antigen-specific IFN-γ and TNF-α production. Compared with diabetic T-bet+/− mice, T-bet−/− mice exhibited an ∼50% decrease in double cytokine–producing (TNF-α and IFN-γ) effector/memory CD8 T cells specific for GP33 and NP396 in the spleen (Fig. 2 B) and pancreatic lymph node (Fig. 2 C) at 1 mo after LCMV infection. IFN-γ and TNF-α production by CD8 T cells was also reduced in T-bet−/− mice after polyclonal stimulation with anti-CD3 and -CD28 1 mo after LCMV infection (Fig. 2, B and C). Thus, effector functions of LCMV-specific autoaggressive lymphocytes were significantly reduced in the pancreatic draining lymph node of T-bet–deficient RIP-GP mice.
Numeric Reduction of Effector/Memory Cells in T-bet−/− Mice.
The aforementioned results demonstrated a reduced frequency of LCMV-specific CD8 T cells in T-bet−/− mice. To determine if this was due to an overall numeric reduction in antigen-specific T cells, the number of LCMV-specific T cells per spleen was determined after peptide restimulation and IFN-γ staining. There were fewer IFN-γ+ LCMV-specific CD8 T cells in spleens of T-bet−/− mice compared with T-bet+/+ or +/− controls 1 mo after infection (Fig. 3 A). In addition, splenocytes were stained with H-2Db tetramers loaded with GP33 or NP396 peptides to ensure that the reduced frequency of IFN-γ+ cells as measured by intracellular cytokine staining reflected a reduction in the actual number of LCMV-specific CD8 memory T cells. As shown in Fig. 3 B, there was also a reduction of GP33 or NP396-specific CD8 T cells in T-bet−/− mice compared with controls after tetramer staining. Thus, deficiency of T-bet results in reduced numbers of CD8 memory lymphocytes in vivo. Splenocytes were also stained for the expression of CCR7 to enumerate the frequency of effector/memory versus central memory T cells. On day 14 after LCMV infection, there was a significantly lower percentage of LCMV-specific effector/memory CD8 T cells (CD8+, NP396 +, CCR7neg) in T-bet−/− mice (Fig. 3 C). CCR7 staining was also performed on day 28. T-bet−/− mice still had a lower percentage of effector/memory T cells, but the difference from controls was not statistically significant, and much less dramatic than observed on day 14 (Fig. 3 C).
Figure 3.
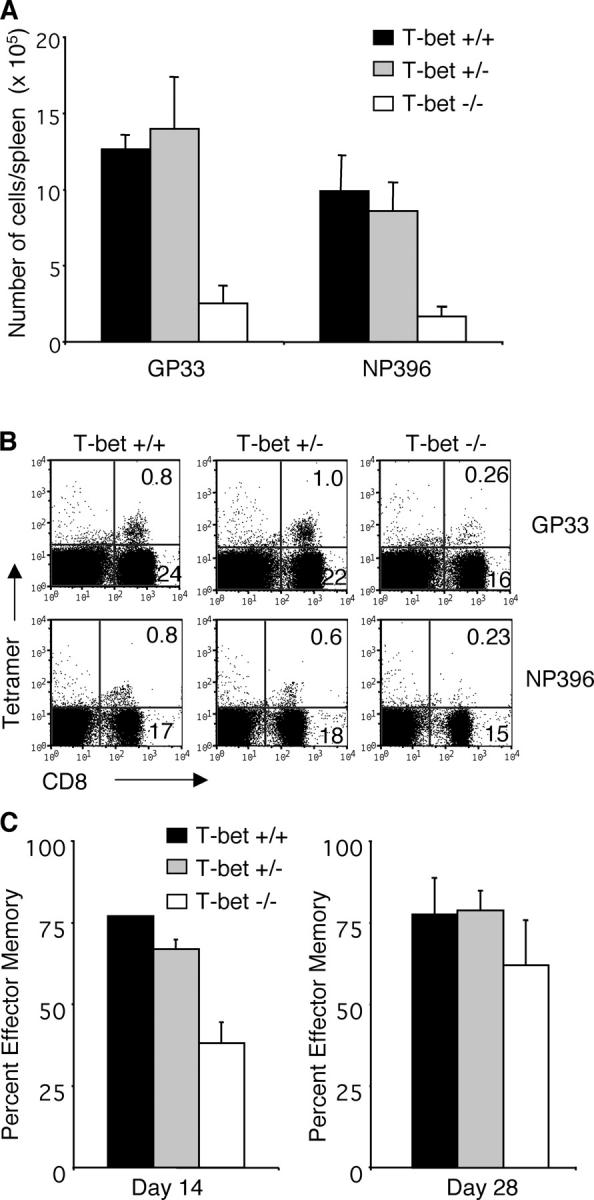
Numeric reduction of IFN-γ–producing CD8 T cells in T-bet−/− mice. T-bet+/−, T-bet+/−, and T-bet−/− mice were infected with 105 PFU LCMV. Spleen cells isolated >30 d after infection were cultured with the indicated LCMV MHC-I peptides before staining for intracellular IFN-γ. The total number of antigen-specific cells per spleen was calculated by multiplying the percent CD8/IFN-γ double positive cells by the total number of cells isolated from the spleen of each mouse. Statistically significant differences (P < 0.005 by Student's t test) were found between +/+ and −/− (A). Cells isolated on day 30 after LCMV were stained with CD8 and Db GP33 or NP396 tetramers. Representative data are from one out of four experiments (B). Cells isolated on day 14 (left) or 28 (right) were stained with Db NP396 tetramers and ELC-Ig to detect CCR7. The data are an average of four to five mice per group and are graphed as the percentage of CCR7 negative cells within NP396 + gated populations (C).
Effector Functions Not Impaired on a Per Cell Basis.
The ability of T-bet−/− memory T cells to expand in response to reexposure to LCMV in vitro was examined. As shown in Fig. 4 A, CFSE-labeled memory splenocytes from T-bet−/− mice do proliferate, although the overall percentage of GP33-specific cells is still reduced compared with controls after 6 d of culture. Because the LCMV-infected T-bet−/− mice typically have approximately threefold fewer LCMV-specific T cells ex vivo, the fold increase in LCMV-specific T cells was determined by dividing the absolute number of GP33-specific T cells present after culture by the number present ex vivo. Using this calculation, the proliferation of T-bet−/− GP33-specific CD8 T cells after 6 d in culture is as good or better than T-bet+/+ T cells (Fig. 4 B). Furthermore, the mean fluorescent intensity of IFN-γ production was equivalent comparing memory CTL from T-bet+/+ and −/− mice (Fig. 4 C). It is interesting that, despite a numeric reduction in CD8 memory T cells in vivo, these cells were still competent to expand in vitro in the presence of antigen (virus or peptide). Thus, although the numbers of effector/memory cells are reduced in T-bet–deficient mice, their ability to respond to antigen with proliferation, cytolytic activity (unpublished data), and IFN-γ production is completely intact when assessed on a per cell basis. Therefore, we conclude that generation of memory CTL in T-bet−/− mice is defective, but that the remaining effector/memory cells found in T-bet−/− mice are functionally competent.
Figure 4.

Normal proliferation and secondary expansion of LCMV-specific effector/memory CD8 cells in T-bet−/− mice. Spleens cells isolated from mice infected with LCMV >30 d previously were labeled with CFSE and were cultured for 6 d on LCMV-infected irradiated peritoneal exudate cells. After a 5-h restimulation with GP33 and brefeldin A, staining for intracellular IFN-γ was performed (A). The fold increase of LCMV-specific CD8 T cells was calculated by dividing the number of IFN-γ+ GP33-specific T cells after 6 d of culture by the number present directly ex vivo (B). Median fluorescence intensity for intracellular IFN-γ was determined after a 5-h culture of spleen cells with GP33 (C).
Defective CD8 Response Does Not Rely on IL-2.
It has been shown previously that one of the functions of T-bet is to repress the IL-2 promoter (1). Accordingly, we examined IL-2 production by CD8 T cells after infection with LCMV. On day 8 after infection, LCMV-specific CD8 T cells from T-bet−/− and T-bet+/− mice produced more IL-2 compared with T-bet+/+. As shown in Fig. 5 A, T-bet−/− mice have a higher frequency of IFN-γ, IL-2+ GP33-specific CD8 T cells compared with T-bet+/+ mice (∼6 vs. 1%, respectively). T-bet+/− mice showed an intermediate phenotype with ∼3% of CD8 T cells producing IL-2 in response to GP33. IL-2 production was also examined at the memory stage. Although the absolute number of LCMV-specific CD8 T cells is reduced in T-bet−/− mice, the ratio of IL-2+ to IL-2− LCMV-specific CD8 T cells is still much higher in T-bet−/− compared with T-bet+/+ (Fig. 5 A). The higher levels of autologous IL-2 production by CD8 T cells in T-bet−/− mice was unable to rescue the memory defect in vivo, despite the fact that IL-2 is a key helper factor for programming CD8 memory (38).
Figure 5.
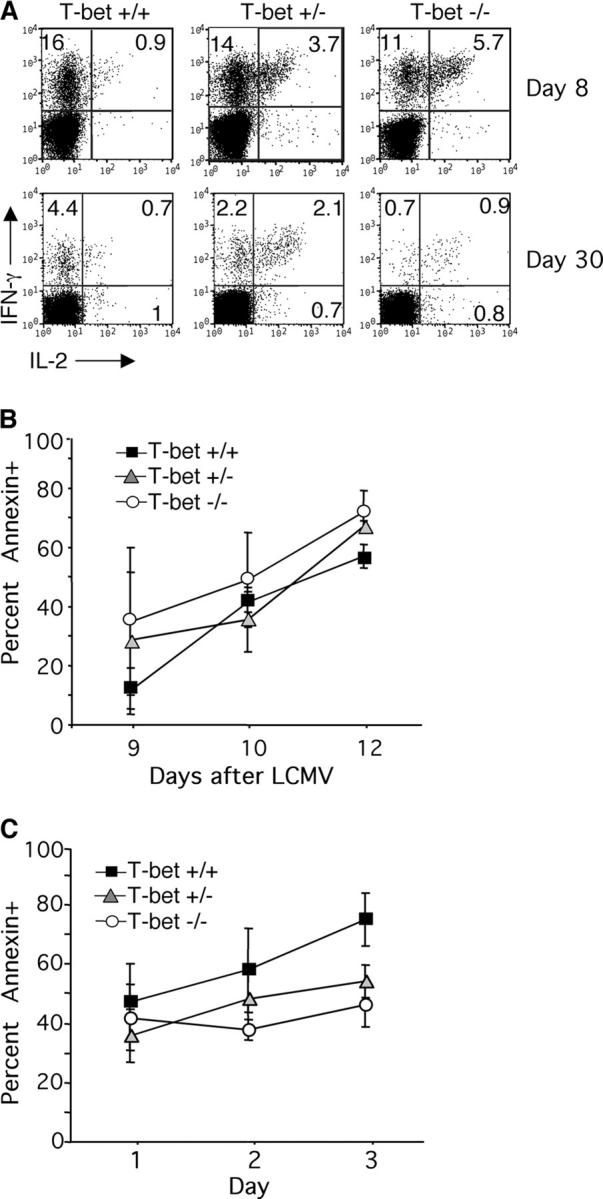
The impaired CD8 memory T cells in T-bet−/− mice are neither caused nor rescued by enhanced autologous IL-2 production. Spleens were harvested at day 8 (top) or day 30 (bottom) after infection, and cells were cultured with the MHC-I LCMV peptides. Cells were stained for intracellular IL-2 and IFN-γ and plots were gated on CD8 T cells. Data are representative of six to eight mice examined per group, with P < 0.005 when comparing IL-2 production by T-bet+/+ and T-bet−/− (A). T-bet+/+, +/−, or −/− mice were infected with LCMV, and peripheral blood was analyzed at the indicated days after infection (B). Alternatively, spleen cells from mice infected with LCMV 8 d previously were cultured in vitro for 1–3 d in medium alone (C). Cells were stained with CD8 and Db NP396 tetramers before staining for annexin V. Data are graphed as the percent annexin V+ of tetramer gated populations and are an average of three to four mice per group.
Interestingly, a recent paper demonstrated that in vivo administration of IL-2 during the expansion phase (days 0–8) resulted in fewer antigen-specific T cells and more apoptosis (39). Thus, it was possible that the enhanced IL-2 production by T-bet−/− CD8 T cells during the acute phase of infection might actually be partially responsible for the defect in effector/memory T cells. To examine this, annexin V staining was performed on LCMV tetramer positive cells at days 9, 10, and 12 after infection with LCMV. Compared with T-bet+/− or +/+ controls, T-bet−/− mice did not have significantly enhanced levels of annexin V+ antigen-specific cells (Fig. 5 B). To ensure that possible differences in apoptosis were not masked by the rapid clearance of apoptotic cells in vivo, these experiments were repeated in vitro. Splenocytes were harvested on day 8 after LCMV infection and were cultured for 1, 2, or 3 d in medium before staining for annexin V on tetramer positive cells. These studies also revealed that T-bet−/− mice did not have elevated levels of apoptosis compared with controls (Fig. 5 C). Thus, increased contraction of the primary immune response by apoptosis is not the cause for reduced levels of effector/memory CD8 CTL in T-bet−/− mice.
Defect in the Effector/Memory Response in T-bet–deficient Mice Is CD8 Intrinsic.
Autoimmune diabetes occurs independently of CD4 help in RIP-GP mice (9, 19). Thus, the resistance to diabetes of T-bet−/− GP+ mice cannot directly be explained by the profound CD4 helper Th1 defect that has been demonstrated in previous studies (2). However, it was still possible that the reduced number of CD4 Th1 lymphocytes in T-bet−/− mice affected generation of CD8 memory. Indeed, memory CD8 responses after LCMV infection depend on CD4 helper activity for maintenance and effector programming (35, 38). We sought to resolve this issue.
As expected, LCMV-specific, IFN-γ–secreting CD4 Th1 cells were decreased in T-bet−/− mice. At both the memory stage (Fig. 6 A), and at the acute phase of infection on day 8 (Fig. 6 C), IFN-γ production by CD4 T cells from T-bet−/− mice was greatly reduced. LCMV-specific CD4 T cells from heterozygous T-bet+/− mice were also substantially reduced in number (Fig. 6, A and C), consistent with the gene-dosage effect observed previously (1, 2). Because both T-bet heterozygous (+/−) and T-bet−/− mice exhibit a similar defect in LCMV-specific CD4 T cells (Fig. 6), but the former maintains completely normal LCMV-specific memory CD8 T cell responses (Fig. 2), it is unlikely that a T-bet–dependent CD4 defect alone can account for the defective α-LCMV CD8 memory response observed. Indeed, attempts to correct the CD8 memory generative defect in T-bet−/− mice by adoptive transfer of 10–30 × 106 naive CD4 T-bet+/+ lymphocytes before LCMV infection failed to correct the memory defect (Fig. 7 A).
Figure 7.
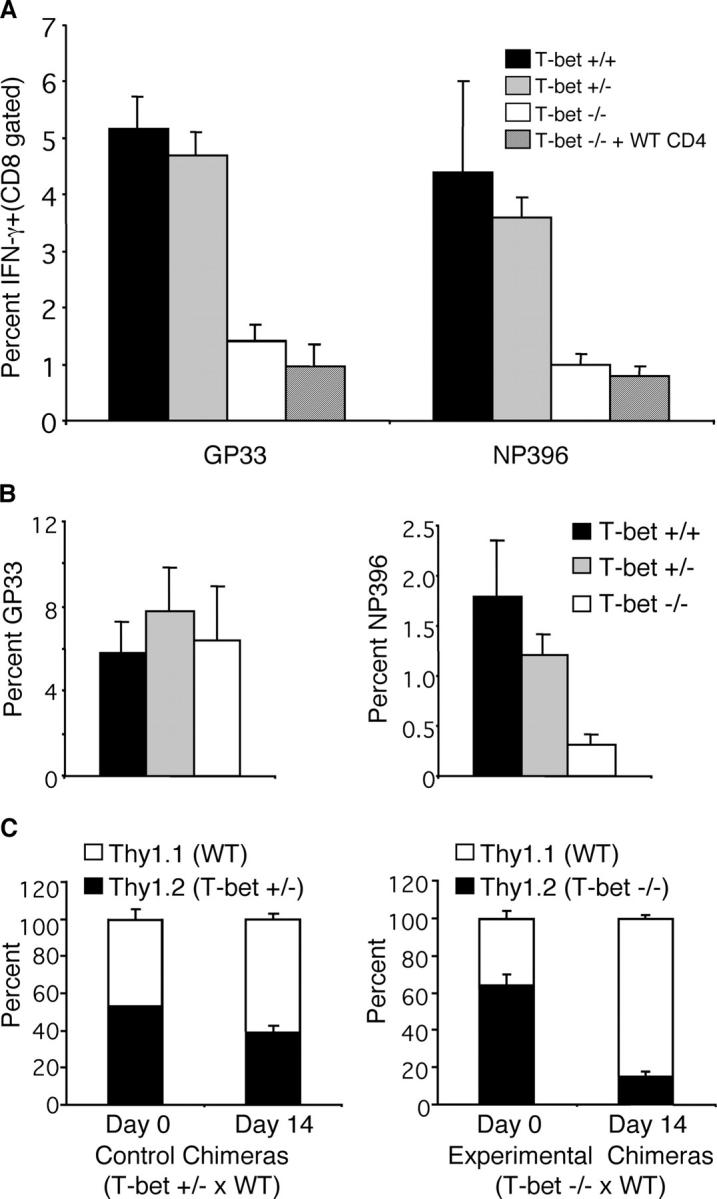
The effector/memory defect in T-bet−/− mice is CD8 intrinsic. T-bet+/+ CD4 T cells were purified by negative selection, and 1–3 × 107 cells were transferred into T-bet−/− recipients. Mice were infected with LCMV and analyzed 30 d later (A). Naive GP33-TCR-tg CD8 T cells were transferred into T-bet+/+, T-bet+/−, or T-bet−/− recipients 1 d before infection with LCMV. The percentage of GP33 or NP396-specific CD8 T cells as determined by intracellular IFN-γ staining is shown 14 d after infection (B). Mixed bone marrow chimeras were created using a 1:1 mixture of C57Bl/6 Thy1.1 congenic bone marrow and Thy1.2 T-bet+/− (C, left) or Thy1.2 T-bet−/− (C, right) bone marrow. After 6 wk to allow reconstitution, the frequency of Thy1.1 (unshaded bars) or Thy1.2 (shaded bars) CD8 T cells in the blood was determined (day 0). Mice were infected with LCMV and splenocytes were isolated 14 d later. The percentage of GP33-specific T cells that expressed Thy1.1 or Thy1.2 was determined after intracellular cytokine staining. Data are an average of three mice per group with indicated standard deviations.
It was possible that in the absence of T-bet, CD4 T cells would default to the Th2 pathway in context of the viral (LCMV) infection as described previously in unprimed mice (3). Therefore, ELISPOT assays for IL-4 or ELISAs for IL-10 and TGF-β were performed using splenocytes stimulated with GP61. Although the CD4 T cells from T-bet−/− and T-bet+/− mice produced reduced levels of IFN-γ compared with controls, there was no significant increase in antigen-specific IL-10, IL-4, or TGF-β production (Fig. 6, B–D). Thus, it is very unlikely that Th2 deviation would affect CD8 memory responses in T-bet−/− via bystander suppression, as described previously in other experimental settings for antigen-induced regulatory cells (11).
To further examine the notion that T-bet−/− CD8 T cells have an intrinsic defect, a series of adoptive transfer experiments was performed. GP33-TCR-tg C57Bl/6 (WT) T cells were transferred into T-bet+/+, +/−, or −/− recipients before infection with LCMV. Although the adoptive transfer of TCR transgenic T cells reduced the overall frequency of the endogenous CD8 T cell response, T-bet−/− mice still had fewer endogenous NP396-specific T cells compared with T-bet+/+ on day 14 (Fig. 7 B). In contrast, the frequency of GP33-specific T cells was normal in T-bet−/− mice (Fig. 7 B). Thus, WT GP33-TCR-tg CD8 T cells were not impaired in an otherwise T-bet–deficient environment. In addition, mixed bone marrow chimeras were created to further demonstrate an intrinsic defect in T-bet−/− CD8 T cells. Bone marrow was isolated from donor T-bet−/−, T-bet+/−, or C57Bl/6 (WT) Thy1.1 congenic mice. A mixture of T-bet−/− and WT Thy1.1 bone marrow was injected into lethally irradiated recipients. As a control, T-bet+/− and WT Thy1.1 mixed bone marrow chimeras were also generated. Mice were rested for 6 wk, and the percentage of Thy1.2+ (T-bet+/− or T-bet−/−) CD8 T cells was determined in peripheral blood. Both T-bet+/− and T-bet−/− chimeras expressed Thy1.2 on 50–60% of peripheral blood CD8 T cells, indicating that the chimerism had been achieved (Fig. 7 C). Mice were subsequently infected with LCMV, and the frequency of Thy1.1 (WT) and Thy1.2 (T-bet+/− or −/−) LCMV-specific CD8 T cells was determined on day 14. The generation of LCMV-specific T-bet−/− CD8 T cells was dramatically impaired in WT/KO mixed bone marrow chimeras. Thy1.2+ (T-bet−/−) CD8 T cells accounted for ∼60% of total CD8 T cells before infection, whereas they represented only ∼16% of GP33-specific T cells on day 14 after infection (Fig. 7 C). In contrast, control WT/T-bet+/− chimeras had a similar fraction of Thy1.2+ LCMV-specific CD8 T cells compared with preinfection. These results demonstrate that the generation of T-bet−/− memory CD8 T cells is impaired in the very same immunological environment that allows the successful generation of WT CD8 memory T cells.
Discussion
Here, we have demonstrated a critical role for the transcription factor T-bet in the autoaggressive behavior of CD8 lymphocytes. It is striking that the absence of T-bet so profoundly affects the development of insulitis and diabetes in a solely CD8-mediated model, as is the case in the RIP-GP transgenic mouse (9). Our data appear to conflict with earlier studies in T-bet−/− mice that failed to demonstrate an IFN-γ production defect in polyclonally activated primary CD8 cells (2). However, we have recently obtained evidence for an essential role for T-bet in antigen-driven as opposed to polyclonal, antigen-nonspecific generation and effector function of CD8 cells. In these studies, a profound defect in CD8 function in the absence of T-bet was revealed in an antigen-specific, TCR-transgenic setting (40). The findings reported here extend these observations, and demonstrate an intrinsic defect in the generation of effector/memory CD8 T cells after viral infection in vivo.
Our data provide novel mechanistic insight as to how T-bet functions in CD8 cells and demonstrate an intrinsic defect in generation of effector/memory CTLs in the absence of this factor. Primary antiviral CD8 responses develop equally in T-bet–competent or –deficient mice. This is conceivable if one considers that primary immune responses to LCMV may follow different rules compared with effector/memory and indeed, have been shown to be independent of many immunological parameters including B7, CD4 T cells, CD28, and CD40. However, in the absence of T-bet, lower numbers of effector/memory CD8 lymphocytes resulting in overall reduced IFN-γ and TNF-α production are found after LCMV infection. Their lack is also evident in the pancreatic draining lymph node and target organ that harbors autoaggressive effector CD8 lymphocytes in RIP-LCMV mice.
The difference between T-bet−/− and T-bet+/+ mice is most dramatic on day 14, which corresponds to the peak of clinical disease in the RIP-GP model. Studies performed on day 14 revealed the largest difference in terms of the number of IFN-γ–producing LCMV-specific T cells, as well as a reduced percentage of CCR7neg effector/memory cells. It is likely that effector/memory cells, as opposed to central memory, are the most important type of cell for mediating immune pathology during T1D, due to their ability to home to peripheral tissues (41).
What is the cause for defective antiviral CD8 memory in T-bet–deficient mice? Although antigen-specific studies performed with transgenic T cells demonstrated a Tc2 shift in terms of IL-4 and IL-10 production (40), we did not observe any significant Tc2 (CD8) or Th2 (CD4) deviation in the LCMV viral system. This discrepancy might be due to the intrinsic Th1 skewing properties of virus infection as compared with peptide stimulation alone. The absence of Tc2/Th2 cytokines in the LCMV system makes the decrease in autoaggressive LCMV-specific CD8 response unlikely to be due to counter-regulation, as has been described previously in the RIP-LCMV system (11).
Because our studies demonstrate that reduced CD4 helped and/or increased IL-2 were likely not the reasons for decreased CD8 memory, we assessed a role for another cytokine important in CTL generation, IL-12. However, systemic administration of IL-12 to T-bet knockout mice did not resuscitate CD8 memory (unpublished data). Along these lines, levels of IL-15, another cytokine critical in the generation and maintenance of CD8 memory cells, was normal in the bone marrow of T-bet−/− mice (unpublished data).
Lastly, some consideration should be given to the role of IFN-γ in our system. IFN-γ is an important factor in β cell destruction and viral clearance (14, 37, 42). Thus, the reduction of T1D in T-bet−/− mice agrees well with reduced levels of CD8 lymphocytes resulting in an overall decrease in IFN-γ and lytic activity. Furthermore, IFN-γ–deficient mice exhibit intact primary CTL responses to LCMV, whereas memory CTLs were reduced (37). If one considers that the memory response is programmed in the LCMV system during the primary antiviral response phase (38), possible reduction of IFN-γ levels at the time of the primary antiviral response might have resulted in an altered program in memory CTL. Indeed, major amounts of IFN-γ by CD8 CTLs are generated early (days 6–9) after LCMV infection. IFN-γ can also be produced by other cell types, such as dendritic cells and macrophages. A recent paper demonstrated that IFN-γ production by myeloid dendritic cells after culture with IL-12/IL-18 is reduced in the absence of T-bet (43); this resulted in a modest reduction in IFN-γ production, but no proliferative defect by T cells after in vivo priming with antigen-pulsed DCs (43). However, we have not detected any difference in the ability of LCMV-infected T-bet+/+ or T-bet−/− APCs to stimulate LCMV-specific CD8 memory or naive T cells in vitro (unpublished data) or in vivo (Fig. 7 B). In addition, because IFN-γ levels made by primary LCMV CTLs in T-bet−/− mice match those of T-bet+/− or +/+ littermates (Fig. 2), we argue that lack of IFN-γ at the acute phase of infection is not responsible for the observed defect in memory CTLs. All of these data make it likely that the defect is intrinsic to the CD8 cell rather than attributable to the cytokine microenvironment. Most convincingly, our studies using bone marrow chimeras demonstrate that the generation of T-bet−/− memory CD8 T cells is impaired in the very same immunological environment that allows the successful generation of WT CD8 memory T cells.
In conclusion, our observations indicate that small molecule inhibitors of crucial transcription factors such as T-bet might offer a unique therapeutic opportunity for preventing autoimmunity. T1D likely depends on both autoaggressive CD4 as well as CD8 lymphocytes. Indeed, a potent role for CD8 effector cells has been found more recently in many T1D models (15–17). Therefore, the intrinsic CD8 defect specific for effector/memory cells but not primary CD8 lymphocytes reported here in the absence of T-bet strengthens the rationale for targeting T1D. It will need to be determined whether control of chronic infections, or long-term immunity after vaccination is sufficient in the absence of T-bet, although our results here demonstrate that lack of T-bet does not render mice susceptible to an acute viral infection.
Acknowledgments
We thank Dr. U. Christen and A. Rhode for the generation of MHC-I tetramers and D. Frye for assistance with the manuscript.
M.G. von Herrath was supported by National Institutes of Health grants AI44451, DK51091, and U-19 AI51973 and a Juvenile Diabetes Research Foundation (JDRF) grant. A.E. Juedes is the recipient of a JDRF postdoctoral fellowship. L. Glimcher was supported by a grant from the JDRF Center for Islet Transplantation at Harvard Medical School. This is manuscript no. 538 from the Department of Developmental Immunology at the La Jolla Institute for Allergy and Immunology.
Abbreviations used in this paper: BGV, blood glucose value; GP, glycoprotein; LCMV, lymphocytic choriomeningitis virus; RIP, rat insulin promoter; T1D, type 1 diabetes.
References
- 1.Szabo, S.J., S.T. Kim, G.L. Costa, X. Zhang, C.G. Fathman, and L.H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 100:655–669. [DOI] [PubMed] [Google Scholar]
- 2.Szabo, S.J., B.M. Sullivan, C. Stemmann, A.R. Satoskar, B.P. Sleckman, and L.H. Glimcher. 2002. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 295:338–342. [DOI] [PubMed] [Google Scholar]
- 3.Finotto, S., M.F. Neurath, J.N. Glickman, S. Qin, H.A. Lehr, F.H. Green, K. Ackerman, K. Haley, P.R. Galle, S.J. Szabo, et al. 2002. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 295:336–338. [DOI] [PubMed] [Google Scholar]
- 4.Neurath, M.F., S. Finotto, and L.H. Glimcher. 2002. The role of Th1/Th2 polarization in mucosal immunity. Nat. Med. 8:567–573. [DOI] [PubMed] [Google Scholar]
- 5.Neurath, M.F., B. Weigmann, S. Finotto, J. Glickman, E. Nieuwenhuis, H. Iijima, A. Mizoguchi, E. Mizoguchi, J. Mudter, P.R. Galle, et al. 2002. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J. Exp. Med. 195:1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohning, M., A. Richter, and A. Radbruch. 2002. Cytokine memory of T helper lymphocytes. Adv. Immunol. 80:115–181. [DOI] [PubMed] [Google Scholar]
- 7.Oldstone, M.B., M. Nerenberg, P. Southern, J. Price, and H. Lewicki. 1991. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 65:319–331. [DOI] [PubMed] [Google Scholar]
- 8.Ohashi, P.S., S. Oehen, K. Buerki, H. Pircher, C.T. Ohashi, B. Odermatt, B. Malissen, R.M. Zinkernagel, and H. Hengartner. 1991. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 65:305–317. [DOI] [PubMed] [Google Scholar]
- 9.von Herrath, M.G., J. Dockter, and M.B. Oldstone. 1994. How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity. 1:231–242. [DOI] [PubMed] [Google Scholar]
- 10.Holz, A., A. Bot, B. Coon, T. Wolfe, M.J. Grusby, and M.G. von Herrath. 1999. Disruption of the STAT4 signaling pathway protects from autoimmune diabetes while retaining antiviral immune competence. J. Immunol. 163:5374–5382. [PubMed] [Google Scholar]
- 11.Homann, D., A. Holz, A. Bot, B. Coon, T. Wolfe, J. Petersen, T.P. Dyrberg, M.J. Grusby, and M.G. von Herrath. 1999. Autoreactive CD4+ T cells protect from autoimmune diabetes via bystander suppression using the IL-4/Stat6 pathway. Immunity. 11:463–472. [DOI] [PubMed] [Google Scholar]
- 12.Christen, U., T. Wolfe, U. Mohrle, A.C. Hughes, E. Rodrigo, E.A. Green, R.A. Flavell, and M.G. von Herrath. 2001. A dual role for TNF-alpha in type 1 diabetes: islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J. Immunol. 166:7023–7032. [DOI] [PubMed] [Google Scholar]
- 13.Slifka, M.K., and J.L. Whitton. 2000. Activated and memory CD8+ T cells can be distinguished by their cytokine profiles and phenotypic markers. J. Immunol. 164:208–216. [DOI] [PubMed] [Google Scholar]
- 14.Seewaldt, S., H.E. Thomas, M. Ejrnaes, U. Christen, T. Wolfe, E. Rodrigo, B. Coon, B. Michelsen, T.W. Kay, and M.G. von Herrath. 2000. Virus-induced autoimmune diabetes: most beta-cells die through inflammatory cytokines and not perforin from autoreactive (anti-viral) cytotoxic T-lymphocytes. Diabetes. 49:1801–1809. [DOI] [PubMed] [Google Scholar]
- 15.Amrani, A., J. Verdaguer, B. Anderson, T. Utsugi, S. Bou, and P. Santamaria. 1999. Perforin-independent beta-cell destruction by diabetogenic CD8(+) T lymphocytes in transgenic nonobese diabetic mice. J. Clin. Invest. 103:1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liblau, R.S., F.S. Wong, L.T. Mars, and P. Santamaria. 2002. Autoreactive CD8 T cells in organ-specific autoimmunity: emerging targets for therapeutic intervention. Immunity. 17:1–6. [DOI] [PubMed] [Google Scholar]
- 17.DiLorenzo, T.P., R.T. Graser, T. Ono, G.J. Christianson, H.D. Chapman, D.C. Roopenian, S.G. Nathenson, and D.V. Serreze. 1998. Major histocompatibility complex class I-restricted T cells are required for all but the end stages of diabetes development in nonobese diabetic mice and use a prevalent T cell receptor alpha chain gene rearrangement. Proc. Natl. Acad. Sci. USA. 95:12538–12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holz, A., T. Dyrberg, W. Hagopian, D. Homann, M. von Herrath, and M.B. Oldstone. 2000. Neither B lymphocytes nor antibodies directed against self antigens of the islets of Langerhans are required for development of virus-induced autoimmune diabetes. J. Immunol. 165:5945–5953. [DOI] [PubMed] [Google Scholar]
- 19.Laufer, T.M., M.G. von Herrath, M.J. Grusby, M.B. Oldstone, and L.H. Glimcher. 1993. Autoimmune diabetes can be induced in transgenic major histocompatibility complex class II–deficient mice. J. Exp. Med. 178:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutko, F.J., and M.B. Oldstone. 1983. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J. Gen. Virol. 64:1689–1698. [DOI] [PubMed] [Google Scholar]
- 21.von Herrath, M.G., S. Guerder, H. Lewicki, R.A. Flavell, and M.B. Oldstone. 1995. Coexpression of B7-1 and viral (“self”) transgenes in pancreatic beta cells can break peripheral ignorance and lead to spontaneous autoimmune diabetes. Immunity. 3:727–738. [DOI] [PubMed] [Google Scholar]
- 22.Gairin, J.E., H. Mazarguil, D. Hudrisier, and M.B. Oldstone. 1995. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J. Virol. 69:2297–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oxenius, A., M.F. Bachmann, P.G. Ashton-Rickardt, S. Tonegawa, R.M. Zinkernagel, and H. Hengartner. 1995. Presentation of endogenous viral proteins in association with major histocompatibility complex class II: on the role of intracellular compartmentalization, invariant chain and the TAP transporter system. Eur. J. Immunol. 25:3402–3411. [DOI] [PubMed] [Google Scholar]
- 24.Manjunath, N., P. Shankar, J. Wan, W. Weninger, M.A. Crowley, K. Hieshima, T.A. Springer, X. Fan, H. Shen, J. Lieberman, and U.H. von Andrian. 2001. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J. Clin. Invest. 108:871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Busch, D.H., I.M. Pilip, S. Vijh, and E.G.P. Am. 1998. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 8:353–362. [DOI] [PubMed] [Google Scholar]
- 26.Pircher, H., K. Burki, R. Lang, H. Hengartner, and R.M. Zinkernagel. 1989. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 342:559–561. [DOI] [PubMed] [Google Scholar]
- 27.von Herrath, M.G., T. Wolfe, U. Mohrle, B. Coon, and A. Hughes. 2001. Protection from type 1 diabetes in the face of high levels of activated autoaggressive lymphocytes in a viral transgenic mouse model crossed to the SV129 strain. Diabetes. 50:2700–2708. [DOI] [PubMed] [Google Scholar]
- 28.Andreasen, S.O., J.E. Christensen, O. Marker, and A.R. Thomsen. 2000. Role of CD40 ligand and CD28 in induction and maintenance of antiviral CD8+ effector T cell responses. J. Immunol. 164:3689–3697. [DOI] [PubMed] [Google Scholar]
- 29.Whitmire, J.K., K. Murali-Krishna, J. Altman, and R. Ahmed. 2000. Antiviral CD4 and CD8 T-cell memory: differences in the size of the response and activation requirements. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomsen, A.R., J. Johansen, O. Marker, and J.P. Christensen. 1996. Exhaustion of CTL memory and recrudescence of viremia in lymphocytic choriomeningitis virus-infected MHC class II-deficient mice and B cell-deficient mice. J. Immunol. 157:3074–3080. [PubMed] [Google Scholar]
- 31.Borrow, P., A. Tishon, S. Lee, J. Xu, I.S. Grewal, M.B. Oldstone, and R.A. Flavell. 1996. CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. J. Exp. Med. 183:2129–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matloubian, M., R.J. Concepcion, and R. Ahmed. 1994. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 68:8056–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tishon, A., H. Lewicki, G. Rall, M. Von Herrath, and M.B. Oldstone. 1995. An essential role for type 1 interferon-gamma in terminating persistent viral infection. Virology. 212:244–250. [DOI] [PubMed] [Google Scholar]
- 34.Suresh, M., J.K. Whitmire, L.E. Harrington, C.P. Larsen, T.C. Pearson, J.D. Altman, and R. Ahmed. 2001. Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J. Immunol. 167:5565–5573. [DOI] [PubMed] [Google Scholar]
- 35.von Herrath, M.G., M. Yokoyama, J. Dockter, M.B. Oldstone, and J.L. Whitton. 1996. CD4-deficient mice have reduced levels of memory cytotoxic T lymphocytes after immunization and show diminished resistance to subsequent virus challenge. J. Virol. 70:1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Homann, D., A. Jahreis, T. Wolfe, A. Hughes, B. Coon, M.J. van Stipdonk, K.R. Prilliman, S.P. Schoenberger, and M.G. von Herrath. 2002. CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity. 16:403–415. [DOI] [PubMed] [Google Scholar]
- 37.von Herrath, M.G., and M.B. Oldstone. 1997. Interferon-γ is essential for destruction of β cells and development of insulin-dependent diabetes mellitus. J. Exp. Med. 185:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janssen, E.M., E.E. Lemmens, T. Wolfe, U. Christen, M.G. von Herrath, and S.P. Schoenberger. 2003. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 421:852–856. [DOI] [PubMed] [Google Scholar]
- 39.Blattman, J.N., J.M. Grayson, E.J. Wherry, S.M. Kaech, K.A. Smith, and R. Ahmed. 2003. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 9:540–547. [DOI] [PubMed] [Google Scholar]
- 40.Sullivan, B.M., A. Juedes, S.J. Szabo, M. von Herrath, and L.H. Glimcher. 2003. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl. Acad. Sci. USA. 100:15818–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wherry, E.J., V. Teichgraber, T.C. Becker, D. Masopust, S.M. Kaech, R. Antia, U.H. von Andrian, and R. Ahmed. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4:225–234. [DOI] [PubMed] [Google Scholar]
- 42.von Herrath, M., B. Coon, D. Homann, T. Wolfe, and L.G. Guidotti. 1999. Thymic tolerance to only one viral protein reduces lymphocytic choriomeningitis virus-induced immunopathology and increases survival in perforin-deficient mice. J. Virol. 73:5918–5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lugo-Villarino, G., R. Maldonado-Lopez, R. Possemato, C. Penaranda, and L.H. Glimcher. 2003. T-bet is required for optimal production of IFN-{gamma} and antigen-specific T cell activation by dendritic cells. Proc. Natl. Acad. Sci. USA. 100:7749–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]