

Abstract
CD1d-restricted Vα24-Jα18–invariant natural killer T cells (iNKTs) are potentially important in tumor immunity. However, little is known about their localization to tumors. We analyzed 98 untreated primary neuroblastomas from patients with metastatic disease (stage 4) for tumor-infiltrating iNKTs using TaqMan® reverse transcription polymerase chain reaction and immunofluorescent microscopy. 52 tumors (53%) contained iNKTs, and oligonucleotide microarray analysis of the iNKT+ and iNKT− tumors revealed that the former expressed higher levels of CCL2/MCP-1, CXCL12/SDF-1, CCL5/RANTES, and CCL21/SLC. Eight tested neuroblastoma cell lines secreted a range of CCL2 (0–21.6 ng/ml), little CXCL12 (≤0.1 ng/ml), and no detectable CCL5 or CCL21. CCR2, the receptor for CCL2, was more frequently expressed by iNKT compared with natural killer and T cells from blood (P < 0.001). Supernatants of neuroblastoma cell lines that produced CCL2 induced in vitro migration of iNKTs from blood of patients and normal adults; this was abrogated by an anti-CCL2 monoclonal antibody. CCL2 expression by tumors was found to inversely correlate with MYCN proto-oncogene amplification and expression (r = 0.5, P < 0.001), and MYCN-high/CCL2-low expression accurately predicted the absence of iNKTs (P < 0.001). In summary, iNKTs migrate toward neuroblastoma cells in a CCL2-dependent manner, preferentially infiltrating MYCN nonamplified tumors that express CCL2.
Keywords: lymphocytes, tumor-infiltrating, T lymphocyte subsets, cell movement, immunologic surveillance
Introduction
Vα24-Jα18–invariant natural killer T cells (iNKTs) are an evolutionarily conserved subset of T cells that depend on CD1d for their development and that specifically recognize α-galactosylceramide (αGalCer) bound to CD1d. iNKTs respond to specific stimulation by rapidly producing a spectrum of cytokines including IL-13 and IL-4 (type 2) and IFN-γ (type 1; references 1, 2). iNKTs may be a part of the innate immune reaction and promote initiation of acquired immune responses (3).
iNKTs are potentially important in tumor immunity (4, 5). They can naturally prevent formation of chemical carcinogen–induced tumors in mice (6, 7) and eradicate established tumors in mice after αGalCer injection by inducing NK cells to produce IL-12 and IFN-γ (8). However, a murine model of tumor growth-regression-recurrence suggested that, without exogenous stimulation, iNKTs may prevent CTL-mediated tumor eradication by producing IL-13 (9, 10). Recent phase I clinical trials with αGalCer-pulsed dendritic cells demonstrated that in vivo stimulation of iNKTs leads to modulation of NK, T, and B cell numbers and increased serum IFN-γ (11, 12). Presumably, iNKT cell effector functions occur within the tumor microenvironment, but little is known about their infiltration into tumors. Recent studies of human myeloma, lung adenocarcinomas, and lung squamous cell carcinomas demonstrated iNKTs in the tumor microenvironment (13, 14), but the mechanisms of their localization were not investigated. Human iNKTs express chemokine receptors that may enable their migration into extra lymphoid tissues (15); therefore, we hypothesized that they infiltrate tumors in response to one or more chemokines.
Similar to many types of cancer, lymphocytes have been detected in neuroblastomas from patients (16), and most are T cells with varying proportions of CD4+, CD8+, and double negative subsets (17). We determined if some of these infiltrating T cells were iNKTs and if their localization was associated with intratumor expression of chemokines. 42% of neuroblastomas from patients with metastatic disease contain iNKTs, and their presence was highly correlated with expression of the chemokine CCL2. Of note, tumors with genomic amplification and high expression of the MYCN oncogene had low expression of CCL2 and little or no iNKT infiltration. Last, we show that human neuroblastoma cell lines can secrete CCL2, which mediates transendothelial migration (TEM) of fresh blood iNKTs in vitro.
Materials and Methods
Neuroblastomas, Blood Specimens, and Cell Lines.
98 primary untreated stage 4 neuroblastomas were analyzed. 79 tumors, which were diagnosed after 1 yr of age, were obtained from patients enrolled in Children's Cancer Group study CCG-3891 for high-risk neuroblastoma. CCG-3891 used a single induction regimen consisting of cisplatin, doxorubicin, etoposide, and cyclophosphamide as well as surgery. This was followed by randomized assignment to either further chemotherapy (ifosfamide, doxorubicin, etoposide, and cisplatin) or myeloablative chemoradiotherapy (carboplatin, etoposide, melphalan, and total body irradiation) supported by purged autologous bone marrow transplantation. Finally, patients were randomized to receive 13-cis retinoic acid beginning 100 d after autologous bone marrow transplantation or no further treatment (18). 19 tumors were obtained from children who were diagnosed before 1 yr of age and who were enrolled in protocol CCG-3881 for intermediate-risk neuroblastoma. After surgery at diagnosis, these patients received cyclophosphamide, cisplatin, etoposide, and doxorubicin at 28-d intervals for 9 mo.
Specimens were snap frozen within 2 h of surgery in foil or after embedding in OCT compound and were maintained at −80°C. PBMCs were isolated from freshly obtained blood from normal adult donors or newly diagnosed patients with stage 4, high-risk neuroblastoma enrolled in Children's Oncology Group study A3973. LA-N-1 and LA-N-2 neuroblastoma cell lines were established in our laboratory (19), and CHLA-15, CHLA-20, SMS-KAN, SMS-KANR, SMS-KCN, and SMS-KCNR neuroblastoma cell lines were provided by C.P. Reynolds (Childrens Hospital, Los Angeles, CA). All cell lines have been identity typed with the AmpliType assay (Applied Biosystems). iNKT cell lines (>99% CD1d/αGalCer-reactive) were established as described previously (20). Informed consent was obtained in accordance with institutional review board policies and procedures for research dealing with tumors and blood specimens.
Real-Time RT-PCR.
TaqMan® real-time RT-PCR was used to detect Vα24-Jα18 RNA with the Sequence Detection System (ABI Prism model 7700; Applied Biosystems). The probe spanned the junction of Vα24-Jα18 TCR. The forward primer was 5′-CCTCCCAGCTCAGCGATTC-3′, and the reverse primer was 5′-TATAGCCTCCCCAGGGTTGA-3′. The probe was FAM-5′-CCTCCTACATCTGTGTGGTGAGCGACA-3′-TAMTph. The probe design was based on the sequence from GenBank/EMBL/DDBJ accession nos. HSTCRA002 for Vα24 and S63026 for Jα18, respectively. The probe for CCL2 was FAM-5′-CCAAGGAGATCTGTGCTGACCCCAA-3′-TAMTph, and the primers were designed between exon 2 and exon 3. The forward primer was 5′-GAAGAATCACCAGCAGCAAGTGT-3′, and the reverse primer was 5′-TGGAATCCTGAACCCACTTCTG-3′. The probe for MYCN was FAM-5′-AGAAGCGGCGTTCCTCCTCCAACA-3′-TAMTph, and the primers were 5′-GAAGAAATCGACGTGGTCACTG-3′ (forward) and 5′-GGTGAATGTGGTGACAGCCTT-3′ (reverse). The probe and primer designs were based on the sequence from GenBank/EMBL/DDBJ accession no. HSMCP-1. All probes and primers were designed using a software program (Primer Express V1.5; Applied Biosystems) and were synthesized by Integrated DNA Technologies.
Oligonucleotide Array Expression Analysis.
Total RNA was precipitated from samples using TRIzol® (Invitrogen) and further purified with the RNeasy Mini kit (QIAGEN). Double-stranded cDNA was synthesized from total RNA using HPLC purified T7-(dT) primer (5′-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG-(dT)24-3′ (Genset Co.) and purified using phase lock gels, phenol/chloroform extraction, and ethanol precipitation. Biotin-labeled cRNA was synthesized from cDNA (in vitro transcription) using the ENZO Bioarray High Yield RNA Transcript Labeling kit (Affymetrix) and purified with RNeasy columns (QIAGEN) and ethanol precipitation. After quantification with a spectrophotometer, cRNA was fragmented and hybridized to Affymetrix U95Av2 arrays (79 patients, diagnosed after 1 yr of age) or U133A and U133B arrays (19 patients, diagnosed before 1 yr of age). Arrays were washed and scanned according to the manufacturer's instructions (Affymetrix). Data from Affymetrix .cel files were preprocessed using dChip® software to normalize arrays, calculate model-based expression values, and identify probe and array outliers (21). Genetrix® software (provided by J. Buckley, University of Southern California, Los Angeles, CA) was used to obtain gene expression levels, which are termed fluorescence units (FU).
MYCN Genomic Analysis.
MYCN was analyzed to identify tumors with genomic amplification by Southern blot, PCR, and/or immunohistochemistry (IHC) as described previously (22–24).
ELISA.
Kits for CCL2, CCL5, CCL21, and CXCL12 were purchased from R&D Systems, and chemokine concentration was determined in neuroblastoma cell supernatants according to the manufacturer's instructions.
Flow Cytometry.
To analyze CCR2 expression by blood iNKTs, PBMCs were sequentially stained with anti-CCR2 48607.121 mAb (R&D Systems), anti–mouse IgG-biotin and Cy-Chrome-streptavidin plus FITC anti-Vβ11 (Beckman Coulter) and PE anti–Vα24-Jα18 6B11 (25). T and NK cells were analyzed with FITC anti-CD3 UCHT1 and PE anti-CD56 B159 (BD Biosciences). Isotype-matching MOPC-21 mAb was used as control (BD Biosciences). Samples were analyzed with a EPICS® Elite flow cytometer (Beckman Coulter). Data were analyzed using EXPO™ 32 Analysis software (Beckman Coulter).
Immunofluorescence Microscopy and IHC.
Frozen 6-μm sections were brought to room temperature, fixed in acetone (10 min), and treated with 5% goat serum supplemented with 2.5% BSA for 20 min. Sections were incubated with Alexa Fluor® 488 anti-CD3 289-13801 mAb (Molecular Probes) for 1 h at room temperature. The sections were washed with PBS containing 0.5% BSA and counterstained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI; Vector Laboratories). Where indicated, tissue sections were sequentially stained with anti–Vα24-Jα18 6B11 mAb (26, 27) followed by Cy™3-anti–mouse IgG (Jackson ImmunoResearch Laboratories) and Alexa Fluor® 488-anti-CD3 mAb. Digital images of fluorescent microscope fields of tumor sections were acquired with a HCX Plan Apochromat 40×/1.25NA oil immersion objective lens with 1.6× optivar on a DM RXA microscope (Leica), Lambda LS175 Xenon arc lamp (Sutter Instrument Co.), using HQ Cy3, HQ fluorescein, and DAPI filter sets (Chroma Technology Corp.) with a SKY™ SD-300/VDS1300 spectral imager and EasyFish software (Applied Spectral Imaging). All nucleated (DAPI+) and T cells (CD3+) were counted in four randomly selected fields and the percentage of T cells was calculated as CD3+/DAPI+ × 100. For immunohistochemical detection of CCL2, we used goat anti–human CCL2 Ab followed by Cell and Tissue Staining Kit (R&D Systems). IHC for CD1d was performed using 42.1 anti-hCD1d mAb (provided by S. Porcelli, Albert Einstein College of Medicine, Bronx, NY).
TEM Assay.
Polycarbonate transwell filters with 5-μm pore size (Costar Corning) were precoated with human umbilical vein endothelial cells (American Type Culture Collection) using F-12K medium (American Type Culture Collection) supplemented with 10% FCS and 0.03 mg/ml endothelial cell growth supplement (BD Biosciences). After rinsing once in RPMI 1640, transwell inserts were transferred to new wells (lower chambers) containing 600 μl RPMI 1640 with 5 mg/ml human serum albumin. Recombinant chemokines or supernatants from neuroblastoma cell lines were added to the lower chambers. An iNKT cell line (20) or PBMCs depleted of monocytes were added to the upper chamber (106 cells in 100 μl), and the transwell chambers were incubated at 37°C in 5% CO2. After 4 h, initial input cells (from a nonpermeable well) and migrated cells from the lower chambers were stained with fluorochrome-conjugated mAbs (see flow cytometry section). iNKTs were identified using FITC anti-Vβ11 C21 mAb (Beckman Coulter) with PE anti-Vα24-Jα18 6B11. After suspension in TruCOUNT™ tubes (Becton Dickinson), multicolor immunofluorescence was analyzed by flow cytometry to determine relative and absolute numbers of cells in subpopulations of interest. The percent migration for each subset was calculated as migrated cells/input cells × 100. Specific migration was calculated by subtracting migration to control medium from migration to the experimental medium.
Data Analyses.
Flow cytometry data were analyzed with EXPO™ 32 Analysis software (Beckman Coulter). SigmaPlot 8.0 (Jandell Scientific) was used to create graphs, and GraphPad Prism™ 4.0 (GraphPad Software) was used to perform Student's t test, Mann-Whitney test, or one-way analysis of variance with the Tukey-Kramer posttest comparison of group means. Correlation was analyzed by the Spearman correlation analysis. Significance was accepted when P < 0.05. To determine the optimum cutoff value, the maximally selected χ2 method of Miller and Halpern was adapted. To determine the p-value associated with the maximum χ2 statistics, we performed 2,000 bootstrap-like simulations. For each simulation, a randomly selected expression value was drawn from the set of observed expression values and assigned to each of the observed responses (i.e., presence of iNKTs). The corrected p-value was calculated as the proportion of the 2,000 simulated maximal χ2 statistics that was larger than the original maximal χ2 test statistic.
Results
Detection and Enumeration of iNKTs Infiltrating Primary Untreated Neuroblastomas.
Because the invariant Vα24-Jα18 rearrangement specifically identifies iNKTs, we designed a Taqman® probe/primer set to span and amplify a Vα24-Jα18 sequence. The strict specificity and high sensitivity of this set were established using αGalCer-reactive iNKT and neuroblastoma cell lines as positive and negative controls, respectively (unpublished data). To standardize RT-PCR–based iNKT cell quantification, purified iNKTs were serially diluted with neuroblastoma cells (LA-N-1 cell line) and analyzed with RT-PCR and flow cytometry to determine iNKT cell RNA concentration and frequency, respectively (Fig. 1 A). iNKT RNA concentration linearly correlated with iNKT cell frequency in the range from 0.01 to 25% (r = 0.99, P < 0.001), which provided a standard curve for subsequent analyses of iNKT cell frequency in tumor specimens by RT-PCR.
Figure 1.
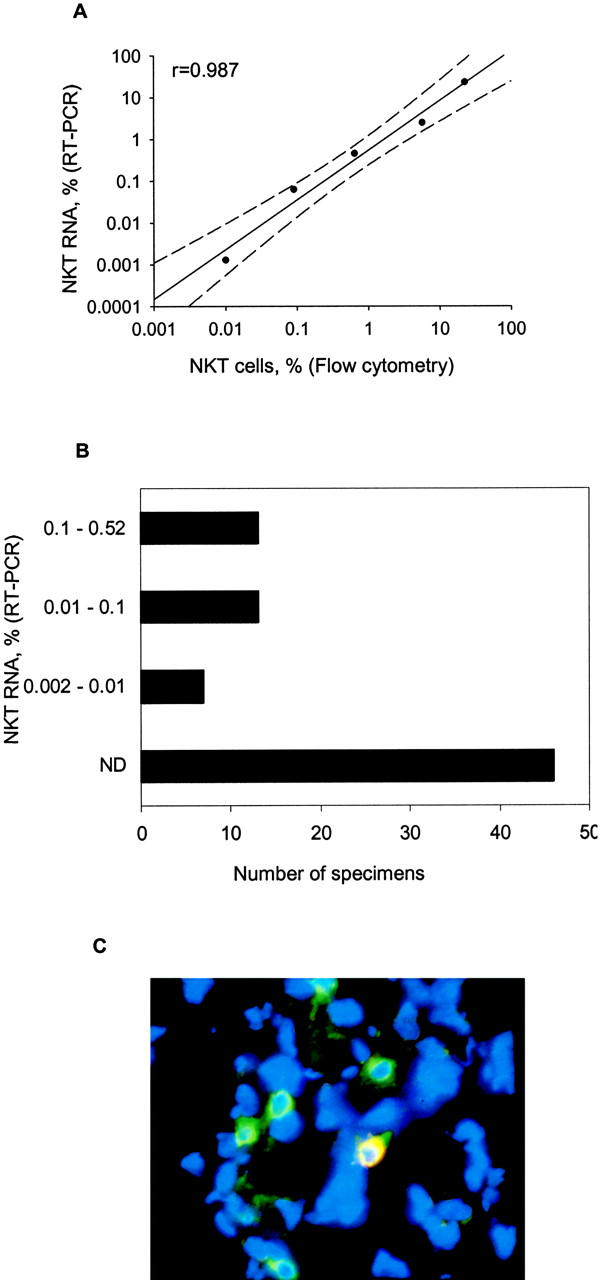
Detection and enumeration of tumor-infiltrating iNKTs. (A) iNKT cells (>99% pure) were serially diluted with neuroblastoma cells (LA-N-1 cell line) from 1:5 to 1:50,000, a iNKT/neuroblastoma cell ratio. RT-PCR for Vα24-Jα18 RNA and flow cytometry for iNKT TCR antigens were performed using cells from the same preparations. iNKT RNA percentage (y axis) is calculated as iNKT RNA ng/500 ng (total sample) × 100 and plotted against iNKT cell frequency detected by flow cytometry (x axis). Solid line is a linear regression, and dashed lines mark 95% confidence interval, P < 0.0001. (B) iNKT cell frequency in neuroblastoma tumors (n = 98) was calculated from detected iNKT RNA amount per 500 ng total sample RNA using the standard curve shown in A. (C) Frozen 6-μm sections were stained with Alexa Fluor® 488 anti-CD3 289-13801 (green), Cy-3 anti–Vα24-Jα18 6B11 (red) mAbs, and DAPI (blue). Digital image of microscopic field of tumor tissue (magnification, 64) is one representative from five analyzed iNKT+ tumors (four to six fields per tumor) with green-circled T cells and yellow (green + red)-circled iNKT among blue nucleated cells.
RT-PCR analysis of 98 primary untreated stage 4 neuroblastomas revealed that 52 (53%) contained iNKTs. Their frequency among all cells was calculated from specific iNKT RNA concentration (Fig. 1 B), and it ranged from <0.01 (not detectable) to 0.52%. Tumors from all 19 patients who were younger than 1 yr at diagnosis contained iNKTs, whereas only 33 of 79 tumors (42%) from older patients were iNKT+. iNKT frequency was similarly distributed in positive tumors regardless of the age of patients with a median of 0.06% and 25th and 75th percentiles of 0.015 and 0.14%. To confirm their presence in tumor tissues, we performed three-color immunofluorescence microscopy on five iNKT+ and five iNKT− specimens (as determined by RT-PCR) using DAPI for nuclear staining, anti-CD3 mAb for T cells, and anti–Vα24-Jα18 CDR3 mAb 6B11 for iNKTs. iNKT− specimens contained only T cells (green fluorescence; not depicted), whereas iNKT+ specimens contained T cells and iNKTs (yellow fluorescence due to green and red fluorescence colocalization; Fig. 1 C).
iNKT Infiltration and Chemokine Expression by Neuroblastoma Tumors and Cell Lines.
To examine whether iNKT cell localization to neuroblastomas was associated with expression of chemokines in tumor tissues, we analyzed oligonucleotide microarray expression data (U95Av2 arrays; Affymetrix) for the 79 tumors diagnosed after 1 yr of age. Among 43 chemokine genes evaluated, expression of CCL2 (r = 0.52, P < 0.001), CXCL12 (r = 0.46, P < 0.001), CCL5 (r = 0.43, P < 0.001), and CCL21 (r = 0.38, P = 0.005) correlated with iNKT infiltration into tumors (Fig. 2 A). In contrast, CD3+ T cell infiltration correlated only with expression of CCL21 (r = 0.42, P < 0.001).
Figure 2.
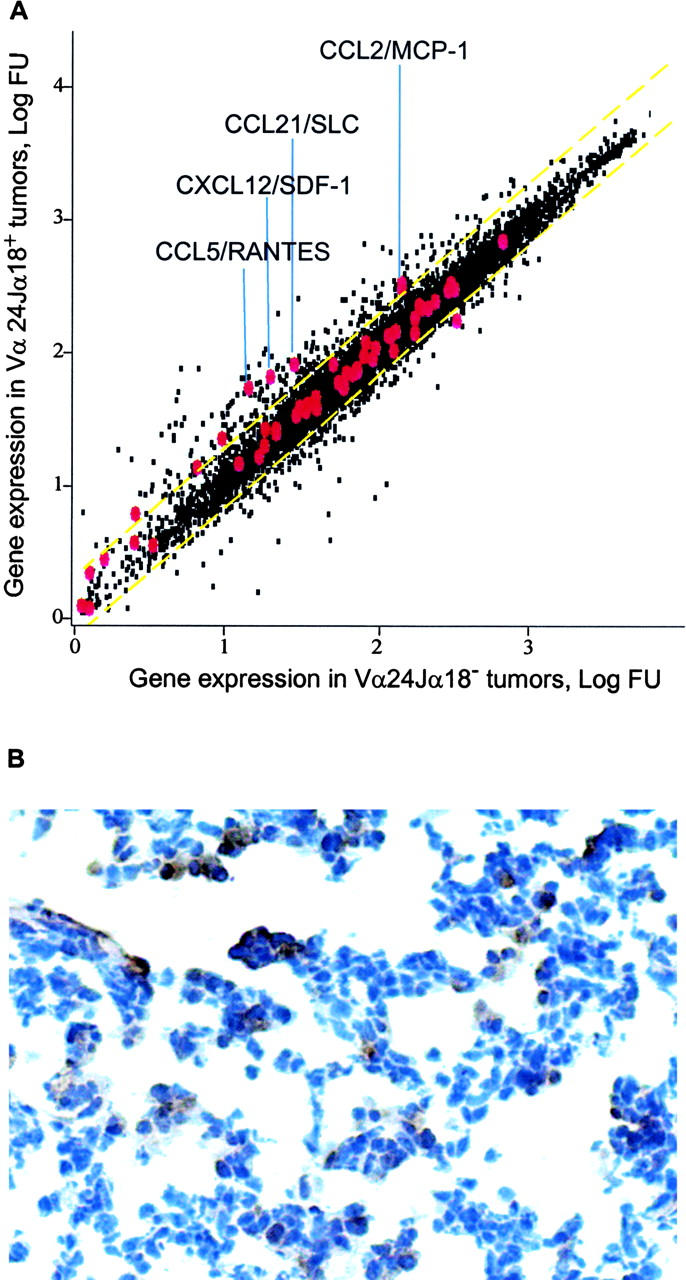
Chemokine expression in primary untreated neuroblastomas. (A) Expression profiles were generated from 79 untreated stage 4 neuroblastomas from patients using Affymetrix U95Av2 oligonucleotide microarrays. All genes (black) are plotted according to their average level of expression in tumors with undetectable (x axis) versus detectable (y axis) Vα24-Jα18 RNA (iNKT TCR). Gene probe sets for 43 chemokines are enlarged and labeled in red. Yellow dashed lines mark 95% confident interval. Names of chemokine genes that positively correlate with iNKT RNA expression are shown. (B) A tumor that expressed CCL2 mRNA was immunostained with goat anti–human CCL2 followed by the Cell and Tissue Staining Kit (R&D Systems). Tumor cells expressing CCL2 have brown cytoplasm.
Supernatants from eight neuroblastoma cell lines were analyzed by ELISA for these four chemokines, and they secreted a range of CCL2 (0–21.6 ng/ml) and little CXCL12 (≤0.1 ng/ml), but no detectable CCL5 or CCL21 (Table I) . Quantitative RT-PCR analysis of CCL2 expression by 17 neuroblastomas diagnosed after 1 yr revealed strong correlation with microarray data for two out of three CCL2 probe sets (r = 0.98, P < 0.001), and CCL2 expression by tumor cells was detected by IHC in two representative tumors with high levels of mRNA (Fig. 2 B). Comparison of iNKT+ versus iNKT− tumors diagnosed after 1 yr demonstrated significantly greater expression of CCL2 in the former (iNKT+: median, 464 microarray FU; 25th and 75th percentiles, 158 and 532 FU; iNKT−: median, 189 FU; 25th and 75th percentiles, 103 and 265 FU; P < 0.001, Wilcoxon test). Importantly, iNKTs could not be detected by RT-PCR in 21 neuroblastoma specimens with CCL2 gene expression <106 FU. For iNKT+ tumors, the level of CCL2 correlated with the extent of infiltration (r = 0.4, P = 0.018).
Table I.
Chemokine Secretion by Neuroblastoma Cell Linesa
| Cell line | CCL2 | CXCL12 | CCL21 | CCL5 |
|---|---|---|---|---|
| pg/ml | pg/ml | pg/ml | pg/ml | |
| SKN-BE-2 | 65 ± 6 | 27 ± 1 | ND | ND |
| SKN-BE-1 | 38 ± 2 | 73 ± 5 | ND | ND |
| SMS-KCNR | ND | 62 ± 2 | ND | ND |
| SMS-KCN | 5,367 ± 8 | 89 ± 3 | ND | ND |
| SMS-KANR | 47 ± 3 | 56 ± 2 | ND | ND |
| SMS-KAN | 72 ± 4 | 103 ± 8 | ND | ND |
| CHLA-20 | 6,608 ± 12 | 61 ± 2 | ND | ND |
| CHLA-15 | 56 ± 5 | 65 ± 1 | ND | ND |
ELISA was performed on supernatants collected from 106 cells after a 24-h incubation.
TEM of iNKTs to Supernatants of Neuroblastoma Cell Lines Producing CCL2.
We examined whether supernatants from neuroblastoma cell lines were chemoattractants for iNKT cell lines. Fig. 3 A demonstrates that supernatants from six out of eight tested neuroblastoma cell lines induced iNKT cell TEM. Almost half of input iNKTs migrated toward supernatants of CHLA-20 and SMS-KCN cell lines, which secreted the highest levels of CCL2. In contrast, iNKTs did not significantly migrate toward supernatant of SMS-KCNR cells, which produced no CCL2 (Table I). These experiments demonstrated a correlation between the quantity of CCL2 in supernatants and the percentage of migrating iNKTs (r = 0.98, P < 0.001). Next, we examined the effect of neutralizing mAbs against the four candidate chemokines (CCL2, CXCL12, CCL5, and CCL21) on iNKT cell TEM toward supernatants from neuroblastoma cell lines CHLA-20 and SMS-KCN (Fig. 3 B). Anti-CCL2 mAb abrogated iNKT cell chemotaxis, whereas anti-CXCL12, anti-CCL21, and anti-CCL5 mAbs had no effect.
Figure 3.

iNKT cell migration to neuroblastoma cell line supernatants. (A) Medium alone or supernatants from neuroblastoma cell lines were used to assess TEM of an iNKT cell line. Percentage of migrating cells is indicated on the x axis. (B) Supernatants from neuroblastoma cell lines mixed with 10 μg/ml of neutralizing mAbs against indicated chemokines, or mouse IgG1 isotype control mAbs were tested for chemotactic activity using the same iNKT cell line and the TEM assay. Specific migration (x axis) is shown as net migration after subtraction of migration to medium alone. Results are mean ± SD from duplicates of four experiments.
iNKT Frequency, CCR2 Expression, and CCL2-dependent TEM of Blood iNKTs from Children with Metastatic Neuroblastoma.
It has been reported recently that CCR2, which is the only known functional receptor for CCL2, is expressed on human iNKTs (15). To examine the effect of metastatic disease (stage 4), we evaluated iNKTs from blood of neuroblastoma patients diagnosed after 1 yr with respect to frequency (Fig. 4 A), expression of CCR2 (Fig. 4, B and C), and TEM in response to CCL2 (Fig. 4 D). We found no significant difference either in iNKT cell frequency or CCR2 expression on the iNKT, NK, and T cells between normal adults and neuroblastoma patients. Combining both groups of subjects demonstrated that iNKTs more frequently expressed CCR2 (median, 78%; range, 40–98%) compared with NK cells (median, 5%; range, 1–11%) and T cells (median, 21%; range, 10–37%; P < 0.001, paired signed rank test; Fig. 4 C). Migration of blood iNKTs from neuroblastoma patients and normal adults in response to CCL2 and CHLA-20 neuroblastoma cell line supernatant also was not different, with both recombinant CCL2 and supernatant from CHLA-20 cells inducing up to 55% TEM of iNKTs from both patients and normal donors. As expected, migration in response to supernatant was specifically prevented by anti-CCL2 neutralizing mAb. In contrast to iNKTs, T and NK cells from patients and normal adults migrated minimally (Fig. 4 D).
Figure 4.
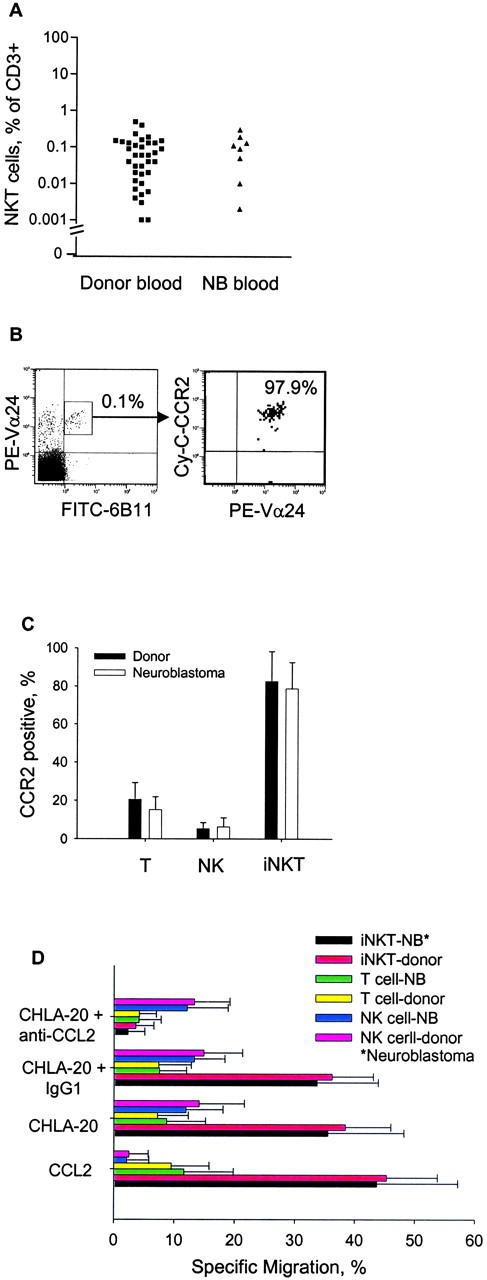
CCR2 expression and migratory response of blood iNKTs from neuroblastoma patients. (A) iNKT cell frequency in blood T cells of normal donors (n = 36) and neuroblastoma (NB) patients (n = 8) was determined by three-color flow cytometry. (B) Representative flow cytometry diagrams demonstrating iNKT frequency in patient PBLs (left) and CCR2 expression in this iNKT subset (right). (C) Mean ± SD of CCR2 expression in indicated lymphocyte subpopulations from normal adults (n = 10) and neuroblastoma patients (n = 6). (D) TEM of monocyte-depleted peripheral blood lymphocytes from neuroblastoma patients (n = 4) and normal adults (n = 4) was evaluated with indicated conditions. Results are mean ± SD from duplicates of four experiments.
Patient Survival, iNKT Infiltration, MYCN Amplification/Expression, and CCL2 Expression.
We evaluated iNKT infiltration in relationship to clinical and molecular biologic variables. Patients diagnosed after 1 yr of age with metastases (stage 4) have high-risk disease, and the 79 studied patients received intensive chemotherapy or myeloablative chemoradiotherapy (18). Their estimated survival 3 and 5 yr after diagnosis was 48 and 33%. Examination of iNKT infiltration and survival demonstrated a trend toward better outcome for those with iNKT-infiltrated tumors, but the difference was not significant (P = 0.32). Of note, therapy received by all patients was immunosuppressive with unknown effects on iNKT cell number and function, which precludes a clear analysis of correlation between only iNKT infiltration and survival. Patients diagnosed before 1 yr of age with stage 4 neuroblastoma without MYCN gene amplification have intermediate-risk disease (28). The 19 studied patients received much less intensive chemotherapy, and all survived for >56 mo after diagnosis. Analysis of outcome for all 98 patients in relationship to iNKT infiltration showed that survival was 64 and 35% at 5 yr for iNKT+ and iNKT− tumors (P = 0.007; Fig. 5 A).
Figure 5.
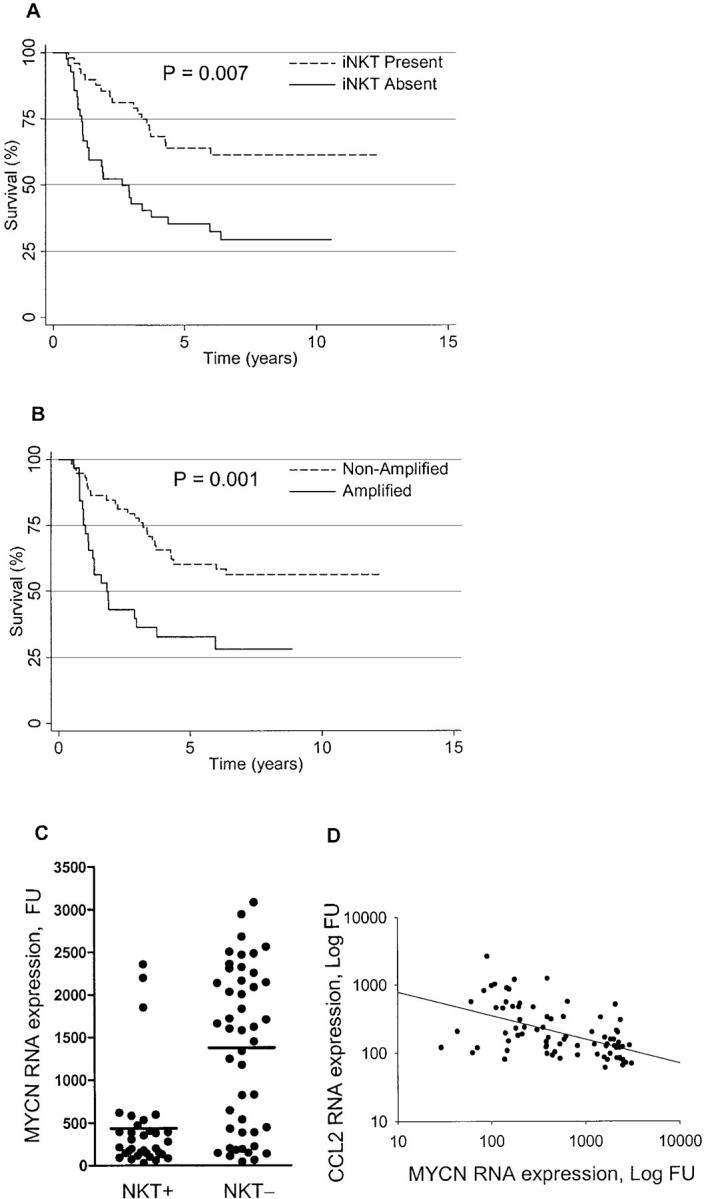
Patient survival, iNKT infiltration, MYCN amplification/expression, and CCL2 expression. (A) Survival for 98 patients whose tumors did or did not have iNKT infiltration (P = 0.007). (B) Survival for 98 patients whose tumors did or did not have MYCN amplification (P = 0.001). (C) MYCN expression and iNKT infiltration in primary neuroblastomas. MYCN RNA expression (y axis) was quantified by Affymetrix U95Av2 oligonucleotide microarrays and Vα24-Jα18 RNA was detected by Taqman RT-PCR in 79 untreated neuroblastomas. Tumors with detectable (NKT+) and undetectable (NKT−). iNKTs expressed 434.6 ± 100.0 (n = 33) and 1,378 ± 139.5 (n = 46) MYCN FU, respectively (Student's t test, P < 0.001). (D) Correlation between MYCN and CCL2 expression. MYCN (x axis) and CCL2 (y axis) RNA were quantified by Affymetrix U95Av2 oligonucleotide microarrays in 79 untreated tumors. Regression line demonstrates an inverse correlation between MYCN and CCL2 (r = 0.5, P < 0.001, Spearman correlation analysis).
Genomic amplification of MYCN is associated with aggressive tumor behavior (22). Survival for all 98 patients in relationship to MYCN gene status was 65 and 30% for MYCN nonamplified and amplified tumors (Fig. 5 B; P = 0.001). The 19 tumors diagnosed before 1 yr that were studied were all nonamplified and all contained iNKTs. Tumors diagnosed after 1 yr with MYCN amplification and high-level expression surprisingly almost always lacked iNKTs, whereas those in this group without amplification accounted for nearly all with infiltration (P < 0.001; Fig. 5 C). Furthermore, for patients diagnosed after 1 yr of age, MYCN and CCL2 expression were inversely correlated (Spearman correlation r = −0.5, P < 0.001; Fig. 5 D). When MYCN and CCL2 RNA expression were categorized according to optimal cut-points, iNKTs infiltrated 76% of MYCN-low/CCL2-high tumors but none of the MYCN-high/CCL2-low tumors (P < 0.001; Table II). The association of iNKT infiltration also was tested with MYCN and CCL2 jointly using a multivariate logistic regression model containing the categorized expression of MYCN and CCL2. In this model, the effects of both genes on iNKT cell infiltration were independently significant (P ≤ 0.01) and were additive but not synergistic. Thus, MYCN-low/CCL2-high and MYCN-high/CCL2-low were estimated to have 76 and 100% accuracy in predicting iNKT presence and absence, respectively.
Table II.
Relationship between MYCN and CCL2 Expression and iNKT Cell Presence in 79 Neuroblastomas Diagnosed after 1 yr of Age
Measured in fluorescence units.
The numerator is the number of tumors having iNKT infiltration, and the denominator is the total number of tumors.
Discussion
This paper demonstrates infiltration of iNKTs into 53% of primary untreated metastatic neuroblastomas and correlation of infiltration with expression of CCL2, CCL5, CCL21, and CXCL12. iNKT localization most likely was primarily due to CCL2 because six out of eight neuroblastoma cell lines secreted CCL2, which was required and sufficient for TEM of iNKTs in vitro. CCL2 expression was inversely associated with MYCN gene amplification and expression, and MYCN-high/CCL2-low expression accurately predicted the absence of iNKTs in tumors. This is the first correlation between oncogene activation, CCL2 expression, and iNKT cell infiltration. Together, our data suggest that iNKTs migrate into neuroblastomas, particularly those without MYCN amplification, in response to CCL2.
The chemokine milieu in the tumor microenvironment is likely to be a major determinant of infiltration and possibly function of specific types of lymphoid and myeloid cells. Although CCL2 alone mediated iNKT cell migration to neuroblastoma cell line supernatants in vitro, tumors expressed three additional chemokines (CXCL12, CCL21, and CCL5) that also correlated with iNKT cell infiltration. These other chemokines could have contributed to localization in vivo because iNKTs express receptors for them. The receptors for all four chemokines are equally expressed among functionally distinct CD4+ and CD4− iNKT subsets, and so it is not likely that a subset of iNKTs preferentially infiltrated neuroblastomas (29–31). Chemokines also may influence the type of immune response in a microenvironment by recruiting specific types of myeloid and lymphoid cells. Indeed, murine iNKTs recruited to lungs by CCL2 after infection with Cryptococcus neoformans were important for development of Th1 immune responses because both lung IFN-γ production and specific delayed-type hypersensitivity were significantly greater in wild-type mice than in Vα14 NKT cell–deficient mice (32). Thus, the repertoire of chemokines expressed by tumors could determine localization of lymphocyte subsets and have a functional effect on immune responses.
Our paper is the first to correlate iNKT cell infiltration into a human tumor with CCL2 expression. Another investigation of 60 squamous cell carcinomas and adenocarcinomas of the lung demonstrated the presence of iNKTs with a PCR assay for invariant Vα24-Jα18, but did not address mechanisms of localization (13). Correlation of CCL2 expression and iNKT infiltration suggests that many types of human tumors that express CCL2 also may contain iNKTs (33). However, effects of CCL2 may be different depending on chemokine level. For example, low expression of CCL2 after gene transfection into a nontumorigenic human melanoma cell line stimulated tumor formation in SCID mice, whereas transfection that caused ∼100-fold higher expression was associated with large infiltration of inflammatory cells (presumably monocytes) and tumor necrosis (34).
Expression of CD1d on APCs is required for iNKT cell functional responses (35), and it has been demonstrated recently that ganglioside GD3 derived from human melanoma cells can be effectively cross-presented to murine iNKTs by syngeneic APCs in a CD1d-dependent manner (36). Similar to melanoma, neuroblastoma cells do not express CD1d (20), but do produce a variety of gangliosides, including GD3 (37). Our oligonucleotide microarray analysis revealed variable, but usually low level CD1d expression, which did not correlate with iNKT infiltration. IHC of two representative tumors expressing CCL2 and containing iNKTs confirmed that tumor cells do not express CD1d, but showed CD1d+ cells, especially in stromal areas (unpublished data). This indicates that iNKTs might function in the tumor microenvironment. Animal models suggest that in the absence of exogenous stimulation, iNKTs may activate (6, 38) or suppress antitumor immune responses (9, 10). However, treatment with the iNKT cell ligand αGalCer (KRN7000), which provides strong stimulation, unequivocally induces potent antitumor responses in mice (4). Thus, understanding mechanisms of localization and stimulation of iNKTs in the tumor microenvironment is important for planning of iNKT cell–mediated immunotherapy.
Disease and therapy could affect infiltration of iNKTs into tumors. In this regard, we found no effect of metastatic neuroblastoma at diagnosis (before therapy) on blood iNKT cell frequency, CCR2 expression, or migratory response to CCL2. Other studies of patients with cancer have evaluated iNKT cell number in peripheral blood but have not investigated their migration. One analysis of patients with advanced gastrointestinal cancers at diagnosis found normal frequencies of iNKTs in blood, which is in agreement with our observations (39). Two others of patients that had been previously treated reported lower than normal iNKT cell frequency in blood of patients with androgen-independent metastatic prostate cancer (26) or with chemotherapy-resistant solid tumors (40). These observations suggest that developing clinical immunotherapy with iNKTs will require examination of effects of disease and therapies on circulating iNKT cell number and function and, when possible, of tumors for chemokine expression and iNKT cell infiltration.
Survival for patients whose tumors did or did not have iNKT cell infiltration at diagnosis was significantly different, but immunosuppressive chemotherapy or myeloablative chemoradiotherapy renders interpretation of this data difficult. However, the survival curves for iNKT+ versus iNKT− groups were similar to those for MYCN amplified versus nonamplified subgroups. This suggested a relationship between iNKT infiltration and MYCN gene status. Our analysis demonstrated for the first time an inverse correlation between MYCN amplification and tumor infiltration by iNKTs that appears to be due, at least in part, to decreased CCL2 production. MYCN amplification, which causes protein overexpression, is independently associated with aggressive tumor behavior in neuroblastoma (22), and laboratory studies demonstrate that MYCN can have a direct role in progression of neuroblastoma by activating or repressing transcription of other genes (41, 42). Because myc family oncoproteins, which include c-myc, N-myc, and L-myc, have roles in the pathogenesis of many human malignancies (43), our finding may have implications for inflammatory and immune responses to several neoplasms.
Acknowledgments
We thank Dr. G. McNamara of the Childrens Hospital Los Angeles Congressman Julian Dixon Cellular Imaging Core for excellent technical assistance with digital image microscopy.
This work was supported in part by National Institutes of Health grants CA81403 and CA22794 (to R.C. Seeger); by grants from Stop Cancer and Concern Foundations (to L.S. Metelitsa); and by the Neil Bogart Memorial Fund of the T.J. Martell Foundation for Leukemia, Cancer, and AIDS Research.
Abbreviations used in this paper: αGalCer, α-galactosylceramide; DAPI, 4′,6-diamidine-2′-phenylindole dihydrochloride; FU, fluorescence units; IHC, immunohistochemistry; iNKT, Vα24-Jα18–invariant natural killer T cell; TEM, transendothelial migration.
References
- 1.Bendelac, A., M.N. Rivera, S.H. Park, and J.H. Roark. 1997. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu. Rev. Immunol. 15:535–562. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey, D.I., K.J. Hammond, L.D. Poulton, M.J. Smyth, and A.G. Baxter. 2000. NKT cells: facts, functions and fallacies. Immunol. Today. 21:573–583. [DOI] [PubMed] [Google Scholar]
- 3.Joyce, S. 2001. CD1d and natural T cells: how their properties jump-start the immune system. Cell. Mol. Life Sci. 58:442–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smyth, M.J., N.Y. Crowe, Y. Hayakawa, K. Takeda, H. Yagita, and D.I. Godfrey. 2002. NKT cells - conductors of tumor immunity? Curr. Opin. Immunol. 14:165–171. [DOI] [PubMed] [Google Scholar]
- 5.Brutkiewicz, R.R., and V. Sriram. 2002. Natural killer T (NKT) cells and their role in antitumor immunity. Crit. Rev. Oncol. Hematol. 41:287–298. [DOI] [PubMed] [Google Scholar]
- 6.Smyth, M.J., K.Y. Thia, S.E. Street, E. Cretney, J.A. Trapani, M. Taniguchi, T. Kawano, S.B. Pelikan, N.Y. Crowe, and D.I. Godfrey. 2000. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 191:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crowe, N.Y., M.J. Smyth, and D.I. Godfrey. 2002. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J. Exp. Med. 196:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smyth, M.J., N.Y. Crowe, D.G. Pellicci, K. Kyparissoudis, J.M. Kelly, K. Takeda, H. Yagita, and D.I. Godfrey. 2002. Sequential production of interferon-gamma by NK1.1(+) T cells and natural killer cells is essential for the antimetastatic effect of alpha-galactosylceramide. Blood. 99:1259–1266. [DOI] [PubMed] [Google Scholar]
- 9.Terabe, M., S. Matsui, N. Noben-Trauth, H. Chen, C. Watson, D.D. Donaldson, D.P. Carbone, W.E. Paul, and J.A. Berzofsky. 2000. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat. Immunol. 1:515–520. [DOI] [PubMed] [Google Scholar]
- 10.Terabe, M., S. Matsui, J.M. Park, M. Mamura, N. Noben-Trauth, D.D. Donaldson, W. Chen, S.M. Wahl, S. Ledbetter, B. Pratt, et al. 2003. Transforming growth factor-β production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte–mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J. Exp. Med. 198:1741–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okai, M., M. Nieda, A. Tazbirkova, D. Horley, A. Kikuchi, S. Durrant, T. Takahashi, A. Boyd, R. Abraham, H. Yagita, T. Juji, and A. Nicol. 2002. Human peripheral blood Valpha24+ Vbeta11+ NKT cells expand following administration of alpha-galactosylceramide-pulsed dendritic cells. Vox Sang. 83:250–253. [DOI] [PubMed] [Google Scholar]
- 12.Nieda, M., M. Okai, A. Tazbirkova, H. Lin, A. Yamaura, K. Ide, R. Abraham, T. Juji, D.J. Macfarlane, and A.J. Nicol. 2004. Therapeutic activation of V{alpha}24+V{beta}11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood. 103:383–389. [DOI] [PubMed] [Google Scholar]
- 13.Motohashi, S., S. Kobayashi, T. Ito, K.K. Magara, O. Mikuni, N. Kamada, T. Iizasa, T. Nakayama, T. Fujisawa, and M. Taniguchi. 2002. Preserved IFN-alpha production of circulating Valpha24 NKT cells in primary lung cancer patients. Int. J. Cancer. 102:159–165. [DOI] [PubMed] [Google Scholar]
- 14.Dhodapkar, M.V., M.D. Geller, D.H. Chang, K. Shimizu, S. Fujii, K.M. Dhodapkar, and J. Krasovsky. 2003. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J. Exp. Med. 197:1667–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim, C.H., B. Johnston, and E.C. Butcher. 2002. Trafficking machinery of NKT cells: shared and differential chemokine receptor expression among V alpha 24(+)V beta 11(+) NKT cell subsets with distinct cytokine-producing capacity. Blood. 100:11–16. [DOI] [PubMed] [Google Scholar]
- 16.Lauder, I., and W. Aherne. 1972. The significance of lymphocytic infiltration in neuroblastoma. Br. J. Cancer. 26:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Facchetti, P., I. Prigione, F. Ghiotto, P. Tasso, A. Garaventa, and V. Pistoia. 1996. Functional and molecular characterization of tumour-infiltrating lymphocytes and clones thereof from a major-histocompatibility-complex-negative human tumour: neuroblastoma. Cancer Immunol. Immunother. 42:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthay, K.K., J.G. Villablanca, R.C. Seeger, D.O. Stram, R.E. Harris, N.K. Ramsay, P. Swift, H. Shimada, C.T. Black, G.M. Brodeur, et al. 1999. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N. Engl. J. Med. 341:1165–1173. [DOI] [PubMed] [Google Scholar]
- 19.Seeger, R.C., S.A. Rayner, A. Banerjee, H. Chung, W.E. Laug, H.B. Neustein, and W.F. Benedict. 1977. Morphology, growth, chromosomal pattern and fibrinolytic activity of two new human neuroblastoma cell lines. Cancer Res. 37:1364–1371. [PubMed] [Google Scholar]
- 20.Metelitsa, L.S., O.V. Naidenko, A. Kant, H.W. Wu, M.J. Loza, B. Perussia, M. Kronenberg, and R.C. Seeger. 2001. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J. Immunol. 167:3114–3122. [DOI] [PubMed] [Google Scholar]
- 21.Li, C., and W.H. Wong. 2001. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA. 98:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seeger, R.C., G.M. Brodeur, H. Sather, A. Dalton, S.E. Siegel, K.Y. Wong, and D. Hammond. 1985. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 313:1111–1116. [DOI] [PubMed] [Google Scholar]
- 23.Crabbe, D.C., J. Peters, and R.C. Seeger. 1992. Rapid detection of MYCN gene amplification in neuroblastomas using the polymerase chain reaction. Diagn. Mol. Pathol. 1:229–234. [PubMed] [Google Scholar]
- 24.Slamon, D.J., T.C. Boone, R.C. Seeger, D.E. Keith, V. Chazin, H.C. Lee, and L.M. Souza. 1986. Identification and characterization of the protein encoded by the human N-myc oncogene. Science. 232:768–772. [DOI] [PubMed] [Google Scholar]
- 25.Thomas, S.Y., R. Hou, J.E. Boyson, T.K. Means, C. Hess, D.P. Olson, J.L. Strominger, M.B. Brenner, J.E. Gumperz, S.B. Wilson, and A.D. Luster. 2003. CD1d-restricted NKT cells express a chemokine receptor profile indicative of Th1-type inflammatory homing cells. J. Immunol. 171:2571–2580. [DOI] [PubMed] [Google Scholar]
- 26.Tahir, S.M., O. Cheng, A. Shaulov, Y. Koezuka, G.J. Bubley, S.B. Wilson, S.P. Balk, and M.A. Exley. 2001. Loss of IFN-gamma production by invariant NK T cells in advanced cancer. J. Immunol. 167:4046–4050. [DOI] [PubMed] [Google Scholar]
- 27.Kukreja, A., G. Cost, J. Marker, C. Zhang, Z. Sun, K. Lin-Su, S. Ten, M. Sanz, M. Exley, B. Wilson, et al. 2002. Multiple immuno-regulatory defects in type-1 diabetes. J. Clin. Invest. 109:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt, M.L., J.N. Lukens, R.C. Seeger, G.M. Brodeur, H. Shimada, R.B. Gerbing, D.O. Stram, C. Perez, G.M. Haase, and K.K. Matthay. 2000. Biologic factors determine prognosis in infants with stage IV neuroblastoma: a prospective Children's Cancer Group study. J. Clin. Oncol. 18:1260–1268. [DOI] [PubMed] [Google Scholar]
- 29.Lee, P.T., K. Benlagha, L. Teyton, and A. Bendelac. 2002. Distinct functional lineages of human V(α)24 natural killer T cells. J. Exp. Med. 195:637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gumperz, J.E., S. Miyake, T. Yamamura, and M.B. Brenner. 2002. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J. Exp. Med. 195:625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim, C.H., E.C. Butcher, and B. Johnston. 2002. Distinct subsets of human Valpha24-invariant NKT cells: cytokine responses and chemokine receptor expression. Trends Immunol. 23:516–519. [DOI] [PubMed] [Google Scholar]
- 32.Kawakami, K., Y. Kinjo, K. Uezu, S. Yara, K. Miyagi, Y. Koguchi, T. Nakayama, M. Taniguchi, and A. Saito. 2001. Monocyte chemoattractant protein-1-dependent increase of V alpha 14 NKT cells in lungs and their roles in Th1 response and host defense in cryptococcal infection. J. Immunol. 167:6525–6532. [DOI] [PubMed] [Google Scholar]
- 33.Graves, D.T., and A.J. Valente. 1991. Monocyte chemotactic proteins from human tumor cells. Biochem. Pharmacol. 41:333–337. [DOI] [PubMed] [Google Scholar]
- 34.Nesbit, M., H. Schaider, T.H. Miller, and M. Herlyn. 2001. Low-level monocyte chemoattractant protein-1 stimulation of monocytes leads to tumor formation in nontumorigenic melanoma cells. J. Immunol. 166:6483–6490. [DOI] [PubMed] [Google Scholar]
- 35.Brossay, L., M. Chioda, N. Burdin, Y. Koezuka, G. Casorati, P. Dellabona, and M. Kronenberg. 1998. CD1d-mediated recognition of an α-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J. Exp. Med. 188:1521–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu, D.Y., N.H. Segal, S. Sidobre, M. Kronenberg, and P.B. Chapman. 2003. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J. Exp. Med. 198:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolfl, M., W.Y. Batten, C. Posovszky, H. Bernhard, and F. Berthold. 2002. Gangliosides inhibit the development from monocytes to dendritic cells. Clin. Exp. Immunol. 130:441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smyth, M.J., N.Y. Crowe, and D.I. Godfrey. 2001. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int. Immunol. 13:459–463. [DOI] [PubMed] [Google Scholar]
- 39.Yanagisawa, K., K. Seino, Y. Ishikawa, M. Nozue, T. Todoroki, and K. Fukao. 2002. Impaired proliferative response of V alpha 24 NKT cells from cancer patients against alpha-galactosylceramide. J. Immunol. 168:6494–6499. [DOI] [PubMed] [Google Scholar]
- 40.Giaccone, G., C.J. Punt, Y. Ando, R. Ruijter, N. Nishi, M. Peters, B.M. von Blomberg, R.J. Scheper, H.J. van der Vliet, A.J. van den Eertwegh, M. Roelvink, J. Beijnen, H. Zwierzina, and H.M. Pinedo. 2002. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin. Cancer Res. 8:3702–3709. [PubMed] [Google Scholar]
- 41.Maris, J.M., and K.K. Matthay. 1999. Molecular biology of neuroblastoma. J. Clin. Oncol. 17:2264–2279. [DOI] [PubMed] [Google Scholar]
- 42.Raetz, E.A., M.K. Kim, P. Moos, M. Carlson, C. Bruggers, D.K. Hooper, L. Foot, T. Liu, R. Seeger, and W.L. Carroll. 2003. Identification of genes that are regulated transcriptionally by Myc in childhood tumors. Cancer. 98:841–853. [DOI] [PubMed] [Google Scholar]
- 43.Nesbit, C.E., J.M. Tersak, and E.V. Prochownik. 1999. MYC oncogenes and human neoplastic disease. Oncogene. 18:3004–3016. [DOI] [PubMed] [Google Scholar]