

Abstract
Contact sensitivity (CS) is an inflammatory disorder characterized by early and late phases of leukocyte recruitment. We used a noninvasive intravital microscopy technique allowing for the direct visualization of leukocyte rolling and adhesion on blood vessel endothelium. By blocking specific adhesion molecules, we elucidated the molecular mechanisms mediating early leukocyte recruitment to be E- and P-selectin and demonstrated that leukocyte recruitment in the late phase had a different adhesive profile (mainly α4-integrin). Complete blockade of E- and P-selectin within the first 2 h of leukocyte–endothelial cell interactions (but not later) eliminated selectin-independent leukocyte recruitment at 24 h. Despite the predominance of neutrophils in the early phase, specific elimination of CD4+ lymphocytes in the early phase eliminated the late response. CD4+ lymphocytes homed to skin via E- and P-selectin within the early phase and induced the late phase response. Addition of these same CD4+ lymphocytes 2 h after antigen challenge was too late for these cells to home to the skin and induce late phase responses. Our data clearly demonstrate that the antigen-challenged microenvironment is only accessible to CD4+ lymphocytes for the first 2 h, and that this process is essential for the subsequent recruitment of other leukocyte populations in late phase responses.
Keywords: delayed hypersensitivity, contact dermatitis, neutrophils, vascular cell adhesion molecule-1, CD4+ T lymphocytes
Introduction
Contact sensitivity (CS) is a form of delayed-type hypersensitivity that is easily reproduced in mice and serves as an excellent model of T cell–mediated inflammation pertinent to autoimmunity and allergy. The basic tenet suggests that after immunization with an antigen, subsequent challenge with the same antigen sets in motion a series of initiating events that include the activation of mast cells (1–3), release of serotonin (4–7), release of TNF (8, 9), and activation of complement via antigen–antibody complexes (10–14). It has been proposed, but never directly demonstrated, that these molecules induce the rapid and selective recruitment of CD4+ lymphocytes into the extravascular space at the challenge site. The CD4+ lymphocytes begin to produce cytokines including IFN-γ that mediate a more profound phase of leukocyte recruitment and edema formation that peaks at ∼24 h (12, 15–19).
There is much indirect evidence to suggest that during the early or initiation phase (first 2 h of challenge), there is selective recruitment of CD4+ lymphocytes into the extravascular space at the challenge site. For example, inhibition of C5a within the first 2 h of antigen challenge reduces both edema formation and the elevated levels of IFN-γ (an index of TH1 cell recruitment) that are characteristic of the 24-h late phase (12). Moreover, inhibition of C5a at 3 h of challenge no longer reduces either the edema formation or the elevated IFN-γ at 24 h. Clearly, the first 2 h are a critical time period for the subsequent late phase response. In addition, because injection of isolated T lymphocytes from immunized mice can completely reconstitute the late phase response (16), the data as a whole infer that C5a recruits TH1 cells within the initiation phase of challenge, and these cells act in a sequential obligate fashion to induce the late-phase response. However, no one to date has directly demonstrated that CD4+ lymphocytes are recruited early in CS. Moreover, C5a has traditionally been thought to be one of the classical chemoattractants for neutrophils and monocytes, and the early recruitment of neutrophils in many inflammatory conditions is thought to modify the microenvironment and/or endothelial surface to induce subsequent mononuclear cell recruitment (20–22). Indeed, there is evidence for early endothelial cell activation and neutrophil recruitment by electron microscopy in CS (23). However, the early initiation phase of challenge is thought to have very little accumulation of leukocytes in the afflicted tissue (based on histology). It is our hypothesis that early transformations at the endothelial–leukocyte interface occur almost immediately, setting in motion the entire series of events known as CS. In other words, it is possible that even a small number of neutrophils and/or lymphocytes rolling and adhering to endothelium and subsequently transmigrating could be important in modifying the microenvironment to allow for the recruitment of effector cells in the late-phase response.
For any leukocyte to exit the circulation in response to an inflammatory stimulus, a sequential cascade of events is followed that involves initial tethering and rolling of leukocytes on endothelial cells. This event is entirely dependent on selectins (E- and P-selectins on endothelium) and is required for subsequent integrin-dependent firm adhesion and transendothelial migration (24–26). Indeed, Staite et al. demonstrated that mice lacking E-selectin and P-selectin have attenuated leukocyte recruitment at 24 h of CS (27). In contrast, other groups have reported that inhibition of α4-integrin can also reduce leukocyte recruitment in the late phase of CS (8, 28, 29). Explanations for these potentially discrepant results include the possibility that selectins mediate rolling, whereas the integrins mediate adhesion. Because leukocyte recruitment is dependent on both rolling and adhesion, inhibition of either would reduce leukocyte recruitment. Interestingly, in the case of lymphocytes, the α4-integrin can also support tethering and rolling (in addition to adhesion), allowing for lymphocytes to entirely bypass any need for selectins (30–32). Most intriguing is the possibility that selectins and α4-integrins mediate leukocyte recruitment at different time points (i.e., the initiation and late phases of CS), and because leukocyte recruitment in the initiation phase of challenge may be important for the late phase of challenge, we hypothesize that inhibition of specific adhesion molecules in the early phase would lead to an abrogated late phase response.
To systematically examine the leukocyte trafficking during the initiation (first 2 h) and late phases (24 h) of challenge, we made use of noninvasive intravital microscopy to visualize leukocyte rolling and adhesion at both time points in the dermal microvasculature. A very profound increase in leukocyte–endothelial cell interactions was noted within the first 2 h with a clear demarcation followed by a second wave of leukocyte recruitment between 4 and 24 h. The early phase was primarily neutrophils and a small subset of lymphocytes. We identified the molecular mechanisms mediating early leukocyte recruitment (E- and P-selectin) that permitted us to demonstrate that leukocyte recruitment in the late phase had a different adhesive profile (primarily of α4-integrin). Additional experiments revealed that complete abrogation of the early phase of leukocyte–endothelial cell interactions with anti–E- and anti–P-selectin eliminated selectin-independent leukocyte recruitment at 24 h. Despite the predominance of neutrophils in the early phase, specific elimination of these cells in the early phase did not affect the late phase response. CD4+ lymphocyte recruitment dependent on E- and P-selectin was demonstrated within the first 2 h and these cells were essential for the subsequent late phase response. Our data suggest that the vascular endothelium is primed by the early recruitment of CD4+ lymphocytes to recruit various leukocyte populations later in CS inflammation.
Materials and Methods
Antibodies.
The antibodies used in this work were as follows: RB40.34, an antimurine P-selectin mAb (50 μg/mouse; BD Biosciences); R1-2, an antimurine α4-integrin mAb (50 μg/mouse; BD Biosciences); RME-1, an antimurine E-selectin Ab (100 μg/mouse; a gift from A.C. Issekutz, Dalhousie University, Halifax, Canada), and RB6-8C5, an antineutrophil mAb (100 μg/mouse; a gift from D.N. Granger, Louisiana State University, Shreveport, LA). Antibodies were administered by either i.v. or i.p. injection. Antibody specificities have been extensively characterized in our laboratory. Antibody isotype control experiments were performed and showed no evidence of significant adhesion molecule blockade.
Animals.
Male C57BL/6 wild-type mice were obtained from Charles River Breeding Laboratories. C57BL/6-Cd4tm4Mak mice were obtained from Jackson ImmunoResearch Laboratories. Mice were kept in sterilized filter-topped cages and fed autoclaved food in the animal facilities of the University of Calgary. The protocols used were in accordance with the guidelines drafted by the University of Calgary Animal Care Committee and the Canadian Council on the Use of Laboratory Animals. All mice weighed between 20 and 32 g and were used between 6 and 10 wk of age.
Oxazolone-induced CS.
Mice were sensitized for CS response by topical application of 50 μL of 5% oxazolone (4-ethoxymethylene-2-oxazolin-5-one; Sigma-Aldrich) in acetone–olive oil vehicle (4:1) to the shaved flank. 6–7 d later, mice received a 10-μL challenge of 1% oxazolone solution on the ventral aspect of the left ear. At various time points after antigen challenge, ear skin venules were visualized via intravital microscopy.
Adoptive Transfer of CD4+ Lymphocytes.
C57BL/6 mice sensitized with oxazolone were killed on day 6, and spleen and peripheral lymph node cells were isolated. CD4+ lymphocytes were isolated by MACS positive selection. Cells were counted, and 107 cells were resuspended in normal saline, fluorescently labeled as described in the next paragraph and injected into oxazolone-sensitized C57BL/6-Cd4tm4Mak mice immediately before antigen challenge or 2 h after antigen challenge.
Intravital Microscopy.
The mouse ear prep was used to study the behavior of leukocytes in the microcirculation (33). Mice were anesthetized by i.p. injection of a mixture of 10 mg/kg xylazine hydrochloride (MTC Pharmaceuticals) and 200 mg/kg ketamine hydrochloride (Rogar/STB). A depilatory solution (Nair; Armkel LLC) was applied to the dorsal and ventral surfaces of the left ear. After 10 min, the solution was gently removed using 0.9% normal saline and cotton swabs. The right jugular vein was cannulated and used to administer additional anesthetic, antibodies, and fluorescent labels. The left ear was mounted against the adjustable plexiglass microscope pedestal and held in place under a coverslip. Mouse rectal temperature was monitored and maintained at 37°C. Rhodamine 6G (0.3 mg/kg body weight; Sigma-Aldrich) and FITC-albumin (0.1 ml of 5% solution in normal saline; Sigma-Aldrich) were administered to fluorescently labeled leukocytes and vessel walls, respectively. Fluorescence was visualized by epi-illumination using 510- and 560-nm filters.
An intravital microscope (Axioskop; Carl Zeiss MicroImaging, Inc.) with a 40× immersion objective lens (Weltzlar; E. Leitz) and a 10× eyepiece were used to examine the ear skin microcirculation. A video camera (HS model 5100; Panasonic) was used to project the images onto a monitor, and the images were recorded for playback analysis using a videocassette recorder. Single unbranched skin venules (20–40 μm in diameter) were selected. The number of rolling and adherent leukocytes was determined offline during video playback analysis. Rolling leukocytes were defined as those cells moving at a velocity slower than that of erythrocytes within a vessel. Leukocyte rolling flux was determined by counting the number of leukocytes that rolled by a fixed point in the venule over 1 min. Leukocyte rolling velocity was determined by measuring the time required for a leukocyte to roll along a 100-μm length of venule. Rolling velocity was determined for 20 leukocytes at each interval. The number of leukocytes rolling per 100 μm of vessel was calculated by dividing leukocyte rolling flux by leukocyte rolling velocity and multiplying by a factor of 100/60. Leukocytes were considered adherent to the venular endothelium if they remained stationary for 30 s or longer.
Measurement of Ear Thickness.
Resulting thickness of antigen-challenged ears was measured using an Engineer's dial micrometer (Mitutoyu Co.). The increase in ear thickness after antigen challenge was calculated by subtracting the thickness of the unchallenged right ear from that of the challenged left ear. Increases in groups of five were expressed as the mean ± SE × 10−3 cm.
Quantitative ELISA for IFN-γ in Ear Homogenates.
In CS-induced mice, three 4-mm punch biopsies were taken per ear. These biopsies were flash frozen in liquid nitrogen, subsequently thawed, and extracted in 300 μL of cold PBS on ice with a tissue microhomogenizer. A commercially available ELISA kit was used to measure tissue homogenate concentrations of IFN-γ (Genzyme). Final concentrations of IFN-γ were corrected for concentrations of protein in each homogenate sample and are represented as picograms of IFN-γ/mg of protein.
Antineutrophil Studies.
Sensitized mice were injected i.p. with RB6-8C5 mAb either 24 h before antigen challenge, or 2 h after antigen challenge. Intravital microscopy was performed at 2 or 24 h to confirm the presence or absence of leukocyte rolling and adhesion. Complete blood counts were performed to confirm the specific depletion of the neutrophil population.
Circulating Leukocyte Counts.
At the end of each experiment, whole blood was drawn via cardiac puncture. Total leukocyte counts were performed using a Bright-line hemocytometer (Hausser Scientific).
Histology.
Tissue samples were fixed in 10% formalin, processed, and H&E stained by members of the Department of Histopathology at the University of Calgary. Data analysis was facilitated by a pathologist (S. Urbanski, University of Calgary, Alberta, Canada). Leukocyte differentials were performed by counting leukocytes seen on the H&E stain; the mean value of 12 high-power (200×) fields was taken.
Statistical Analysis.
All data are displayed as mean ± SEM. Data were analyzed using standard statistical analysis (analysis of variance and Student's t test, with Bonferroni's correction for multiple comparisons where appropriate). Statistical significance was set at P < 0.05.
Online Supplemental Material.
Online supplemental video clips of our noninvasive intravital microscopy ear prep are available at http://www.jem.org/cgi/content/full/jem.20032016/DC1. Shown is baseline leukocyte rolling in the ear in the absence of any inflammatory stimulus (Video S1). Very few adherent cells can be seen. In contrast, leukocyte rolling and adhesion can be seen at 2 h of CS (Video S2).
Results
Two Distinct Populations of Leukocytes Are Recruited in the Initiation and Late Phases of CS.
Fig. 1 summarizes an extensive time scale of leukocyte rolling and adhesion at 0, 1, 2, 3, 4, 8, 16, and 24 h after antigen challenge. It has been reported previously that basal rolling occurs in skin primarily because of constitutive expression of P-selectin and E-selectin (34–36). Indeed, our data also demonstrate basal rolling (Video S1, available at http://www.jem.org/cgi/content/full/jem.20032016/DC1) and, furthermore, reveal that although a small peak of rolling occurred over the first 2 h (Video S2, available at http://www.jem.org/cgi/content/full/jem.20032016/DC1), the rolling remained relatively constant throughout the remainder of the CS time course (Fig. 1 A). Leukocyte rolling velocity was observed to decrease at ∼4 h (Fig. 1 B), which was consistent with previous findings suggesting that the slow leukocyte rolling is mediated by an increased expression of E-selectin on endothelial cells at that time point (37). The rolling leukocytes accumulate in the vessels as they roll very slowly such that, even though flux data do not change, the number of rolling cells in a given vessel is increased after 4 h (Fig. 1 C). Fig. 1 D reveals that leukocyte adhesion displayed a bimodal distribution with a peak centered around 2 h and a broader peak starting at 4 h and continuing past 24 h. Of note, although leukocytes continued to roll at the 3 h time point, the vessels were devoid of any leukocyte adhesion. These data suggest that there are two, temporally very distinct, nonoverlapping populations of leukocytes recruited within the vasculature in the early (2 h) and late (24 h) phases of CS. Clearly, there is a very distinct demarcation at 3 h between early and late phase of CS.
Figure 1.
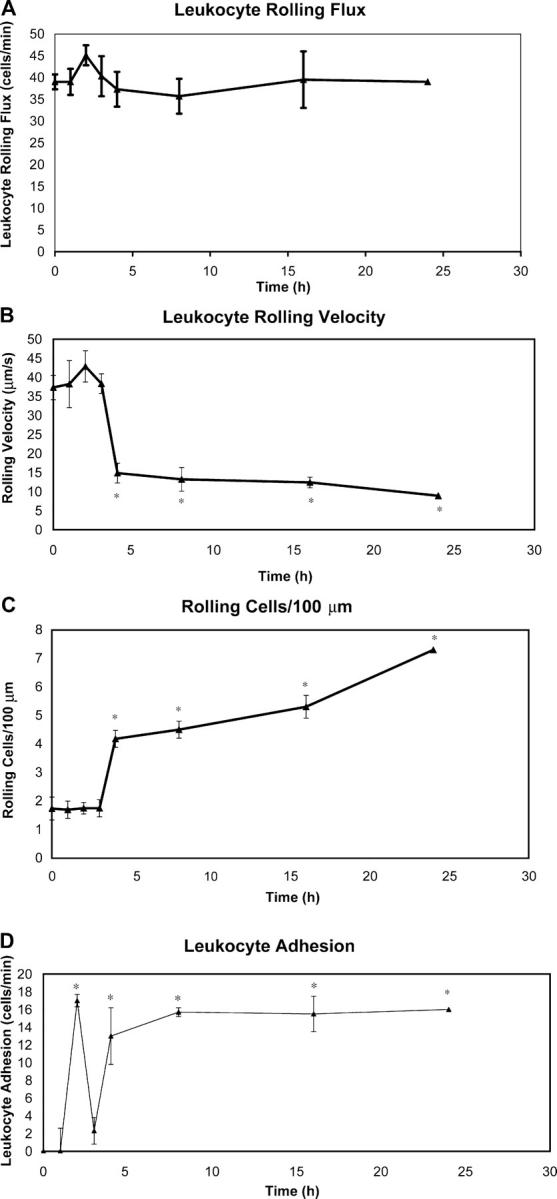
CS time course. Intravital microscopy was used to assess leukocyte–endothelial interactions over the first 24 h of the clinical course of CS. Time points were taken at 0, 1, 2, 3, 4, 8, 16, and 24 h after antigen challenge. Leukocyte rolling flux (A), leukocyte rolling velocity (B), the number of rolling cells (C), and the number of adherent cells (D) were measured. A minimum of five animals was used at each time point (*, P < 0.05 relative to time 0).
Tissue samples were taken at 2 and 24 h after antigen challenge and sections were stained with hemotoxylin and eosin (Fig. 2, A and B). At 2 h of CS, epidermal and dermal layers were clearly defined with no signs of erosion, microabscess formation, or ulceration. In contrast, at 24 h, there were obvious erosions in the epidermal layer and formation of microabscesses as evidenced by the deposition of cellular debris around the hair follicles. Both CS phases were characterized by a mixed leukocytosis; however, the two profiles were quite distinct. Close to 70% of the leukocytes noted in the extravascular space at 2 h were neutrophils, with the remaining 30% made up of lymphocytes with a few mast cells and eosinophils (Fig. 2 C). By 24 h of challenge, the number of neutrophils decreased to 30% of leukocytes and the majority of infiltrating leukocytes were lymphocytes (Fig. 2 D). Data were assessed in a blinded fashion by a pathologist (S. Urbanski). In addition, more conventional indicators of a CS reaction, including ear thickness and IFN-γ, were measured and correlated well with the leukocyte–endothelial cell parameters (see Fig. 7).
Figure 2.

Leukocyte infiltration at 2 and 24 h of CS. Inflammation at 2 (A) and 24 h (B) of CS was assessed by histology. Differential leukocyte counts on histology confirmed differing leukocyte infiltrate composition at 2 (C, magnification, 200) and 24 h (D, magnification, 100) of CS. Leukocyte counts on histology were an average of 12 high-power fields ± SEM. A minimum of three animals was used at each time point.
Figure 7.

Late phase inflammation in CS is dependent on early leukocyte recruitment. Changes in ear thickness (A) and IFN-γ concentration in tissue homogenates (B) were measured at 2 and 24 h time points of CS as well as at the 24-h time point with treatments of anti–E- and anti–P-selectin antibodies either immediately before antigen challenge or 2 h after antigen challenge. A minimum of five animals was used in each experimental group (*, P < 0.05 relative to control).
Endothelial Selectins Are Essential for the Initial Phase But Not the Late Phase of Leukocyte Recruitment in CS.
Administration of monoclonal antibodies against E-selectin and/or P-selectin within the first 2 h of antigen challenge revealed an essential role for both molecules in early leukocyte recruitment (Fig. 3 A). Treatment with anti–P-selectin antibody attenuated leukocyte flux by 63%. Treatment with anti–E-selectin antibody did not significantly reduce leukocyte rolling; however, leukocyte flux was reduced by >97% with a combination of anti–E- and anti–P-selectin antibodies. These results suggest that E- and P-selectin are necessary for leukocyte rolling in the early phase of CS, but that their roles are overlapping; P-selectin is able to fully compensate for the absence of E-selectin, whereas E-selectin seems to only partially compensate for the absence of P-selectin. Of note, α4-integrin was not found to have any effect on leukocyte rolling flux in the early phase of CS. Isotype control antibodies did not affect any leukocyte–endothelial cell interactions (unpublished data).
Figure 3.

Leukocyte rolling flux and adhesion at 2 h of CS. Animals were treated with specific antibodies against E-selectin, P-selectin, and α4-integrin i.v. at the 2-h time point. Leukocyte rolling flux (A) and leukocyte adhesion (B) were assessed using intravital microscopy at 2 h of CS. A minimum of three animals was used at each time point (*, P < 0.05 relative to no treatment group).
We also observed leukocyte adhesion in the early phase of CS. We found that leukocyte adhesion was not significantly reduced by individual i.v. treatments of either anti–E-selectin or anti–P-selectin antibodies (Fig. 3 B). However, combined treatments of both antibodies completely eliminated leukocyte adhesion. α4-integrin antibody had no effect on leukocyte recruitment.
The molecular mechanisms of leukocyte rolling differed during the late phase of CS. In these experiments, antibodies were administered at 24 h of CS. Single treatments with either anti–E-selectin or anti–P-selectin antibodies were not found to reduce leukocyte rolling flux at 24 h of CS (Fig. 4 A). Most unexpectedly, a combination therapy of anti–E- and anti–P-selectin antibodies (which completely inhibited early leukocyte rolling) did not in any way reduce leukocyte rolling flux at 24 h. These observations are quite different from reports that E-selectin and P-selectin double deficient mice have significantly reduced late phase of CS (27). Anti–α4-integrin antibody treatment alone reduced leukocyte flux by 65%. Addition of anti–α4-integrin antibody to the two antiselectin antibodies significantly reduced leukocyte flux by 91%. Anti–α4-integrin antibody in combination with either anti–P- or anti–E-selectin antibody reduced leukocyte flux by 72 and 67%, respectively (unpublished data). These results suggest that α4-integrin is the dominant rolling molecule at 24 h of CS.
Figure 4.

Leukocyte rolling flux and adhesion at 24 h of CS. Antibodies against P-selectin, E-selectin, and α4-integrin were injected i.v. at 24 h of CS. Leukocyte rolling (A) and leukocyte adhesion (B) were assessed. A minimum of three animals was used at each time point (*, P < 0.05 relative to no treatment group).
Leukocyte adhesion at 24 h of CS was not significantly reduced by individual treatments of anti–E-selectin, anti–P-selectin, or even anti–α4-integrin antibodies (Fig. 4 B). However, a combination of the three antibodies completely eliminated leukocyte adhesion. The lack of inhibition of adhesion with anti–α4-integrin antibody despite a 65% inhibition of rolling is entirely consistent with our previous observation that one has to inhibit >90% of rolling leukocytes to affect adhesion significantly (24).
Late Phase CS Is Dependent on Leukocyte Recruitment in the Early Phase.
Because the early phase of CS was dependent on the two endothelial selectins and the late phase was not, it permitted us to explore the importance of inhibiting leukocyte recruitment in the early phase on leukocyte recruitment in the late phase. First, we wanted to ensure that the antibodies had a 24-h functional half-life in vivo. Tandem injection of the E-selectin and P-selectin antibody 24 h before oxazolone challenge revealed complete inhibition of leukocyte rolling and adhesion at what was equivalent to 2 h of challenge (unpublished data). Clearly, the antibodies had biological activity for 24 h in vivo. Next, E-selectin and P-selectin antibody were given at the time of challenge. Fig. 5 A demonstrates that neither P-selectin nor E-selectin antibody given alone at the time of challenge had any effect on leukocyte rolling at 24 h of antigen challenge. However, tandem administration of P-selectin and E-selectin antibodies at the time of challenge completely abolished leukocyte rolling at 24 h of challenge (Fig. 5 A) despite the fact that some of the rolling was α4-integrin–dependent when antibodies were given at 24 h (Fig. 4 A). Fig. 5 B illustrates that administration of both P-selectin and E-selectin antibodies together (but not separately) completely inhibited the subsequent adhesion of leukocytes within the vessels. This lack of inflammatory cell infiltrate was confirmed using histology that revealed no signs of inflammation or leukocyte recruitment (Fig. 5 C). These data clearly demonstrate that the leukocyte recruitment in the initiation phase of CS modifies the microenvironment to allow the α4-integrin bearing leukocytes to roll and adhere at 24 h independent of selectins. Inhibiting the selectin-dependent leukocyte recruitment in the early phase completely blocked the α4-integrin–dependent leukocyte recruitment in the late phase.
Figure 5.
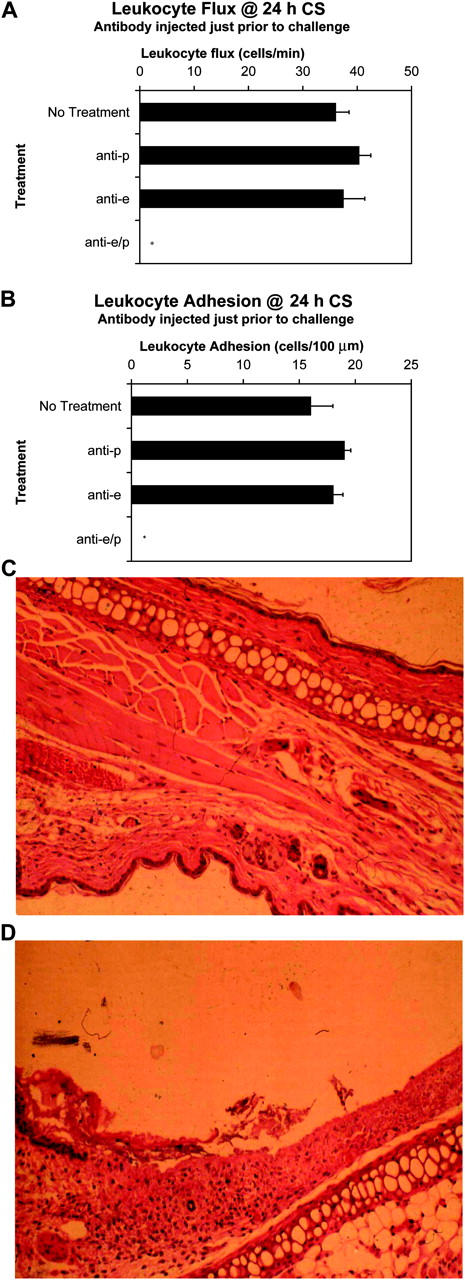
Inhibition of leukocyte recruitment within 2 h of challenge abrogates later inflammation. Antibodies against E- and/or P-selectin were injected 24 h before antigen challenge. Subsequent leukocyte rolling flux (A) and leukocyte adhesion (B) were assessed by intravital microscopy at 24 h of CS. A minimum of three animals was used at each time point (*, P < 0.05 relative to no treatment group). Histological sections of each animal were taken. Shown is a representative H&E–stained ear (at 24 h of CS) of a mouse treated with both antiselectin antibodies (C, magnification, 100) compared with that of an untreated control at 24 h of CS (D, magnification, 100).
To further demonstrate that the inhibition of leukocyte recruitment at 24 h was absolutely dependent on the leukocyte rolling and adhesion that occur at 2 h, we challenged mice for 2 h and administered anti–E- and anti–P-selectin antibodies at 2 h after antigen challenge. Fig. 6 (A and B) demonstrates that this approach yielded no significant reduction in either leukocyte flux or leukocyte adhesion at 24 h. Together, these results confirm that the first 2 h of leukocyte rolling and adhesion in the early phase modified the challenged tissue in a manner absolutely necessary for leukocyte recruitment in the late phase.
Figure 6.

Inhibition of leukocyte recruitment after 2 h of challenge does not abrogate later inflammation. Anti–E-selectin and anti–P-selectin antibodies were injected immediately before antigen challenge or 2 h after challenge. Leukocyte rolling flux (A) and leukocyte adhesion (B) were assessed at 24 h of CS. A minimum of three animals was used at each time point (*, P < 0.05 relative to E-/P-selectin antibody at 2 h after of challenge).
In addition to the leukocyte–endothelial measures of CS observed by intravital microscopy, experiments were performed to measure more conventional indicators of CS reactions. These included changes in ear thickness (Fig. 7 A) and tissue concentrations of the inflammatory cytokine IFN-γ (Fig. 7 B). Notably, results of these experiments paralleled those performed with intravital microscopy. Both increases in ear thickness and increased levels of IFN-γ at 24 h of CS could be attenuated by the addition of anti–E- and anti–P-selectin antibodies before antigen challenge, but not by the same antibody treatment 2 h after antigen challenge.
Early Neutrophil Recruitment Is Not Required for Late Phase Inflammation.
In a recent paper, we reported that neutrophil depletion prevented subsequent lymphocyte recruitment in a model of hepatitis (38). To test for a similar mechanism, neutrophils were depleted through the use of an antineutrophil antibody that was injected i.p. 24 h before antigen challenge. Differential leukocyte counts performed at 2 and 24 h after antigen challenge confirmed that the neutrophil population had been depleted to 98–99% versus controls. Intravital microscopy revealed that a significant inhibition in leukocyte rolling and adhesion occurred at 2 h (Fig. 8 A). The inhibition was ∼70–80%, which was entirely consistent with the fact that ∼70% of recruited leukocytes at 2 h are neutrophils. However, at 24 h, a very minor decrease in the late phase was noted, which was consistent with the view that the early recruitment of neutrophils was not responsible for the subsequent transformation of the microenvironment that permits later mononuclear cell recruitment (Fig. 8 B).
Figure 8.
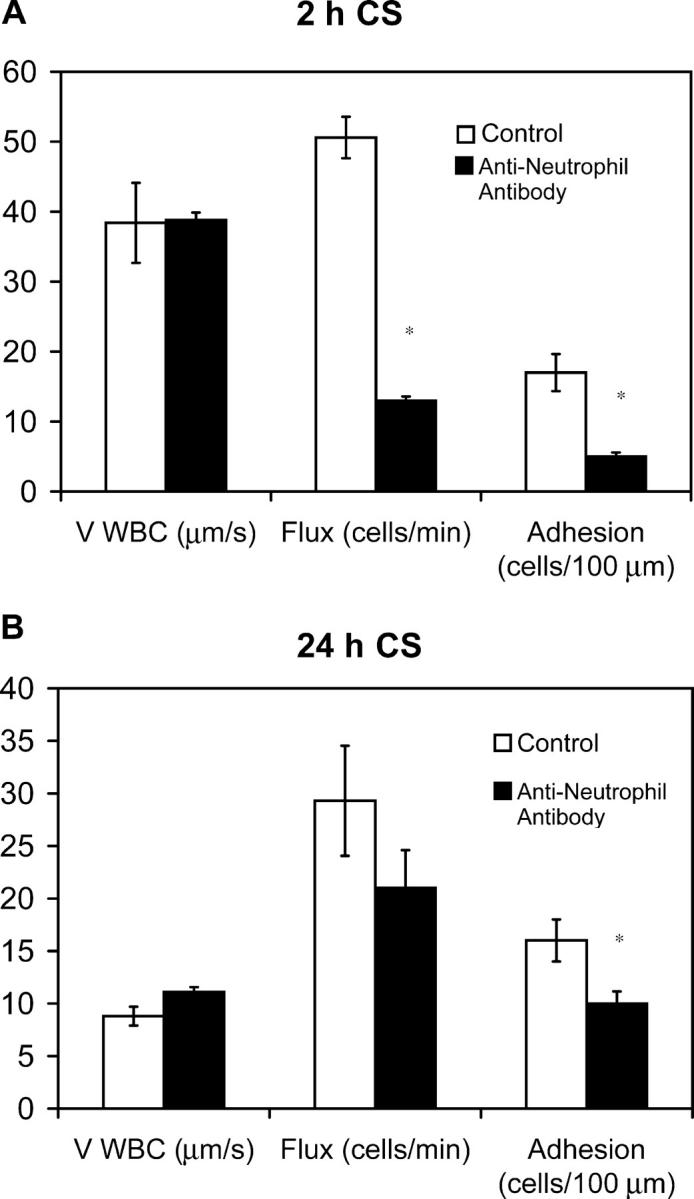
Early neutrophil recruitment is not required for late phase inflammation. Mice were injected with an antineutrophil mAb i.p. 24 h before antigen challenge. Leukocyte–endothelial interactions (rolling flux, rolling velocity, and cell adhesion) were assessed by intravital microscopy at 2 (A) and 24 h (B) of CS. A minimum of three animals was used at each time point (*, P < 0.05 relative to control group).
Early Recruitment of CD4+ Lymphocytes Is Essential for Late Phase Inflammation.
Because a second, albeit much more minor, population of leukocytes seen at 2 h were lymphocytes, we examined the importance of recruitment of these cells to the late-phase response. C57BL/6-Cd4tm4Mak mice lack CD4+ lymphocytes (39). We showed that leukocyte adhesion at 24 h of CS in C57BL/6-Cd4tm4Mak mice was greatly attenuated (Fig. 9 A). Adoptive transfer of CD4+ lymphocytes from unsensitized C57BL/6 mice into sensitized C57BL/6-Cd4tm4Mak mice just before antigen challenge did not confer the ability to mount a significant CS reaction at 24 h. However, the adoptive transfer of CD4+ lymphocytes from sensitized C57BL/6 mice into sensitized C57BL/6-Cd4tm4Mak mice just before antigen challenge restored leukocyte adhesion at 24 h of CS to levels seen in wild-type mice. However, when these same CD4+ lymphocytes from sensitized C57BL/6 mice were adoptively transferred into sensitized C57BL/6-Cd4tm4Mak mice 2 h after antigen challenge, no leukocyte adhesion was seen with intravital microscopy at 24 h of CS. The data support that late-phase leukocyte adhesion at 24 h of CS is dependent on the recruitment of CD4+ lymphocytes within the initial 2 h after antigen challenge.
Figure 9.
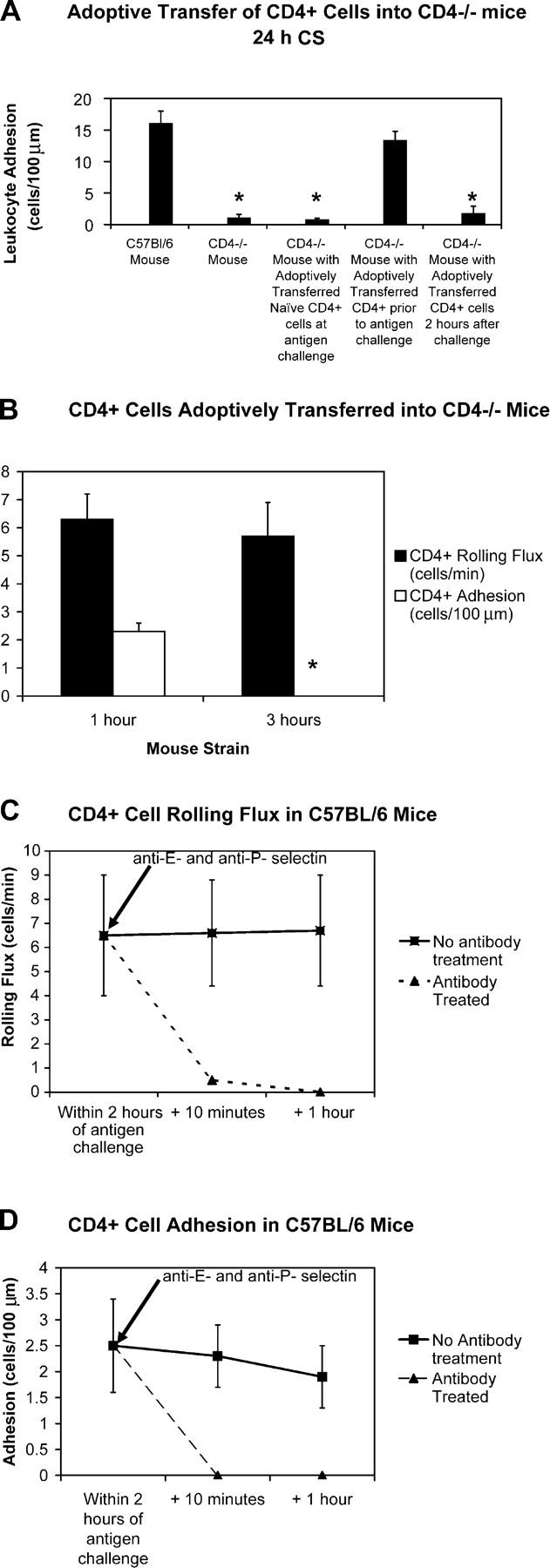
CD4+ lymphocytes must be recruited within 2 h of challenge for late phase inflammation to occur. C57BL/6-Cd4tm4Mak lack CD4+ cells. Leukocyte adhesion was assessed by intravital microscopy at 24 h of CS in wild-type mice (C57BL/6), C57BL/6-Cd4tm4Mak mice, C57BL/6-Cd4tm4Mak mice with CD4+ cells from unsensitized C57BL/6 mice adoptively transferred at the time of antigen challenge, C57BL/6-Cd4tm4Mak mice with CD4+ cells from previously sensitized C57BL/6 mice adoptively transferred at the time of antigen challenge, and C57BL/6-Cd4tm4Mak mice with CD4+ cells from previously sensitized C57BL/6 mice adoptively transferred 2 h after antigen challenge (A). Also, sensitized CD4+ cells from wild-type mice were adoptively transferred into sensitized and challenged C57BL/6-Cd4tm4Mak mice either 1 or 3 h after antigen challenge (B). A minimum of three animals was used in each experimental group (*, P < 0.05 relative to control group). Rhodamine 6G–labeled CD4+ cells from previously sensitized C57BL/6 mice were adoptively transferred into different C57BL/6 mice within the first 2 h of CS, and leukocyte rolling flux (C) and adhesion (D) were assessed by intravital microscopy. Leukocyte rolling flux was completely abrogated by the addition of anti–P- and anti–E-selectin antibodies.
To further elaborate this process, fluorescently labeled CD4+ lymphocytes from sensitized C57BL/6 mice were injected i.v. into sensitized C57BL/6-Cd4tm4Mak mice at 1 h (within 2 h of antigen challenge) or 3 h (outside 2 h of antigen challenge) after antigen challenge and assessed by intravital microscopy (Fig. 9 B). A small number of CD4+ lymphocytes injected at 1 h were observed to roll with 1–2 cells adhering per minute. Although a similar number of CD4+ lymphocytes was observed to roll when injected at 3 h, no cells were adherent. Our data suggest that CD4+ lymphocytes recruited within 2 h of antigen challenge are capable of being recruited into the extravascular space; however, if these same CD4+ lymphocytes are prevented from interacting with endothelium for >2 h after antigen challenge, they are no longer able to home to skin.
We further demonstrated that early endothelial interactions by CD4+ lymphocytes were mediated by E- and P-selectin (Fig. 9, C and D). We fluorescently labeled CD4+ lymphocytes isolated from sensitized C57BL/6 mice and adoptively transferred the cells into different C57BL/6 mice within the first 2 h of CS. These cells were observed to immediately roll and adhere in the dermal microvasculature. This CD4+ lymphocyte rolling and adhesion was completely abrogated by the addition of anti–E- and anti–P-selectin antibodies.
Discussion
By visualizing the microvasculature during CS, we identified the trafficking and recruitment of two temporally distinct nonoverlapping populations of leukocytes (one present at 0–2 h after challenge, and the other present from 4 to 24 h) separated by ∼1 h in which leukocyte recruitment was absent. The two phases demonstrated different adhesive profiles; leukocyte rolling during the initial 2 h of CS was dependent on E- and P-selectin, whereas the dominant rolling molecule at 24 h was α4-integrin. This difference allowed us to study the importance of inhibiting leukocyte recruitment in the early phase on leukocyte recruitment in the late phase. We established that inhibition of the initiation phase of leukocyte–endothelial cell interactions by immunoneutralizing E- and P-selectin led to the abrogation of the later phase inflammatory response despite its lack of absolute dependence on the selectins. We further demonstrated that despite an early predominance of neutrophils in the leukocyte infiltrate, elimination of neutrophils early in CS did not prevent leukocyte infiltration at 24 h. Finally, our data reveal that during the initiation phase, E/P-selectin–dependent endothelial cell interactions with CD4+ lymphocytes are required for leukocyte recruitment later in CS. Moreover, the CD4+ lymphocytes only had access to the tissue for the first 2 h of challenge, after which the lymphocytes were no longer able to infiltrate the afflicted tissue.
Over the last 20 yr, Askenase and colleagues in elegant experiments have highlighted that there exists an initiation phase, which, when abrogated, leads to inhibition of later inflammation (3, 16, 19). At the time of secondary antigen challenge to elicit CS, antigen binds IgM (14) derived from B-1 cells at the time of primary antigen immunization (10). These IgM–antigen complexes activate the complement cascade, leading to the local elaboration of C5a (12, 13). This classic anaphylotoxin is thought to interact with C5a receptors on mast cells (and/or platelets) that release vasoactive mediators, including TNF-α and serotonin (8, 11, 12). In addition to increasing vascular permeability, these mediators may activate postcapillary venules, inducing the expression of adhesion molecules such as VCAM-1, ICAM-1, P-selectin, and E-selectin on the luminal surface (8, 37). In fact, it has been generally postulated, but never demonstrated, that de novo synthesis or up-regulation of presynthesized endothelial adhesion molecules was critical for both the early and later phase of leukocyte recruitment. However, in our ear model, which required no invasive surgery and therefore no trauma-induced adhesion molecule expression, our data reveal that constitutive P-selectin and E-selectin are responsible for the early CD4+ lymphocyte recruitment. In fact, we provide direct evidence that constitutively expressed endothelial selectins recruit these CD4+ lymphocytes, which produce factors that either directly or indirectly modify the endothelium to permit for subsequent late phase recruitment. This is consistent with the view that VCAM-1, P-selectin, and E-selectin are not up-regulated sooner than 3–4 h (8, 37) and that cytokines such as TNF require this time frame to induce these adhesion molecules (8). Clearly, these data are also important from a therapeutic perspective. Indeed, targeting either endothelial selectin alone will not suffice, and targeting selectins after challenge will not inhibit the late phase of CS. However, if selectins can be targeted prophylactically, this may be a potent therapeutic intervention.
It has been noted previously that very few cells are recruited into the extravascular space within the first 2 h of CS (12). However, our results clearly demonstrate that inhibition of the early recruitment of CD4+ lymphocytes via inhibition of both endothelial selectins disrupted the ability of cells to infiltrate afflicted tissue in the late phase. The early recruitment of CD4+ lymphocytes had to modify the microenvironment to allow for the later recruitment of α4-integrin–dependent leukocytes. One possibility is that the early leukocyte–endothelial cell interaction activated the endothelium to express VCAM-1 (the α4-integrin ligand). Indeed, it has been shown that VCAM-1 expression is elevated by 4 h after CS elicitation (8), the same time that we observed the beginning of a later peak of leukocyte recruitment (Fig. 1 D). In addition, it is possible that the early infiltration of leukocytes induced the local production of chemokines that specifically recruited α4-integrin–bearing lymphocytes. Although the mechanism by which the CD4+ lymphocytes appear to affect endothelium is unclear, we speculate that production of IFN-γ, TNF-α, and perhaps IL-4 from CD4+ T lymphocytes and NKT cells would induce expression of adhesion molecules and chemokines necessary for the late phase. Alternatively, the CD4+ lymphocytes could release factors to activate mast cells to induce endothelial changes.
Interestingly, our data would strongly suggest that, regardless of the identity of the chemoattractant that recruited CD4+ lymphocytes, it had a very limited life span; the addition of CD4+ lymphocytes at challenge (but not at 2 h after challenge) induced the 24-h late-phase response. Numerous chemoattractants have been identified as important in CS. Indeed, Askenase and colleagues reported that the early recruitment of leukocytes in CS was C5a dependent (12, 13). Homey et al. reported that CCL27 (CTACK) partially (37%) inhibits T cell homing to skin after allergen challenge (40). Finally, MCP-1 (41, 42), IP-10 (41, 43, 44), MIP-1α, and MIP-1β (45) have all been detected in inflamed skin at both 2 and 24 h of allergen. We would argue that the latter chemokines did not have the necessary limited 2-h profile. Although at this stage it is unclear which chemokine was activated and rapidly inactivated to induce lymphocyte recruitment for only 2 h, C5a has been shown to be a lymphocyte chemoattractant. Tsuji et al. reported that addition of C5a inhibitors at the time of challenge, but not at a later time after challenge, was sufficient to inhibit the late phase response (12) entirely consistent with our lymphocyte homing data. Whether the C5a directly recruits lymphocytes or whether it activates endothelium, mast cells, or macrophages to release a chemokine remains to be elucidated.
An interesting question is why is there such a small 2-h window of opportunity for lymphocytes to infiltrate the inflamed tissue. Although many investigators have argued that this is a simple progression of inflammation, our intravital data reveal that at 2–3 h, there is complete cessation of leukocyte adhesion and therefore inhibition of recruitment of cells into the tissues. By 4 h, a second wave of leukocytes is initiated. This could be a very important regulatory mechanism, such that the early 2-h recruitment is quite nonspecific with multiple cell types recruited, but unless appropriate lymphocytes are recruited, the inflammation is rapidly terminated. However, if appropriate CD4+ lymphocytes are recruited, expression of the α4-integrin ligand, VCAM-1, initiates the recruitment of α4-integrin–dependent lymphocytes, and subsequent late phase is initiated.
Our data are partly consistent with studies suggesting that oxazolone-induced CS is attenuated in mice deficient in both E- and P-selectin (27). However, in those studies, it was not possible to discern that it was only in the first 2 h of CS that P-selectin and E-selectin were critical. Using intravital microscopy and visualizing the microcirculation allowed us to discern two very distinct microvascular phases in CS and that only the first phase was inhibitable with E-/P-selectin antibody. We were surprised to find that in E-/P-selectin double deficient mice, only 50% of the late phase was inhibited (27), whereas in our analysis >90% of leukocyte recruitment was blocked in mice receiving anti–E- and anti–P-selectin antibodies. Indeed, if in the initiation phase, leukocyte recruitment is dependent on E- and P-selectin, and recruitment of these cells is required for later phase inflammation, no CS should be seen in E-/P-selectin double knockout mice. However, when we used E-/P-selectin–deficient mice (unpublished data), we found that they had basal rolling dependent on α4-integrin and, unlike with antibodies in wild-type mice, some cells were recruited during the initial phase in E-/P-selectin–deficient mice. This recruitment was entirely inhibitable with α4-integrin antibody (unpublished data). Clearly, the expression pattern of VCAM-1 in E-/P-selectin double knockout mice differs from that of wild-type mice.
The early recruitment of neutrophils in many inflammatory conditions is thought to modify the microenvironment and/or endothelial surface to induce subsequent mononuclear cell recruitment (20–22). We found that neutrophils were the predominant leukocyte cell type recruited at 2 h of CS and hypothesized that they might modify the local microenvironment to allow for later leukocyte infiltration. In fact, we have observed exactly this mechanism in concavalin A–induced hepatitis (38). However, our results clearly showed that elimination of the neutrophil population early in CS did not abrogate later inflammation (Fig. 8 B). The recruitment of neutrophils in this particular case is arguably incidental, perhaps a result of the elaboration of C5a.
In conclusion, we have established that in the initiation phase, leukocyte–endothelial cell interaction is mandatory for late-phase inflammation to occur. Inhibition of early selectin-dependent leukocyte rolling led to the abrogation of late phase leukocyte recruitment. Despite a predominance of neutrophils in the early cellular infiltrate, elimination of neutrophils did not abrogate late phase CS; rather, we believe that it is the early recruitment of CD4+ lymphocytes through E-selectin and P-selectin that modify the local microenvironment for the future recruitment of other α4-integrin–bearing lymphocytes.
Acknowledgments
We thank Dr. S. Urbanski for his help in analyzing histology and the Canadian Institutes of Health for operating funds.
J. Hwang is supported in part by the Surgeon Scientist Program at the University of Calgary, the Alberta Heritage Foundation for Medical Research, and the Canadian Institutes of Health Training Program in Immunology, Immunopathogenesis, and Inflammation at the University of Calgary. P. Kubes is a Canada Research Chair and a Scientist of the Alberta Heritage Foundation for Medical Research.
The online version of this article contains supplemental material.
Abbreviation used in this paper: CS, contact sensitivity.
References
- 1.van Loveren, H., R. Meade, and P.W. Askenase. 1983. An early component of delayed-type hypersensitivity mediated by T cells and mast cells. J. Exp. Med. 157:1604–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Loveren, H., S. Kraeuter-Kops, and P.W. Askenase. 1984. Different mechanisms of release of vasoactive amines by mast cells occur in T cell-dependent compared to IgE-dependent cutaneous hypersensitivity responses. Eur. J. Immunol. 14:40–47. [DOI] [PubMed] [Google Scholar]
- 3.Matsuda, H., H. Ushio, V. Paliwal, W. Ptak, and P.W. Askenase. 1995. Adoptive cell transfer of contact sensitivity-initiation mediated by nonimmune cells sensitized with monoclonal IgE antibodies. Dependence on host skin mast cells. J. Immunol. 154:5080–5092. [PubMed] [Google Scholar]
- 4.Geba, G.P., W. Ptak, G.M. Anderson, V. Paliwal, R.E. Ratzlaff, J. Levin, and P.W. Askenase. 1996. Delayed-type hypersensitivity in mast cell-deficient mice: dependence on platelets for expression of contact sensitivity. J. Immunol. 157:557–565. [PubMed] [Google Scholar]
- 5.Matsuda, H., H. Ushio, G.P. Geba, and P.W. Askenase. 1997. Human platelets can initiate T cell-dependent contact sensitivity through local serotonin release mediated by IgE antibodies. J. Immunol. 158:2891–2897. [PubMed] [Google Scholar]
- 6.Ptak, W., G.P. Geba, and P.W. Askenase. 1991. Initiation of delayed-type hypersensitivity by low doses of monoclonal IgE antibody. Mediation by serotonin and inhibition by histamine. J. Immunol. 146:3929–3936. [PubMed] [Google Scholar]
- 7.Meade, R., H. Van Lovern, H. Parmentier, G.M. Iverson, and P.W. Askenase. 1988. The antigen-binding T cell factor PCl-F sensitizes mast cells for in vitro release of serotonin. Comparison with monoclonal IgE antibody. J. Immunol. 141:2704–2713. [PubMed] [Google Scholar]
- 8.McHale, J.F., O.A. Harari, D. Marshall, and D.O. Haskard. 1999. Vascular endothelial cell expression of ICAM-1 and VCAM-1 at the onset of eliciting contact hypersensitivity in mice: evidence for a dominant role of TNF-alpha. J. Immunol. 162:1648–1655. [PubMed] [Google Scholar]
- 9.Ferreri, N.R., I. Millet, V. Paliwal, W. Herzog, D. Solomon, R. Ramabhadran, and P.W. Askenase. 1991. Induction of macrophage TNF alpha, IL-1, IL-6, and PGE2 production by DTH-initiating factors. Cell. Immunol. 137:389–405. [DOI] [PubMed] [Google Scholar]
- 10.Paliwal, V., R.F. Tsuji, M. Szczepanik, I. Kawikova, R.A. Campos, M. Kneilling, M. Rocken, J. Schuurman, F.A. Redegeld, F.P. Nijkamp, and P.W. Askenase. 2002. Subunits of IgM reconstitute defective contact sensitivity in B-1 cell-deficient xid mice: kappa light chains recruit T cells independent of complement. J. Immunol. 169:4113–4123. [DOI] [PubMed] [Google Scholar]
- 11.Askenase, P.W. 2001. Yes T cells, but three different T cells (alphabeta, gammadelta and NK T cells), and also B-1 cells mediate contact sensitivity. Clin. Exp. Immunol. 125:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuji, R.F., I. Kawikova, R. Ramabhadran, M. Akahira-Azuma, D. Taub, T.E. Hugli, C. Gerard, and P.W. Askenase. 2000. Early local generation of C5a initiates the elicitation of contact sensitivity by leading to early T cell recruitment. J. Immunol. 165:1588–1598. [DOI] [PubMed] [Google Scholar]
- 13.Tsuji, R.F., M. Kikuchi, and P.W. Askenase. 1996. Possible involvement of C5/C5a in the efferent and elicitation phases of contact sensitivity. J. Immunol. 156:4444–4450. [PubMed] [Google Scholar]
- 14.Tsuji, R.F., M. Szczepanik, I. Kawikova, V. Paliwal, R.A. Campos, A. Itakura, M. Akahira-Azuma, N. Baumgarth, L.A. Herzenberg, and P.W. Askenase. 2002. B cell–dependent T cell responses: IgM antibodies are required to elicit contact sensitivity. J. Exp. Med. 196:1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garssen, J., H. van Loveren, K. Kato, and P.W. Askenase. 1994. Antigen receptors on Thy-1+ CD3− CS-initiating cell. In vitro desensitization with hapten-amino acid or hapten-Ficoll conjugates, versus hapten-protein conjugates, suggests different antigen receptors on the immune cells that mediate the early and late components of murine contact sensitivity. J. Immunol. 153:32–44. [PubMed] [Google Scholar]
- 16.Ptak, W., W.R. Herzog, and P.W. Askenase. 1991. Delayed-type hypersensitivity initiation by early-acting cells that are antigen mismatched or MHC incompatible with late-acting, delayed-type hypersensitivity effector T cells. J. Immunol. 146:469–475. [PubMed] [Google Scholar]
- 17.Ptak, W., M. Bereta, M. Ptak, and P.W. Askenase. 1986. Isotype-like suppression of T cell-mediated immunity in vivo. I. Delayed-type hypersensitivity specificity of T cell suppression induced by antigen-binding T cell factors that initiate contact sensitivity. J. Immunol. 136:1554–1563. [PubMed] [Google Scholar]
- 18.van Loveren, H., K. Kato, R. Meade, D.R. Green, M. Horowitz, W. Ptak, and P.W. Askenase. 1984. Characterization of two different Ly-1+ T cell populations that mediate delayed-type hypersensitivity. J. Immunol. 133:2402–2411. [PubMed] [Google Scholar]
- 19.van Loveren, H., and P.W. Askenase. 1984. Delayed-type hypersensitivity is mediated by a sequence of two different T cell activities. J. Immunol. 133:2397–2401. [PubMed] [Google Scholar]
- 20.Scapini, P., J.A. Lapinet-Vera, S. Gasperini, F. Calzetti, F. Bazzoni, and M.A. Cassatella. 2000. The neutrophil as a cellular source of chemokines. Immunol. Rev. 177:195–203. [DOI] [PubMed] [Google Scholar]
- 21.Kudo, C., T. Yamashita, A. Araki, M. Terashita, T. Watanabe, M. Atsumi, M. Tamura, and F. Sendo. 1993. Modulation of in vivo immune response by selective depletion of neutrophils using a monoclonal antibody, RP-3. I. Inhibition by RP-3 treatment of the priming and effector phases of delayed type hypersensitivity to sheep red blood cells in rats. J. Immunol. 150:3728–3738. [PubMed] [Google Scholar]
- 22.Chertov, O., D.F. Michiel, L. Xu, J.M. Wang, K. Tani, W.J. Murphy, D.L. Longo, D.D. Taub, and J.J. Oppenheim. 1996. Identification of defensin-1, defensin-2, and CAP37/azurocidin as T-cell chemoattractant proteins released from interleukin-8-stimulated neutrophils. J. Biol. Chem. 271:2935–2940. [DOI] [PubMed] [Google Scholar]
- 23.Kops, S.K., H. van Loveren, R.W. Rosenstein, W. Ptak, and P.W. Askenase. 1984. Mast cell activation and vascular alterations in immediate hypersensitivity-like reactions induced by a T cell-derived antigen-binding factor. Laboratory Investigation. 50:421–434. [PubMed] [Google Scholar]
- 24.Kubes, P., and S.M. Kerfoot. 2001. Leukocyte recruitment in the microcirculation: the rolling paradigm revisited. News Physiol. Sci. 16:76–80. [DOI] [PubMed] [Google Scholar]
- 25.Granger, D.N., and P. Kubes. 1994. The microcirculation and inflammation: modulation of leukocyte-endothelial cell adhesion. J. Leukoc. Biol. 55:662–675. [PubMed] [Google Scholar]
- 26.Kubes, P., and P.A. Ward. 2000. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 10:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staite, N.D., J.M. Justen, L.M. Sly, A.L. Beaudet, and D.C. Bullard. 1996. Inhibition of delayed-type contact hypersensitivity in mice deficient in both E-selectin and P-selectin. Blood. 88:2973–2979. [PubMed] [Google Scholar]
- 28.Issekutz, A.C., and T.B. Issekutz. 2002. The role of E-selectin, P-selectin, and very late activation antigen-4 in T lymphocyte migration to dermal inflammation. J. Immunol. 168:1934–1939. [DOI] [PubMed] [Google Scholar]
- 29.Chisholm, P.L., C.A. Williams, and R.R. Lobb. 1993. Monoclonal antibodies to the integrin alpha-4 subunit inhibit the murine contact hypersensitivity response. Eur. J. Immunol. 23:682–688. [DOI] [PubMed] [Google Scholar]
- 30.Johnston, B., and P. Kubes. 1999. The alpha4-integrin: an alternative pathway for neutrophil recruitment? Immunol. Today. 20:545–550. [DOI] [PubMed] [Google Scholar]
- 31.Hickey, M.J., D.N. Granger, and P. Kubes. 1999. Molecular mechanisms underlying IL-4-induced leukocyte recruitment in vivo: a critical role for the alpha 4 integrin. J. Immunol. 163:3441–3448. [PubMed] [Google Scholar]
- 32.Grabovsky, V., S. Feigelson, C. Chen, D.A. Bleijs, A. Peled, G. Cinamon, F. Baleux, F. Arenzana-Seisdedos, T. Lapidot, Y. van Kooyk, R.R. Lobb, and R. Alon. 2000. Subsecond induction of α4 integrin clustering by immobilized chemokines stimulates leukocyte tethering and rolling on endothelial vascular cell adhesion molecule 1 under flow conditions. J. Exp. Med. 192:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robert, C., R.C. Fuhlbrigge, J.D. Kieffer, S. Ayehunie, R.O. Hynes, G. Cheng, S. Grabbe, U.H. von Andrian, and T.S. Kupper. 1999. Interaction of dendritic cells with skin endothelium: a new perspective on immunosurveillance. J. Exp. Med. 189:627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayrovitz, H.N. 1992. Leukocyte rolling: a prominent feature of venules in intact skin of anesthetized hairless mice. Am. J. Physiol. 262:H157–H161. [DOI] [PubMed] [Google Scholar]
- 35.Nolte, D., P. Schmid, U. Jager, A. Botzlar, F. Roesken, R. Hecht, E. Uhl, K. Messmer, and D. Vestweber. 1994. Leukocyte rolling in venules of striated muscle and skin is mediated by P-selectin, not by L-selectin. Am. J. Physiol. 267:H1637–H1642. [DOI] [PubMed] [Google Scholar]
- 36.Janssen, G.H., G.J. Tangelder, M.G. Oude Egbrink, and R.S. Reneman. 1994. Spontaneous leukocyte rolling in venules in untraumatized skin of conscious and anesthetized animals. Am. J. Physiol. 267:H1199–H1204. [DOI] [PubMed] [Google Scholar]
- 37.Harari, O.A., J.F. McHale, D. Marshall, S. Ahmed, D. Brown, P.W. Askenase, and D.O. Haskard. 1999. Endothelial cell E- and P-selectin up-regulation in murine contact sensitivity is prolonged by distinct mechanisms occurring in sequence. J. Immunol. 163:6860–6866. [PubMed] [Google Scholar]
- 38.Bonder, C.S., M.N. Ajuebor, L.D. Zbytnuik, P. Kubes, and M.G. Swain. 2004. Essential role for neutrophil recruitment to the liver in concavalin A-induced hepatitis. J. Immunol. 172:45–53. [DOI] [PubMed] [Google Scholar]
- 39.Rahemtulla, A., W.P. Fung-Leung, M.W. Schilham, T.M. Kundig, S.R. Sambhara, A. Narendran, A. Arabian, A. Wakeham, C.J. Paige, and R.M. Zinkernagel. 1991. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature. 353:180–184. [DOI] [PubMed] [Google Scholar]
- 40.Homey, B., H. Alenius, A. Muller, H. Soto, E.P. Bowman, W. Yuan, L. McEvoy, A.I. Lauerma, T. Assmann, E. Bunemann, et al. 2002. CCL27-CCR10 interactions regulate T cell-mediated skin inflammation. Nat. Med. 8:157–165. [DOI] [PubMed] [Google Scholar]
- 41.Enk, A.H., and S.I. Katz. 1992. Early molecular events in the induction phase of contact sensitivity. Proc. Natl. Acad. Sci. USA. 89:1398–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luster, A.D., and M.E. Rothenberg. 1997. Role of the monocyte chemoattractant protein and eotaxin subfamily of chemokines in allergic inflammation. J. Leukoc. Biol. 62:620–633. [DOI] [PubMed] [Google Scholar]
- 43.Baggiolini, M., and C.A. Dahinden. 1994. CC chemokines in allergic inflammation. Immunol. Today. 15:127–133. [DOI] [PubMed] [Google Scholar]
- 44.Gautam, S., J. Battisto, J.A. Major, D. Armstrong, M. Stoler, and T.A. Hamilton. 1994. Chemokine expression in trinitrochlorobenzene-mediated contact hypersensitivity. J. Leukoc. Biol. 55:452–460. [DOI] [PubMed] [Google Scholar]
- 45.Goebeler, M., A. Trautmann, A. Voss, E.V. Brocker, A. Toksoy, and R. Gillitzer. 2001. Differential and sequential expression of multiple chemokines during elicitation of allergic contact hypersensitivity. Am. J. Pathol. 158:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]