

Abstract
Legionella pneumophila is a bacterial pathogen that infects eukaryotic host cells and replicates inside a specialized organelle that is morphologically similar to the endoplasmic reticulum (ER). To better understand the molecular mechanisms governing transport of the Legionella-containing vacuole (LCV), we have identified host proteins that participate in the conversion of the LCV into a replicative organelle. Our data show that Rab1 is recruited to the LCV within minutes of uptake. Rab1 recruitment to the LCV precedes remodeling of this compartment by ER-derived vesicles. Genetic inhibition studies demonstrate that Rab1 is important for the recruitment of ER-derived vesicles to the LCV and that inhibiting Rab1 function abrogates intracellular growth of Legionella. Morphological studies indicate that the Sec22b protein is located on ER-derived vesicles recruited to the LCV and that Sec22b is delivered to the LCV membrane. Sec22b function was found to be important for biogenesis of the specialized organelle that supports Legionella replication. These studies demonstrate that Legionella has the ability to subvert Rab1 and Sec22b function to facilitate the transport and fusion of ER-derived vesicles with the LCV, resulting in the formation of a specialized organelle that can support bacterial replication.
Keywords: brefeldin A, ARF1, Sar1, phagosome, endoplasmic reticulum
Introduction
After uptake by a eukaryotic cell, the bacterial pathogen Legionella pneumophila has the ability to modulate transport of the vacuole in which it resides. The initial compartment in which virulent Legionella resides is derived from the plasma membrane and we refer to it as the Legionella-containing vacuole (LCV). Unlike avirulent microorganisms, which typically reside in vacuoles that interact sequentially with endocytic vesicles resulting in their delivery to lysosomes, the LCV avoids endocytic maturation and intercepts secretory vesicles as they exit the ER (1–5). To modulate transport of their vacuole, Legionella requires a specialized secretion apparatus called the Dot/Icm system, which injects bacterial proteins into the cytosol of host cells during infection (6–9). The functions of most bacterial proteins injected by the Dot/Icm system and the host factors that participate in LCV transport remain unknown.
The LCV transport pathway has been defined primarily by morphological studies. Fluorescence microscopy indicates that the LCV diverges from the default endocytic pathway within the first 5 min of infection (4). Although endocytic markers are excluded (10, 11), proteins residing in secretory vesicles cycling between the ER and Golgi apparatus are associated with most LCVs within 30 min, and resident ER proteins such as calnexin are found associated with the LCV within 1–2 h (3). Vacuoles containing dot/icm mutants of Legionella do not stain positive for secretory proteins or ER markers (3, 12–14). Electron microscopy has been used to confirm and extend observations made by fluorescence microscopy. Electron micrographs show that ER-derived vesicles attach to LCVs within the first 30 min of infection (12). Several hours later, attached ER-derived vesicles are less frequent, and LCVs at this time have ribosomes decorating the cytoplasmic surface of their membrane (12, 15). It is within this ER-derived organelle that Legionella begins to replicate (15), which is why this specialized compartment is called a replicative organelle. These data demonstrate that the Legionella Dot/Icm system is necessary for intercepting secretory vesicles. However, it is unknown how these ER-derived vesicles are recruited to a LCV and whether these vesicles participate directly in the remodeling of this compartment into a replicative organelle.
Herein, to better understand the cell biology of LCV transport, we examine host proteins that regulate the transport and fusion of ER-derived vesicles to see if they are important for biogenesis of the Legionella replicative organelle. Rab1 is a small guanosine triphosphatase (GTPase) that plays an important role in fusion of ER-derived vesicles with preGolgi intermediate compartments and the Golgi apparatus (16–18). Rab1 recruits factors necessary for the tethering and fusion of ER-derived vesicles with target membranes (16, 17). This membrane fusion process requires the pairing of soluble N-ethylmaleimide–sensitive factor attachment protein receptors (SNAREs) found on the vesicle membrane (v-SNARE) and target membrane (t-SNARE; references 19–21). The Sec22b protein in mammalian cells is found on ER-derived vesicles and functions as a v-SNARE (22–24). The proteins Membrin, Syntaxin 5, and Bet1 comprise the cognate t-SNARE found on preGolgi intermediate compartments (23, 24).
The Sar1 protein is a small GTPase that initiates the formation of ER-derived vesicles (25). When GTP bound, the Sar1 protein recruits the Sec23/24 subunits of the COPII coat to the ER membrane (26, 27). The Sec23/24 complex interacts directly with cytosolic regions of cargo proteins to be packaged into ER-derived vesicles and the Sec13/31 subunits of the COPII coat and binds to this prebudding complex to generate transport vesicles (28–32). The COPII coat plays an important role in the recruitment of SNARE proteins Sec22b, Syntaxin 5, Membrin, and Bet1 into ER-derived vesicles, allowing these SNARE proteins to participate in both homotypic fusion of ER-derived vesicles and heterotypic fusion with Golgi membranes (33).
After the COPII coat is removed from ER-derived vesicles, it is rapidly replaced by a COPI coat (34–36). The recruitment of COPI to vesicles is mediated by proteins belonging to the ADP ribosylation factor (ARF) family of GTPases (37). Vesicle production from the ER is dependent on the sequential activities of the Sar1/COPII and ARF/COPI systems and can be disrupted using inhibitors that prevent the cycling of GDP and GTP on either the Sar1 or ARF proteins (34–36).
Previous data has shown that inhibitors that block Sar1 or ARF function disrupt Legionella replicative organelle biogenesis by preventing the production of ER-derived vesicles (3). Here, we set out to determine the cellular pathways used for transport and fusion of these ER-derived vesicles with the LCV. Our data indicate that the host proteins Rab1 and Sec22b play important roles in converting LCVs into organelles that support the replication of Legionella.
Materials and Methods
Bacterial Strains and Tissue Culture.
Growth and macrophage infection by L. pneumophila strains derived from serogroup 1 strain LP01 have been described previously (6, 38, 39). Bone marrow–derived macrophages (BMMs) from the A/J mouse were prepared as described previously (40). Chinese hamster ovary (CHO) cells (41) and CHO FcγRII cells (42) were maintained in minimal Eagle's media α (GIBCO BRL) supplemented with 10% FBS.
Plasmids.
The cDNA encoding ARF1T31N (43) was ligated into pCLXSN (44). Plasmids producing myc-tagged versions of the mammalian Sec22b and Membrin were described previously (23, 24). Rab1 fusion proteins with an amino terminal green fluorescent protein (GFP) tag were created in pEGFP-C1 (CLONTECH Laboratories, Inc.) using plasmids described previously that encode Rab1 (45). Plasmids encoding ARF1T31N-GFP and Sar1H79G were described previously (3). Plasmid encoding GFP-tagged VAMP4 was provided by N. Andrews (Yale University School of Medicine, New Haven, CT). Fugene-6 (Roche) was used to transfect CHO and CHO FcγRII cells with the indicated plasmids according to the manufacturer's instructions. BMMs were transfected with plasmids encoding Sec22b and Membrin using the CD34 cell Nucleofector™ kit (Amaxa Biosystems).
Immunofluorescence Microscopy.
To examine LCV localization of the Rab1b, Rab2, and Rab6 proteins in BMMs, cells were infected with Legionella for 30 min as described previously (3). Where indicated, 10 μg ml−1 brefeldin A (Molecular Probes) was added to host cells 45 min before infection and remained in the medium during the course of the experiment. Cells were fixed in 2% paraformaldehyde in PBS for 20 min, permeabilized with ice-cold methanol, and blocked with 2% goat serum in PBS. Samples were stained with Rab-specific antibodies (Santa Cruz Biotechnology, Inc.) and fluorescein-labeled anti–rabbit IgG secondary antibodies (Molecular Probes). Bacteria and the host cell DNA were labeled using 0.1 μg ml−1 4,6-diamidino-2-phenylindole. The same fixation and DNA staining procedure was used to localize GFP-Rab1a in transfected CHO FcγRII cells. To localize Sec22b and Membrin in CHO FcγRII cells, Legionella were opsonized with rabbit anti-Legionella antibody (1:1,000), and cells were infected for the times indicated. Cells were fixed in 2% paraformaldehyde in PBS for 20 min. Cells were permeabilized in 0.4% saponin for 15 min. Sec22b and Membrin were identified by anti-myc staining using the monoclonal antibody 9E10 (46) and a fluorescein-labeled anti–mouse secondary antibody. Bacteria were identified using a rhodamine-labeled anti–rabbit secondary antibody. Images were collected on an inverted microscope (model TE200; Nikon) equipped with a digital Hamamatsu ORCA ER camera. Images were exported, and color was added using Photoshop 7.0 (Adobe). When measuring the percentage of vacuoles that stained positive for Sec22b in CHO FcγRII cells producing dominant-interfering proteins, it was important to distinguish between intracellular and extracellular bacteria. This was done by differential labeling where the extracellular bacteria were first rhodamine-labeled with an anti–rabbit secondary antibody before permeabilization and 4,6-diamidino-2-phenylindole staining was used to identify all bacteria.
Intracellular Growth Assays.
CHO cells were plated in 24-well dishes at a density of 3 × 104 cells/well. After an overnight incubation, cells were cotransfected with plasmids encoding the FcγRII receptor (42) and the other indicated proteins. 16 h later, cells were infected with Legionella that were opsonized with rabbit polyclonal anti-Legionella antibodies (1:1,000 dilution). Plates were centrifuged at 150 g for 5 min and placed in a 37°C water bath for 5 min. Cells were washed three times with PBS, and fresh medium containing gentamicin (20 μg ml−1) was added for 3 h to kill remaining extracellular bacteria. Cells were washed in PBS and either lysed to determine the efficiency of Legionella uptake or refreshed with 1 ml of antibiotic-free medium and lysed after an additional incubation period of 24 h. Bacterial numbers were determined in each lysate by measuring CFUs on charcoal yeast extract agar plates. Intracellular replication was measured as the fold increase in bacteria recovered after 24 h.
Secretion Assays.
CHO FcγRII cells were plated in 24-well dishes at 3 × 104 cells/well. After an overnight incubation, the cells were cotransfected with plasmids encoding the proteins indicated and a plasmid encoding a secreted alkaline phosphatase (SEAP) protein. After a 24-h incubation period, cells were washed, and fresh tissue culture medium was added. SEAP activity was measured in triplicate wells 7 h later using the Phospha-light SEAP kit (Applied Biosystems). Data are presented as a secretion index, which is the ratio of SEAP activity detected in the culture medium to the cell-associated SEAP activity.
Immunoelectron Microscopy.
CHO cells were plated in six-well dishes at a density of 2 × 105 cells/well. After an overnight incubation, cells were cotransfected with plasmids encoding FcγRII and Sec22b. Cells were infected 16 h after transfection with Legionella that were opsonized with rabbit polyclonal anti-Legionella antibodies (1:1,000 dilution). Plates were centrifuged at 150 g for 5 min and placed in a 37°C water bath for 10 min. Cells were washed three times with PBS and refreshed with prewarmed media for 1 h at 37°C. Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in 0.25 M Hepes, pH 7.4, for 1 h at room temperature, followed by 8% paraformaldehyde in the same buffer overnight at 4°C. Cells were scraped, pelleted, and embedded in 10% bovine skin gelatin in PBS. Pieces of the pellet were infiltrated overnight with 2.3 M sucrose in PBS at 4°C, mounted on aluminum studs, and frozen in liquid nitrogen. Blocks were sectioned at −108°C in an Ultracut cryo-ultramicrotome (Leica). 60-nm thick sections were collected using a 1:1 mixture of 2.3 M sucrose and 2% methyl cellulose and transferred onto formvar- and carbon-coated nickel grids. Sections were incubated for 10 min with 0.1 M NH4Cl in PBS and for 20 min with 0.5% fish skin gelatin (FSG; Sigma-Aldrich) in PBS (PBS-FSG) and incubated for 30 min at room temperature with rabbit anti-myc polyclonal antibody (Santa Cruz Biotechnology, Inc.) diluted in PBS-FSG. After four washes in PBS (5 min total), the sections were labeled with 10 nm protein A–gold conjugate (Utrecht University, Netherlands) diluted in PBS-FSG for 30 min. Sections were washed again in PBS and fixed with 1% glutaraldehyde in PBS. The sections were rinsed in distilled water and incubated with 1.8% methyl cellulose (25 centipoises; Sigma-Aldrich) and 0.5% uranyl acetate for 10 min on ice. The sections were dried and examined in a Philips Tecnai 12-electron microscope.
Online Supplemental Material.
An unbiased assay to measure the effect dominant-interfering proteins have on Legionella intracellular growth is provided in Fig. S1. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20031706/DC1.
Results
Specific Recruitment of Rab1 to the LCV.
Because Rab proteins are key regulators of most vesicle transport and fusion processes, it is predicted that a subset of Rab proteins should be involved in biogenesis of the Legionella replicative organelle. Given that the LCV is remodeled into an ER-derived organelle, we asked whether Rab proteins that function in the transport of secretory vesicles between the ER and Golgi apparatus are recruited to the LCV. Immunolocalization studies demonstrated that the LCV acquires Rab1 within the first 30 min of infection (Fig. 1, A and D). There was no detectable staining of LCVs using antibodies specific for either Rab2 or Rab6 (Fig. 1, B and C), which are two other Rab proteins that function in vesicular transport between the ER and Golgi apparatus (47–51). Rab1 staining was not observed on LCVs harboring a dotA mutant of Legionella (Fig. 2 A and Fig. 3), indicating that Rab1 recruitment to the LCV requires factors delivered into host cells by the Dot/Icm system. Rab1 recruitment to LCVs was also observed in CHO-FcγRII cells (Figs. 2 D and 3), a stable cell line that produces the FcγRII protein. Rab1 staining in CHO-FcγRII cells is consistent with previous work showing that the LCV transport pathway in these cells mirrors the pathway defined in BMMs (3).
Figure 1.
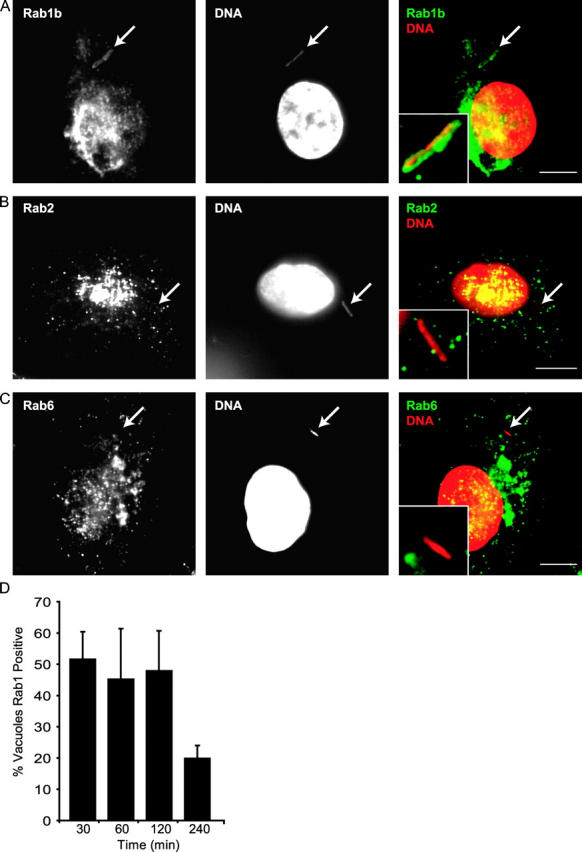
Specific recruitment of Rab1 to the LCV. (A–C) Rab-specific antibodies were used to stain BMMs infected for 30 min with wild-type Legionella. LCVs are indicated using arrows. Images show Rab1b (A) staining of the LCV. LCV staining was not observed using antibodies specific for Rab2 (B) or Rab6 (C). (D) BMMs were fixed at the indicated times after infection by wild-type Legionella and Rab1b staining of the LCV was determined using fluorescence microscopy. Values represent the mean ± SE of three independent experiments in which 50 vacuoles were scored for each time point. Bars, 5 μm.
Figure 2.
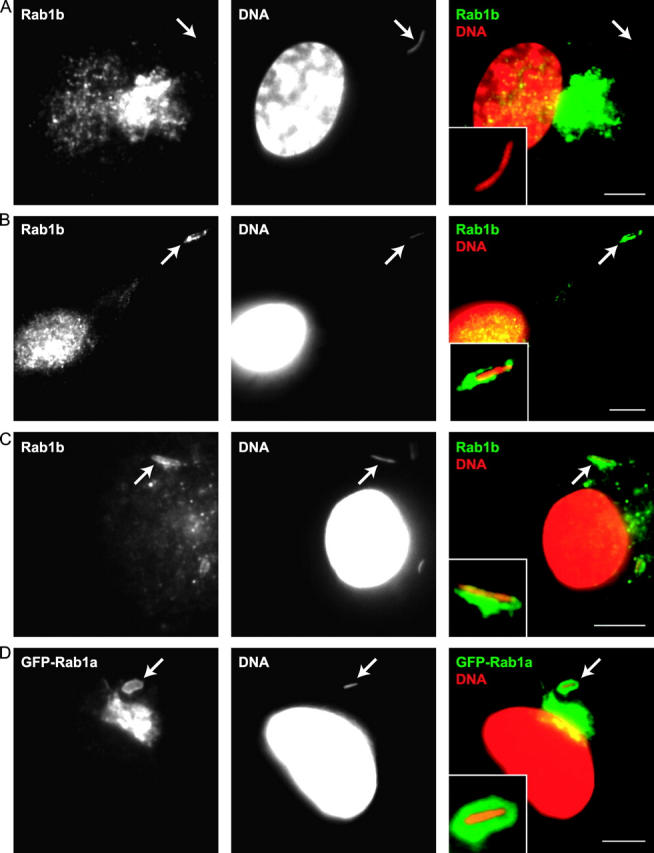
Recruitment of Rab1 to the LCV requires the Dot/Icm system and is independent of vesicular transport from the ER. (A and B) BMMs were fixed 30 min after infection with a Legionella dotA mutant (A) or ralF mutant (B) and stained with a Rab1b-specific antibody. Arrows indicate the location of the LCV that is magnified in the inset of each panel. (C) Rab1b staining is detected on an LCV containing wild-type Legionella in BMMs treated with brefeldin A. Notice that the expected Golgi fragmentation resulting from brefeldin A treatment has eliminated the intense Rab1b-specific signal observed in the untreated cells in A and B. (D) GFP-Rab1a localizes to an LCV containing wild-type Legionella in CHO FcγRII cells. Bars, 5 μm.
Figure 3.

Requirements for Rab1 recruitment to the LCV are similar in macrophages and CHO FcγRII cells. BMMs (gray bars) or CHO FcγRII cells (black bars) were infected with wild-type Legionella (WT), dotA mutants, or ralF mutants as indicated on the x axis. Cells treated with brefeldin A during infection with wild-type Legionella are indicated (WT + BFA). Cells were fixed 30 min after infection, and the percent of LCVs staining positive for Rab1 was measured by fluorescence microscopy. Values are the mean ± SE of three independent experiments in which 50 vacuoles were scored for each condition.
The majority of LCVs stained positive for Rab1 at 30 min after infection (Fig. 1 D). Rab1 staining diminished over time with fewer LCVs staining positive for Rab1 at 4 h after infection (Fig. 1 D). The kinetics of Rab1 staining of the LCV were more rapid than that observed previously for ARF1 (3) and correlated with a stage of vacuole maturation in which LCVs have intercepted ER-derived vesicles, but have not yet taken on the appearance of an ER-like organelle (3, 12, 15). The Legionella protein RalF, which plays an important role in the recruitment of ARF to the LCV by functioning as an guanine nucleotide exchange factor for ARF (6), was not required for Rab1 recruitment to the LCV (Figs. 2 B and 3). Treatment of BMMs with brefeldin A, which blocks delivery of early secretory markers to the LCV by disrupting vesicular transport from ER exit sites, did not affect Rab1 recruitment to the LCV (Figs. 2 C and 3). These data indicate that recruitment of Rab1 to the LCV is a Dot/Icm-mediated process that does not require remodeling of the LCV by early secretory vesicles.
Inhibition of Rab1 Function Interferes with Intracellular Replication of Legionella.
To determine whether Rab1 function is important for intracellular replication of Legionella, a plasmid encoding the GDP-locked dominant-interfering protein Rab1S25N was cotransfected into CHO cells along with a plasmid encoding the FcγRII protein. Because CHO cells are normally nonphagocytic, the uptake of Legionella is inefficient. However, cotransfection of a plasmid encoding FcγRII allowed uptake of IgG-opsonized Legionella preferentially into cells that were transiently transfected (Supplemental Results, available at http://www.jem.org/cgi/content/full/jem.20031706/DC1). Using this approach, we consistently found >90% of the internalized bacteria residing in transfected cells; even after intracellular growth of Legionella in untransfected cells over a 24-h period, the total number of bacteria in these untransfected cells did not equal the starting number of bacteria in the transfected cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20031706/DC1). Using this assay, we have confirmed previous studies based on single cell analysis to show that the interfering variants ARF1T31N and Sar1H79G both disrupt intracellular replication of Legionella (Fig. 4 A). Intracellular growth of Legionella in cells producing Rab1S25N was significantly reduced compared with control cells producing GFP alone (Fig. 4 A). These data indicate that Rab1 function is important for conversion of the LCV into an organelle that supports intracellular replication of Legionella.
Figure 4.
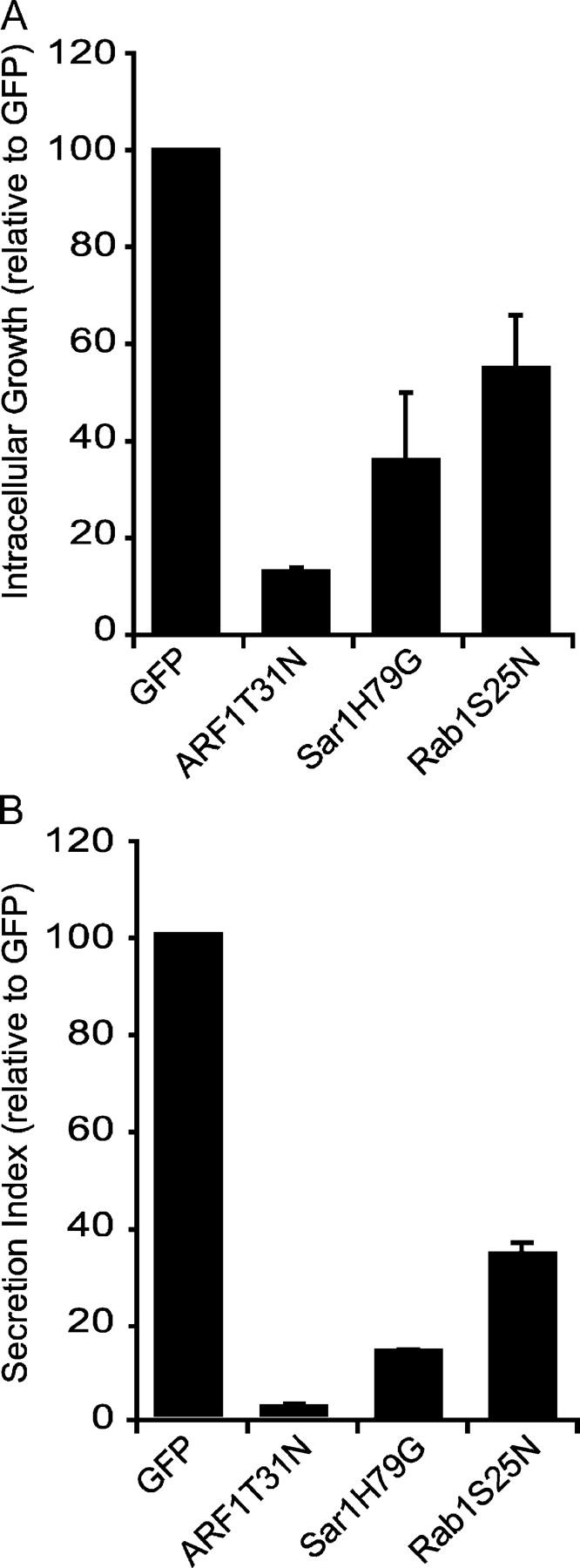
Rab1 is important for intracellular growth of Legionella. (A and B) CHO cells were cotransfected with plasmids encoding FcγRII and the proteins indicated on the x axis of the graphs. (A) Growth of IgG-opsonized Legionella was calculated over a 24-h period by measuring the fold increase in CFUs. Because absolute values will vary in independent experiments, data for cells producing a dominant-interfering protein are presented as percents of an internal control, which were CHO cells that received a plasmid encoding GFP. Values are the mean ± SE from three independent experiments in which Legionella growth was averaged for each condition from three independent wells. These data indicate that Rab1S25N production significantly interferes with the intracellular growth of Legionella (Student's unpaired t test, P = 0.0074). (B) SEAP assays were performed on CHO cells transfected with plasmids encoding the indicated proteins, and the protein secretion index is displayed. Values are the mean ± SE for three independent assay wells.
The degree of Legionella growth inhibition observed using Rab1S25N was not as pronounced as that seen using ARF1T31N or Sar1H79G. To determine how effective these proteins were at disrupting secretory traffic, plasmids encoding SEAP were cotransfected into CHO cells along with plasmids encoding the proteins being used to disrupt the secretory pathway. These data show that ARF1T31N, Sar1H79G, and Rab1S25N all disrupt protein secretion (Fig. 4 B). The degree to which interfering variants affect protein secretion correlates with the severity of defects observed for intracellular replication of Legionella.
Rab1 Function Is Important for the Delivery of the Sec22b Protein to the LCV.
The primary role of Rab1 is to facilitate the transport and docking of ER-derived vesicles with the cis-Golgi apparatus. Docking of these vesicles promotes SNARE-mediated vesicle fusion. To determine whether Rab1 is playing a similar role on the LCV, we examined whether SNARE proteins that mediate fusion of ER-derived vesicles with target membranes are delivered to the LCV. Toward this end, we examined the localization of both the v-SNARE protein Sec22b and the t-SNARE protein Membrin. Distinct colocalization of Sec22b was observed on the LCV in CHO-FcγRII cells (Fig. 5 A). Vacuoles containing dotA mutant Legionella did not acquire Sec22b (Fig. 5 B). Interestingly, Membrin colocalization was not detected on LCVs (Fig. 5 C). Similar results were obtained in transfected BMMs (unpublished data). Sec22b colocalization was inhibited by brefeldin A (Fig. 5 D). Consistent with results obtained with brefeldin A, inhibiting the production of vesicles from ER exit sites using Sar1H79G or ARF1T31N also had a potent effect on delaying the delivery of Sec22b to the LCV (Fig. 5 D). In contrast with what was observed for Rab1, Sec22b recruitment to the LCV requires vesicular transport from the ER. These data indicate that Rab1 binding to the LCV precedes the delivery of Sec22b.
Figure 5.
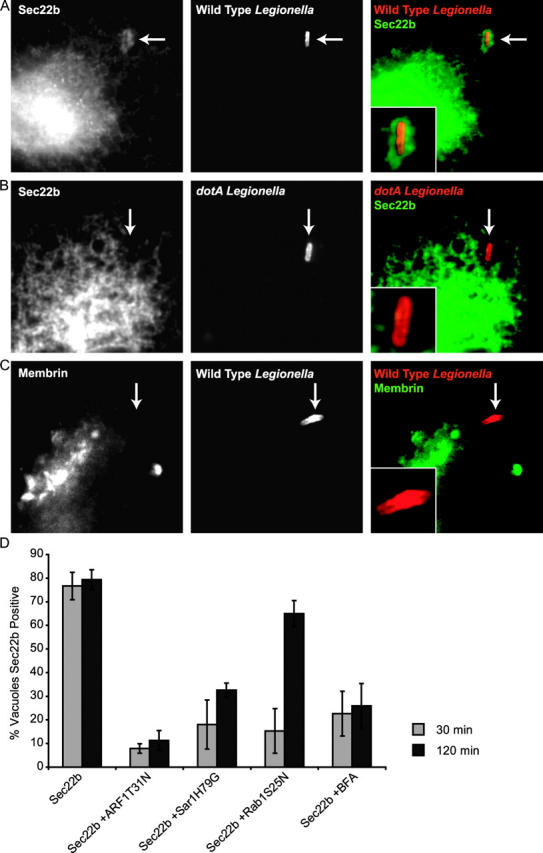
ER-derived vesicles recruited to the LCV contain Sec22b. Sec22b and Membrin staining was examined in CHO FcγRII cells fixed 30 min after Legionella infection. (A) Sec22b staining was observed on vacuoles containing wild-type Legionella. (B) Sec22b staining was absent from vacuoles containing dotA mutants. (C) Membrin staining was absent from vacuoles containing wild-type Legionella. (D) Sec22b staining of vacuoles containing wild-type Legionella was measured in CHO FcγRII cells by immunofluorescence microscopy at 30 and 120 min after infection. The percentage of LCVs that stained positive for Sec22b at these times was determined in CHO FcγRII cells transfected with plasmids encoding the indicated proteins. Data show that Sec22b transport to the LCV requires production of ER-derived vesicles and Rab1 function. Values are the mean ± SE of three independent experiments in which 50 vacuoles were scored for each condition.
Because Rab1 coordinates the tethering and fusion of ER-derived vesicles with target membranes, next we asked whether inhibiting Rab1 function had any effect on the kinetics of Sec22b localization to the LCV. Ectopic production of the GDP-locked Rab1S25N protein potently inhibited Sec22b localization to LCVs examined 30 min after infection (Fig. 5 D). Within 2 h, most LCVs had acquired Sec22b in cells producing Rab1S25N, which indicates that there is a kinetic defect in Sec22b transport to the LCV but not an absolute block, consistent with Rab1 being involved in vesicle tethering rather than vesicle production. These data suggest that Rab1 recruitment to the LCV promotes the transport and fusion of ER-derived vesicles containing Sec22b.
The Sec22b Protein Is Delivered from ER-derived Vesicles to the Vacuole Membrane Surrounding Legionella.
To determine whether ER-derived vesicles containing Sec22b fuse with the LCV, immunogold labeling studies were conducted. Electron micrographs clearly show Sec22b-specific gold particles labeling both vesicles attached to the LCV (Fig. 6, A and B, arrowheads) and on the vacuole membrane surrounding Legionella (Fig. 6, A and B, arrows). Sec22b-specific gold particles were not detected on the vacuole membrane surrounding dotA mutant Legionella (Fig. 6, C and D), even though gold particles were in abundance on ER membranes in close proximity to these vacuoles (Fig. 6, C and D, asterisks). These data indicate that Sec22b is transferred from the attached vesicles to the limiting membrane of the vacuole that surrounds Legionella. Because Sec22b is a transmembrane protein, the presence of Sec22b on the LCV membrane indicates that attached ER-derived vesicles have undergone fusion with the LCV.
Figure 6.
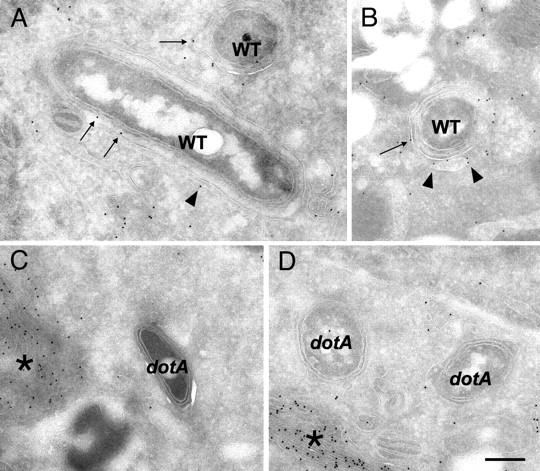
ER-derived vesicles deliver Sec22b to the LCV. (A–D) CHO cells cotransfected with plasmids encoding FcγRII and Sec22b were infected with Legionella and vacuoles containing wild-type (A and B) or dotA mutant (C and D) bacteria were examined by immunoelectron microscopy. (A and B) Sec22b-specific gold particles are found surrounding vacuoles containing wild-type Legionella (WT). Arrowheads identify Sec22b on the vesicles attached to LCVs. Long arrows identify Sec22b on the vacuole membrane. (C and D) Vacuoles harboring dotA mutant Legionella are devoid of Sec22b-specific gold particles. Although there is clear Sec22b-specific labeling of ER membrane (asterisk) in these infected cells, gold particles are notably absent around and on vacuoles containing dotA mutant Legionella. Bar = 0.5 μm.
Sec22b Function Is Important for Establishment of the Legionella Replicative Organelle.
Overproduction of an individual SNARE protein can have a dominant negative effect on specific vesicular transport processes by affecting the proper distribution and functioning of cognate protein binding partners, including other SNARE proteins (24, 52). Previous studies have shown that overproduction of Membrin, and to a lesser extent overproduction of Sec22b, can interfere with the transport of early secretory vesicles (24). We found that overproduction of Membrin interfered with the intracellular growth of Legionella; however, overproduction of Sec22b did not affect Legionella replication (Fig. 7 A). Overproduction of the trans-Golgi SNARE protein VAMP4 did not affect intracellular growth of Legionella (Fig. 7 A). Defects in secretion of an alkaline phosphatase reporter were observed in cells overproducing either Membrin or VAMP4, but not in cells overproducing Sec22b (Fig. 7 B). These data suggest that overproduction of Membrin might specifically interfere with intracellular growth of Legionella by titration of a host factor that is important for replicative organelle biogenesis.
Figure 7.
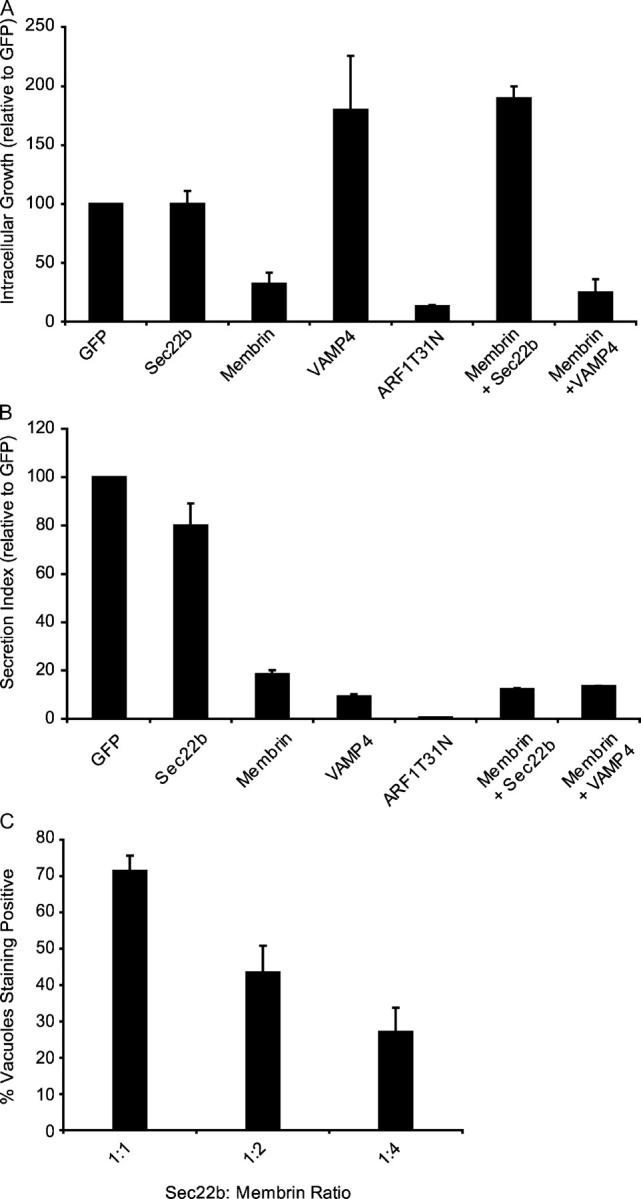
Sec22b function is important for biogenesis of the Legionella replicative vacuole. (A and B) CHO cells were cotransfected with plasmids encoding FcγRII and the proteins indicated on the x axis of the graph. (A) Growth of IgG-opsonized Legionella was measured over a 24-h period by measuring the fold increase in CFUs. Values were normalized to control cells that received a plasmid encoding GFP. Values are the mean ± SE for three independent assay wells. (B) SEAP assays demonstrate that overproduction of either Membrin or VAMP4 interferes with alkaline phosphatase secretion by a process that is not suppressed by Sec22b overproduction. Values are the mean ± SE for three independent assay wells. (C) CHO FcγRII cells were cotransfected with plasmids encoding Sec22b and Membrin. The ratio of Sec22b:Membrin plasmid DNA used in each transfection is indicated on the x axis. Data show that delivery of Sec22b to the LCV is inhibited as the amount of plasmid encoding Membrin increases. Data represent the mean ± SE for three independent assay wells in which at least 20 vacuoles were scored in each assay.
To test whether this effect might be due to titration of Sec22b, we overexpressed both Membrin and Sec22b in cells by cotransfection and assayed intracellular replication of Legionella. These data show that ectopic production of Sec22b can suppress Legionella growth restriction mediated by Membrin overproduction (Fig. 7 A). VAMP4 overproduction did not suppress the Legionella growth restriction caused by ectopic production of Membrin. Importantly, coproduction of Sec22b did not suppress the defect in secretion of alkaline phosphatase caused by Membrin overproduction. These data strongly suggest that Membrin overproduction restricts the growth of Legionella by titration of Sec22b as opposed to the more general effect Membrin overproduction has on protein secretion. Consistent with the hypothesis that Membrin overproduction titrates available Sec22b, we found that increasing the amounts of transfected plasmid DNA encoding Membrin resulted in a decrease in the percent of LCVs that stained positive for Sec22b (Fig. 7 C). These data indicate that Membrin overproduction prevents the delivery of Sec22b to the LCV. Thus, both morphological and functional data indicate that Sec22b plays an important role in the creation of the Legionella replicative organelle.
Discussion
The goal of this paper was to define transport pathways and host factors involved in the creation of the Legionella replicative organelle. Toward this end, first we examined the localization of Rab protein family members that are involved in vesicle transport between the ER and Golgi apparatus. These studies revealed that Rab1 is recruited to vacuoles containing wild-type Legionella and is not found on vacuoles containing dotA mutants (Fig. 1). The proteins Rab2 and Rab6, which (like Rab1) are involved in vesicular transport between the ER and Golgi apparatus, were not found on LCVs. These data indicate that Rab1 recruitment to the LCV is specific and controlled by the Dot/Icm system.
Rab1 recruitment to the LCV was not inhibited by brefeldin A (Fig. 2). This finding is significant because brefeldin A has been shown to block the recruitment of other early secretory proteins that display LCV colocalization (3). Because brefeldin A inhibits production and transport of ER-derived vesicles, these data demonstrate that Rab1 recruitment to the LCV is not coincident with remodeling of this compartment by ER-derived vesicles, suggesting a role for Rab1 on the LCV before the binding of these vesicles. Interference of Rab1 function by the Rab1S25N protein inhibited intracellular replication of Legionella (Fig. 4). These data indicate that Rab1 is playing an important role in biogenesis of the organelle in which Legionella replicates.
Because Rab1 is involved in the recruitment of factors required for the tethering and fusion of ER-derived vesicles with target membranes (16, 17), we examined whether the SNARE proteins involved in this fusion reaction were recruited to the LCV. Our data show that the Sec22b protein was recruited to LCVs (Figs. 5 and 6). Sec22b recruitment to the LCV was blocked by inhibiting the production of ER-derived vesicles with either Sar1H79G, ARF1T31N, or brefeldin A, demonstrating that Sec22b is transported to the LCV in secretory vesicles. The finding that Sec22b recruitment to the LCV is delayed in cells producing Rab1S25N indicates that Rab1 function is important for delivering cargo in ER-derived vesicles to the LCV. These data indicate that the principle role for Rab1 on the LCV is to promote transport and fusion of ER-derived vesicles with this organelle.
Immunogold labeling was used to determine whether ER-derived vesicles fuse with the LCV. These studies show that the Sec22b protein is present both on the membrane of vesicles attached to the LCV and also on the membrane surrounding Legionella (Fig. 6). Because Sec22b is a transmembrane protein that initially inserts in the ER membrane and exits the ER in early secretory vesicles, detection of the Sec22b protein on the LCV membrane provides evidence that the ER-derived vesicles attached to the LCV are fusing with this organelle. The absence of Sec22b from the membrane of vacuoles containing dotA mutants of Legionella is consistent with previous data showing that the transport and attachment of ER-derived vesicles to the LCV requires the Dot/Icm system (3, 12–14). Although this is the first direct evidence that ER-derived vesicles fuse with the LCV, these data are consistent with previous electron microsopy data, which had suggested that fusion of attached vesicles could account for a thinning of the LCV membrane that was observed shortly after bacterial uptake (12).
Given that cognate SNAREs form cis–SNARE complexes that colocalize in the cell, it was surprising to find that the Sec22b protein was enriched on the LCV, but that the cognate SNARE protein Membrin was undetectable on these vacuoles (Fig. 5). Recent studies examining COPII-directed recruitment of Saccharomyces cerevisiae homologues of these mammalian SNAREs might provide an explanation for the specific recruitment of Sec22b to the LCV (29, 53, 54). Yeast Sec22p can exit the ER in either a monomeric form or in a cis complex with other SNARE proteins. The recruitment of Sec22p is directed by the Sec23/24p subcomplex, which can bind to a motif in Bet1p and recruit cis–SNARE complexes that contain Sec22p into ER-derived vesicles. Additionally, there is a spatially distinct binding site on the Sec23/24p subcomplex that can recruit a monomeric form of Sec22p into ER-derived vesicles. Because the Sec22p binding site in the Sec23/24p complex is likely to be conserved between yeast and mammalian homologues, the packaging of Sec22b into ER-derived vesicles should be regulated similarly. Given the distinct binding site for Sec22b in COPII, it is possible that the LCV preferentially recruits COPII-generated vesicles enriched for the monomeric form of Sec22b, which would explain the absence of cognate SNARE proteins that bind to Sec22b.
It has been shown that dominant negative effects on membrane transport sometimes result from the overproduction of a single SNARE protein (24, 52), presumably by affecting the proper subcellular distribution of cognate proteins to which that SNARE protein binds, including other SNAREs. Overproduction of the SNARE protein Membrin was found to inhibit the intracellular growth of Legionella by a process that could be suppressed by overproduction of Sec22b (Fig. 7). Although secretion of alkaline phosphatase from cells was inhibited upon overproduction of the SNARE proteins Membrin or VAMP4, only Membrin overproduction interfered with Legionella replication. Additionally, overproduction of Sec22b did not suppress the secretory defect caused by overproduction of Membrin, indicating that inhibition of Legionella replication was not simply a consequence of a general perturbation in the secretory pathway caused by overproduction of a SNARE protein. These data are consistent with monomeric Sec22b being an important SNARE protein that is used for biogenesis of the Legionella replicative organelle. Accordingly, overproduction of Membrin interferes with replicative organelle biogenesis because it effectively titrates monomeric Sec22b. Overproduction of Sec22b can suppress the inhibition of Legionella intracellular growth caused by Membrin overproduction by restoring cellular pools of monomeric Sec22b but does not restore secretion defects caused by the titration of the other proteins to which Membrin binds. Direct evidence in support of this hypothesis comes from data showing that increasing the amount of Membrin produced in host cells interferes with the delivery of Sec22b to the LCV. These data provide the first functional evidence that host ER–Golgi SNARE proteins play an important role in formation of the Legionella replicative organelle.
Based on these data, we hypothesize that LCVs preferentially recruit vesicles created by COPII that contain the monomeric form of the Sec22b protein. We demonstrate that Rab1 plays an important role in the recruitment of these Sec22b-containing vesicles to the LCV. Because the LCV is derived from the plasma membrane, the t-SNARE complex composed of Membrin, Bet1, and Syntaxin5 would not be present on this compartment. To initiate fusion with ER-derived vesicles, it is possible that Legionella proteins injected into the vacuole membrane by the Dot/Icm system could serve as a functionally equivalent t-SNARE complex for a Sec22b-dependent fusion reaction. Alternatively, a t-SNARE complex present at the plasma membrane might pair with Sec22b and promote the fusion of ER-derived organelles with the LCV. In this second model, the main role for Legionella proteins would be to recruit proteins such as Rab1 that can facilitate vesicle tethering and facilitate membrane fusion. Interestingly, previous in vitro studies using purified components have shown that the S. cerevisiae Sec22 protein can promote membrane fusion with a plasma membrane t-SNARE complex consisting of the proteins Sso1 and Sec9p (55). Although it has not been shown that the mammalian Sec22b protein can function as a v-SNARE for fusion with a plasma membrane t-SNARE, recent studies that implicate direct fusion between the ER and phagosomes suggest that such interactions are possible (56). Thus, it is likely that determining the mechanism of membrane fusion between ER-derived vesicles and the LCV will reveal new insights into the cell biology of phagosome maturation and vesicular transport in the early secretory pathway.
Acknowledgments
We thank J. Hay, N. Andrews, G. Warren, and J. Donaldson for generously providing plasmids; M. Beard and A. Neild for helpful discussions; and K. Zichichi for technical support.
This work was supported by National Institutes of Health grant AI41699 to C.R. Roy and an Anna Fuller postdoctoral fellowship award to M.P. Stein.
The online version of this article contains supplemental material.
Abbreviations used in this paper: ARF, ADP ribosylation factor; BMM, bone marrow–derived macrophage; CHO, Chinese hamster ovary; FSG, fish skin gelatin; GFP, green fluorescent protein; GTPase, guanosine triphosphatase; LCV, Legionella-containing vacuole; SEAP, secreted alkaline phosphatase; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; t-SNARE, SNARE found on target membrane; v-SNARE, SNARE found on vesicle membrane.
References
- 1.Horwitz, M.A. 1983. The Legionnaires' disease bacterium (Legionella pneumophila) inhibits phagosome lysosome fusion in human monocytes. J. Exp. Med. 158:2108–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horwitz, M.A., and F.R. Maxfield. 1984. Legionella pneumophila inhibits acidification of its phagosome in human monocytes. J. Cell Biol. 99:1936–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kagan, J.C., and C.R. Roy. 2002. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4:945–954. [DOI] [PubMed] [Google Scholar]
- 4.Roy, C.R., K. Berger, and R.R. Isberg. 1998. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol. Microbiol. 28:663–674. [DOI] [PubMed] [Google Scholar]
- 5.Wiater, L.A., K. Dunn, F.R. Maxfield, and H.A. Shuman. 1998. Early events in phagosome establishment are required for intracellular survival of Legionella pneumophila. Infect. Immun. 66:4450–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagai, H., J.C. Kagan, X. Zhu, R.A. Kahn, and C.R. Roy. 2002. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. 295:679–682. [DOI] [PubMed] [Google Scholar]
- 7.Segal, G., M. Purcell, and H.A. Shuman. 1998. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. USA. 95:1669–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogel, J.P., H.L. Andrews, S.K. Wong, and R.R. Isberg. 1998. Conjugative transfer by the virulence system of Legionella pneumophila. Science. 279:873–876. [DOI] [PubMed] [Google Scholar]
- 9.Luo, Z.Q., and R.R. Isberg. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. USA. 101:841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clemens, D.L., and M.A. Horwitz. 1996. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J. Exp. Med. 184:1349–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clemens, D.L., B.Y. Lee, and M.A. Horwitz. 2000. Deviant expression of rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infect. Immun. 68:2671–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tilney, L.G., O.S. Harb, P.S. Connelly, C.G. Robinson, and C.R. Roy. 2001. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114:4637–4650. [DOI] [PubMed] [Google Scholar]
- 13.Berger, K.H., and R.R. Isberg. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7:7–19. [DOI] [PubMed] [Google Scholar]
- 14.Swanson, M.S., and R.R. Isberg. 1995. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63:3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horwitz, M.A. 1983. Formation of a novel phagosome by the Legionnaires' disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158:1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moyer, B.D., B.B. Allan, and W.E. Balch. 2001. Rab1 interaction with a GM130 effector complex regulates COPII vesicle cis–Golgi tethering. Traffic. 2:268–276. [DOI] [PubMed] [Google Scholar]
- 17.Allan, B.B., B.D. Moyer, and W.E. Balch. 2000. Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science. 289:444–448. [DOI] [PubMed] [Google Scholar]
- 18.Zahraoui, A., N. Touchot, P. Chardin, and A. Tavitian. 1989. The human Rab genes encode a family of GTP-binding proteins related to yeast YPT1 and SEC4 products involved in secretion. J. Biol. Chem. 264:12394–12401. [PubMed] [Google Scholar]
- 19.Rothman, J.E., and G. Warren. 1994. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr. Biol. 4:220–233. [DOI] [PubMed] [Google Scholar]
- 20.Lian, J.P., S. Stone, Y. Jiang, P. Lyons, and S. Ferro-Novick. 1994. Ypt1p implicated in v-SNARE activation. Nature. 372:698–701. [DOI] [PubMed] [Google Scholar]
- 21.Sogaard, M., K. Tani, R.R. Ye, S. Geromanos, P. Tempst, T. Kirchhausen, J.E. Rothman, and T. Sollner. 1994. A rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell. 78:937–948. [DOI] [PubMed] [Google Scholar]
- 22.Zhang, T., S.H. Wong, B.L. Tang, Y. Xu, and W. Hong. 1999. Morphological and functional association of Sec22b/ERS-24 with the pre-Golgi intermediate compartment. Mol. Biol. Cell. 10:435–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu, D., A.P. Joglekar, A.L. Williams, and J.C. Hay. 2000. Subunit structure of a mammalian ER/Golgi SNARE complex. J. Biol. Chem. 275:39631–39639. [DOI] [PubMed] [Google Scholar]
- 24.Hay, J.C., D.S. Chao, C.S. Kuo, and R.H. Scheller. 1997. Protein interactions regulating vesicle transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Cell. 89:149–158. [DOI] [PubMed] [Google Scholar]
- 25.Nakano, A., and M. Muramatsu. 1989. A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J. Cell Biol. 109:2677–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bi, X., R.A. Corpina, and J. Goldberg. 2002. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 419:271–277. [DOI] [PubMed] [Google Scholar]
- 27.Barlowe, C. 2002. Three-dimensional structure of a COPII prebudding complex. Dev. Cell. 3:467–468. [DOI] [PubMed] [Google Scholar]
- 28.Haucke, V. 2003. Vesicle budding: a coat for the COPs. Trends Cell Biol. 13:59–60. [DOI] [PubMed] [Google Scholar]
- 29.Miller, E., B. Antonny, S. Hamamoto, and R. Schekman. 2002. Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J. 21:6105–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aridor, M., J. Weissman, S. Bannykh, C. Nuoffer, and W.E. Balch. 1998. Cargo selection by the COPII budding machinery during export from the ER. J. Cell Biol. 141:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barlowe, C., L. Orci, T. Yeung, M. Hosobuchi, S. Hamamoto, N. Salama, M.F. Rexach, M. Ravazzola, M. Amherdt, and R. Schekman. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77:895–907. [DOI] [PubMed] [Google Scholar]
- 32.Barlowe, C. 1995. COPII: a membrane coat that forms endoplasmic reticulum-derived vesicles. FEBS Lett. 369:93–96. [DOI] [PubMed] [Google Scholar]
- 33.Barlowe, C. 2003. Signals for COPII-dependent export from the ER: what's the ticket out? Trends Cell Biol. 13:295–300. [DOI] [PubMed] [Google Scholar]
- 34.Scales, S.J., R. Pepperkok, and T.E. Kreis. 1997. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 90:1137–1148. [DOI] [PubMed] [Google Scholar]
- 35.Aridor, M., S.I. Bannykh, T. Rowe, and W.E. Balch. 1995. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 131:875–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ward, T.H., R.S. Polishchuk, S. Caplan, K. Hirschberg, and J. Lippincott-Schwartz. 2001. Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 155:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lippincott-Schwartz, J., N.B. Cole, and J.G. Donaldson. 1998. Building a secretory apparatus: role of ARF1/COPI in Golgi biogenesis and maintenance. Histochem. Cell Biol. 109:449–462. [DOI] [PubMed] [Google Scholar]
- 38.Coers, J., J.C. Kagan, M. Matthews, H. Nagai, D.M. Zuckman, and C.R. Roy. 2000. Identification of Icm protein complexes that play distinct roles in the biogenesis of an organelle permissive for Legionella pneumophila intracellular growth. Mol. Microbiol. 38:719–736. [DOI] [PubMed] [Google Scholar]
- 39.Zuckman, D.M., J.B. Hung, and C.R. Roy. 1999. Pore-forming activity is not sufficient for Legionella pneumophila phagosome trafficking and intracellular growth. Mol. Microbiol. 32:990–1001. [DOI] [PubMed] [Google Scholar]
- 40.Celada, A., P.W. Gray, E. Rinderknecht, and R.D. Schreiber. 1984. Evidence for a γ-interferon receptor that regulates macrophage tumoricidal activity. J. Exp. Med. 160:55–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puck, T.T., S.J. Cieciura, and A. Robinson. 1958. Genetics of somatic mammalian cells. III. Long-term cultivation of euploid cells from human and animal subjects. J. Exp. Med. 108:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joiner, K.A., S.A. Fuhrman, H.M. Miettinen, L.H. Kasper, and I. Mellman. 1990. Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor-transfected fibroblasts. Science. 249:641–646. [DOI] [PubMed] [Google Scholar]
- 43.Peters, P.J., V.W. Hsu, C.E. Ooi, D. Finazzi, S.B. Teal, V. Oorschot, J.G. Donaldson, and R.D. Klausner. 1995. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J. Cell Biol. 128:1003–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naviaux, R.K., E. Costanzi, M. Haas, and I.M. Verma. 1996. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70:5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satoh, A., Y. Wang, J. Malsam, M.B. Beard, and G. Warren. 2003. Golgin-84 is a rab1 binding partner involved in Golgi structure. Traffic. 4:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evan, G.I., G.K. Lewis, G. Ramsay, and J.M. Bishop. 1985. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5:3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tisdale, E.J. 1999. A Rab2 mutant with impaired GTPase activity stimulates vesicle formation from pre-Golgi intermediates. Mol. Biol. Cell. 10:1837–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mallard, F., B.L. Tang, T. Galli, D. Tenza, A. Saint-Pol, X. Yue, C. Antony, W. Hong, B. Goud, and L. Johannes. 2002. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J. Cell Biol. 156:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White, J., L. Johannes, F. Mallard, A. Girod, S. Grill, S. Reinsch, P. Keller, B. Tzschaschel, A. Echard, B. Goud, and E.H. Stelzer. 1999. Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J. Cell Biol. 147:743–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Girod, A., B. Storrie, J.C. Simpson, L. Johannes, B. Goud, L.M. Roberts, J.M. Lord, T. Nilsson, and R. Pepperkok. 1999. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat. Cell Biol. 1:423–430. [DOI] [PubMed] [Google Scholar]
- 51.Chavrier, P., R.G. Parton, H.P. Hauri, K. Simons, and M. Zerial. 1990. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 62:317–329. [DOI] [PubMed] [Google Scholar]
- 52.Dascher, C., and W.E. Balch. 1996. Mammalian Sly1 regulates syntaxin 5 function in endoplasmic reticulum to Golgi transport. J. Biol. Chem. 271:15866–15869. [DOI] [PubMed] [Google Scholar]
- 53.Miller, E.A., T.H. Beilharz, P.N. Malkus, M.C. Lee, S. Hamamoto, L. Orci, and R. Schekman. 2003. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. 114:497–509. [DOI] [PubMed] [Google Scholar]
- 54.Mossessova, E., L.C. Bickford, and J. Goldberg. 2003. SNARE selectivity of the COPII coat. Cell. 114:483–495. [DOI] [PubMed] [Google Scholar]
- 55.McNew, J.A., F. Parlati, R. Fukuda, R.J. Johnston, K. Paz, F. Paumet, T.H. Sollner, and J.E. Rothman. 2000. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407:153–159. [DOI] [PubMed] [Google Scholar]
- 56.Gagnon, E., S. Duclos, C. Rondeau, E. Chevet, P.H. Cameron, O. Steele-Mortimer, J. Paiement, J.J. Bergeron, and M. Desjardins. 2002. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 110:119–131. [DOI] [PubMed] [Google Scholar]