

Abstract
Somatic hypermutation (SHM) and class switch recombination (CSR) are initiated in activated B lymphocytes by activation-induced deaminase (AID). AID is thought to make lesions in DNA by deaminating cytidine residues in single-stranded DNA exposed by RNA polymerase during transcription. Although this must occur in the nucleus, AID is found primarily in the cytoplasm. Here we show that AID is actively excluded from the nucleus by an exportin CRM1-dependent pathway. The AID nuclear export signal (NES) is found at the carboxyl terminus of AID in a region that overlaps a sequence required for CSR but not SHM. We find that AID lacking a functional NES causes more hypermutation of a nonphysiologic target gene in transfected fibroblasts. However, the NES does not impact on the rate of mutation of immunoglobulin genes in B lymphocytes, suggesting that the AID NES does not limit AID activity in these cells.
Keywords: somatic hypermutation, activation-induced deaminase, nucleo-cytoplasmic trasnport, B lymphocytes, Ig class switching
Introduction
In the course of immune responses, two distinct DNA modification reactions, somatic hypermutation (SHM) and class switch recombination (CSR), diversify antibody genes. SHM introduces mutations into the variable region exons of antibody genes thereby producing a set of variant antibodies that can be selected on the basis of antigen-binding affinity (1). CSR is a DNA recombination reaction that joins two Ig switch regions by deletion of intervening sequences. This diversifies antibody responses by producing related clones of B cells where a single variable region exon is combined with different constant region exons with unique effector functions (2–6).
Despite the clear differences in the DNA modification processes that result in SHM and CSR, both reactions appear to be initiated by a single enzyme, activation-induced deaminase (AID). AID is expressed specifically in activated B cells that undergo CSR and SHM and it is essential for both reactions in humans and mice (7–9). Further, AID is the only B cell–specific factor required for SHM and CSR because both reactions can be induced in fibroblasts transfected with AID (10–12).
AID is homologous to APOBEC-1, an mRNA editing cytidine deaminase, and it was first proposed that AID might initiate SHM or CSR indirectly by editing mRNA to create a novel nuclease (7, 8). An alternative possibility is that AID initiates SHM and CSR by deaminating cytidines in DNA (13). Cytidine deaminated to uridine could be replicated to produce a transition mutation (13) or be repaired by the mismatch repair pathway (14–20). Alternatively, the mismatched uracil could be removed by uracil-DNA glycosylase to produce an abasic site that might be processed by nucleases and error-prone polymerases to cause SHM or CSR (19–24).
The DNA deamination hypothesis is supported by cell biologic, genetic, and biochemical studies (13, 19, 20, 25–30). AID–estrogen receptor fusion proteins are only active when they are targeted to the nucleus (29), and AID is physically associated with Ig switch region DNA in B cells undergoing CSR (31). Mutations that result in loss of DNA uracil-DNA glycosylase activity interfere with SHM and CSR in Escherichia coli, chicken DT40 cell lines, and mice (13, 19, 20). Biochemical experiments show that AID binds single-stranded DNA (ssDNA), that it deaminates cytidine residues in ssDNA in vitro (26–28), and that it is required for switch region break formation (30). Finally, experiments in E. coli have shown that AID deaminates ssDNA exposed by RNA polymerase during transcription, consistent with the known link between transcription and SHM (25). Thus, the available evidence favors the DNA deamination hypothesis but fails to account for the cytoplasmic localization of AID.
Appropriate cellular localization is a key event for the proper physiological function of proteins. Movement of proteins to and from the nucleus occurs through nuclear pore complexes (for review see references 32 and 33). The nuclear pore complexes allow passive diffusion of molecules up to ∼60 kD in size into and out of the nucleus, whereas transport of larger proteins requires soluble shuttling receptors that recognize nuclear localization signals (NLSs) or nuclear export signals (NESs) for movement into or out of the nucleus, respectively (34, 35). CRM1 (chromosome region maintenance/exportin 1) is a ubiquitous soluble shuttling receptor that binds leucine-rich NESs and translocates them out of the nucleus (34, 35). CRM1 facilitates active export by interacting directly with the Ran GTPase and nuclear pore components.
AID has been observed to localize to the cytoplasm with exclusion from the nucleus (36). Here we report that AID contains an NES recognized by CRM1 that prevents it from accumulating in the nucleus. The AID NES is found at the C terminus of AID in a region that is required for CSR but not SHM.
Materials and Methods
Mouse B Cell Cultures and Retroviral Infection.
Wild-type or AID−/− B cells were purified from mouse spleen by depletion with anti-CD43 magnetic beads (Miltenyi Biotec) and cultured in RPMI 1640 medium with 5 ng/ml IL-4 plus LPS (Sigma-Aldrich). Retroviral supernatants for mouse B cell infection were obtained by cotransfecting BOSC 23 cells with pMX-PIE and pCL-ECO plasmids using Fugene 6 (Roche; reference 37). Supernatants were harvested 24 h after transfection and used to infect B cells that had been cultured in LPS plus IL-4 for 1 d. 1 d after infection, the retroviral supernatant was replaced. Infection efficiency was in the range of 30% as determined by green fluorescent protein (GFP) expression. Retroviruses for infecting 3T3-NTZ cells (12) were produced using pQCXIP and pCL-ECO plasmids (CLONTECH Laboratories, Inc.). 3T3-NTZ were cultured in the absence of tetracycline and selected in puromycin starting 1 d after infection. All assays on 3T3-NTZ cells were performed on puromycin-selected cells.
Flow Cytometry and Cell Sorting.
Mouse B cells were stained with biotinylated anti–mouse IgG1 antibodies and visualized with streptavidin PE-Cy7 (Becton Dickinson) 72 h after infection with retrovirus. Data was acquired on a FACSCalibur™ and analyzed with the CELLQuest™ software, FACSCalibur™ (both from Becton Dickinson). GFP+ and IgG1+ cells were sorted on a FACS Vantage™ SE cell sorter (BD Immunocytometry Systems). GFP expression in 3T3-NTZ cells was analyzed on a FACSCalibur™ as previously described (12).
PCR and Mutation Analysis.
Switch μ region genomic DNA was amplified from 100,000 sorted cells by PCR using Pfu Turbo DNA polymerase (Stratagene). Amplification conditions were 25 cycles at 94°C for 30 s, 60°C for 30 s, and 72°C for 40 s. Primers were μ switch region: 5μ-570 (5′-GGGTTTGAATTTTGAATCTATTCTG-3′) and SμR5 (5′-GCGGCCCGGCTCATTCCAGTTCATTACAG-3′; reference 38). These amplify GenBank/EMBL/DDBJ accession number 109111111–109111733 of mouse chromosome 12 and do not discriminate between germline and recombined switch μ region. For mutational analysis, the GFP gene from NTZ-3T3 DNA was amplified 3 d after infection using Pfu Turbo polymerase. Amplification conditions were 25 cycles at 94°C for 30 s, 57°C for 30 s, and 72°C for 1 min (12) using the primers CMVPF (5′-TGACCTCCATAGAAGACACCG-3′) and TetGFPR (5′-TTATGTTTCAGGTTCAGGGGG-3′). PCR products were cloned using TOPO-TA cloning kit (Invitrogen) and sequenced using T7 universal primers. Sequence analysis was performed using Sequence Manager II software (DNASTAR).
Microscopy.
293 cells were plated on glass coverslips, transfected with GFP fusion constructs, and evaluated 48 h after transfection. Cells were fixed with 4% formaldehyde and made permeable with 0.1% Triton X-100. Nuclear membranes were stained with mAb-414 (39) and visualized by donkey anti–mouse Alexa Fluor 555 (Molecular Probes). Micrographs were produced using a Carl Zeiss MicroImaging, Inc. LSM 510 confocal microscope. AID−/− B cells were infected with pQCXIP AID-GFP retrovirus 48 h before staining and evaluation. Leptomycin B (LMB; Sigma-Aldrich) treatment was for 1 h at 10 ng/ml before fixation.
Plasmid Constructs and E. coli Assays.
Pyruvate kinase (PK) or PK fused to the NLS from the human K protein (PKNLS; reference 40) was fused to the amino terminus of GFP and cloned into pCDNA3 (Invitrogen). Murine AID or its variants were fused to the carboxyl terminus of GFP with a GGST linker to produce PK-GFP-AID or PKNLS-GFP. To produce NES-GFP or AID188–198-GFP, a double-stranded oligonucleotide corresponding to MAPKK NES (MDLQKKLEELELDE) or AID NES (MDLRDAFRMLGF) was fused to the amino terminus of GFP. Retroviral expression constructs were produced by inserting AID or AID fused to the amino terminus of GFP (AID-GFP) into pMX-PIE or pQCXIP (CLONTECH Laboratories, Inc.). E. coli assays were performed exactly as previously described (25).
Protein Analysis.
Rabbit AID antibody was produced using a peptide derived from the carboxyl terminus of AID (EVDDLRDAFRMLGF) coupled to keyhole limpet haemocyanin. For Western blot assays, cells were lysed in 50 mM Tris, pH 8.0, 400 mM NaCl, 0.5% NP-40, 10% glycerol. Western blots were performed on 40 ug protein using the anti-AID antibody or anti-PK antibody (Polysciences Inc.), and normalized based on PK and GFP expression in infected B cells, PK in puromycin-selected 3T3-NTZ cells, or total cell protein in E. coli.
Results
AID Contains an NES.
To determine whether cellular localization of AID is actively regulated, we made use of a PK-GFP reporter (PK-GFP; reference 40). PK-GFP is found in the cytoplasm of transfected 293 cells because it lacks an NLS and is too large to pass through nuclear pores by diffusion (Fig. 1; references 40 and 41). We found that addition of AID to PK-GFP (PK-GFP-AID) produced a protein that like PK-GFP, localizes to the cytoplasm (Fig. 1). Addition of an exogenous NLS to the PK-GFP indicator (PKNLS-GFP) targets the chimeric protein to the cell nucleus where it remains because PKNLS-GFP lacks an NES (40). However, PKNLS-GFP-AID differed from PKNLS-GFP in that it remained cytoplasmic despite the NLS, suggesting that AID either anchors the chimeric protein to the cytoplasm or that it is actively exported from the nucleus (Fig. 1).
Figure 1.
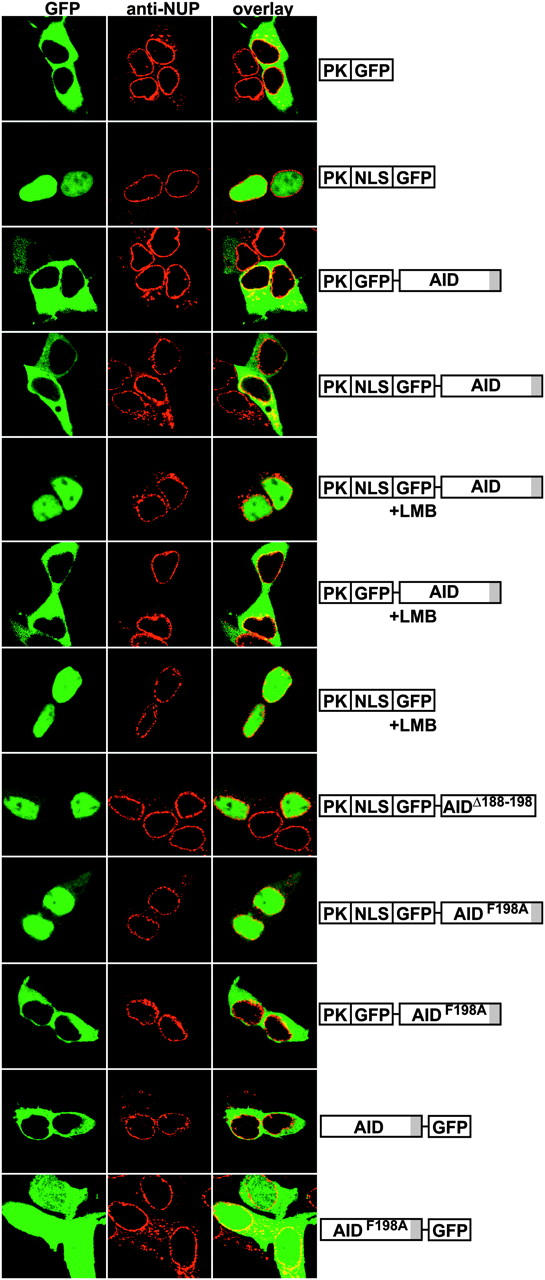
AID has an NES. Diagrams at right (not to scale) show transfected indicator constructs. +LMB indicates that transfected cells were treated with 10 nM LMB for 1 h. Representative confocal micrographs show 293 cells expressing green GFP and red nuclear membranes stained with fluorescent anti-nuclear pore (NUP) antibodies.
To determine if nuclear export controls PKNLS-GFP-AID exclusion from the nucleus, we treated cells with the CRM1 exportin inhibitor LMB (42). Treatment of PKNLS-GFP-AID–expressing cells with LMB for 1 h caused a dramatic shift in reporter localization from cytoplasm to nucleus (Fig. 1). However, LMB treatment had no effect on cells expressing control reporters without the NLS or AID (Fig. 1, PK-GFP-AID and PKNLS-GFP, respectively). The nearly complete nuclear accumulation of PKNLS-GFP-AID after LMB treatment indicates that cytoplasmic localization of PKNLS-GFP-AID is due to AID-dependent CRM1-mediated nuclear export. Thus, AID contains an NES that actively regulates its subcellular localization.
To define the AID NES, we systematically deleted amino acids from the carboxyl and amino terminus of AID and tested the mutant proteins in combination with the PKNLS-GFP reporter (unpublished data). We found that the last 10 residues of AID are essential for export and that deletion of these residues (PKNLS-GFP-AIDΔ188–198) resulted in nuclear accumulation of the reporter as opposed to cytoplasmic localization of the intact reporter (Fig. 1, compare PKNLS-GFP-AIDΔ188–198 and PKNLS-GFP-AID). Proteins transported by CRM1 have an NES characterized by a short peptide sequence rich in leucine or hydrophobic residues, and alignment of authentic NESs with residues 188–198 of AID reveals that the sequence conforms to an NES consensus (Table I; reference 43). To confirm that AID188–198 is an NES, we made a point mutation changing phenylalanine198 to alanine AIDF198A and tested the mutant AID in our nuclear export assay as a fusion protein (PKNLS-GFP-AIDF198A). We found that PKNLS-GFP-AIDF198A had no export activity in transfected 293 cells and showed nuclear accumulation similar to PKNLS-GFP-AIDΔ188–198 (Fig. 1). We conclude that the sequence between amino acids 188–198 in AID contains an active NES.
Table I.
Alignment of AID Residues with Authentic NESs
| AID | 188 | D | L | R | D | A | F | R | M | L | G | F | 198 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAPKK | A | L | Q | K | K | L | E | E | L | E | L | ||
| PKIα | E | L | A | L | K | L | A | G | L | D | I | ||
| STAT1 | S | L | A | A | E | F | R | H | L | Q | L | ||
| Cyclin B | D | L | C | Q | A | F | S | D | V | L | I | ||
| HIV-Rev | L | Q | L | P | P | L | E | R | L | T | L |
Conserved leucines and hydrophobic amino acids critical to NES function are in bold.
To determine whether AID has an endogenous NLS, we removed the exogenous NLS from PKNLS-GFP-AIDF198A to produce PK-GFP-AIDF198A (Fig. 1). In contrast to PKNLS-GFP-AIDF198A, which is constitutively imported and trapped in the nucleus because it lacks an NES, PK-GFP-AIDF198A was found in the cytoplasm of transfected cells. Consistent with this finding, inhibition of CRM1 by LMB in cells transfected with PK-GFP-AID also failed to show nuclear accumulation of the indicator (Fig. 1). We conclude that AID does not have a constitutive NLS capable of actively transporting PK fusion proteins into the nucleus.
To ensure that the amino terminal fusions of GFP to AID did not interfere with normal localization, we fused GFP to the carboxyl terminus of AID (AID-GFP) and expressed it in 293 cells (Fig. 1). As reported by others, AID-GFP was found in the cytoplasm (36, 44). Mutation of the NES (AIDF198A-GFP) resulted redistribution to the cytoplasm and nuclear compartments. This localization is consistent with passive diffusion because the size of this chimeric protein (50 kD) is less then the nuclear pore exclusion limit. Thus, we do not detect a constitutive NLS in AID regardless of the GFP tag location.
To determine whether AID188–198 can function as an autonomous NES, we fused it directly to GFP. GFP is small enough to diffuse freely through nuclear pores and is found in both nucleus and cytoplasm (Fig. 2). Addition of AID188–198 peptide to GFP (AID188–198-GFP) resulted in nuclear exclusion similar to that of GFP fused with an authentic NES peptide from MAPKK (Fig. 2, NES-GFP), and a point mutation changing phenylalanine 198 to alanine inactivated nuclear exclusion (Fig. 2, AID188–198, F198A-GFP). Finally, the AID NES mediates nuclear export by the CRM1 pathway because LMB treatment reversed the nuclear exclusion of AID188–198-GFP (Fig. 2). We conclude that AID188–198 constitutes an NES recognized by CRM1.
Figure 2.
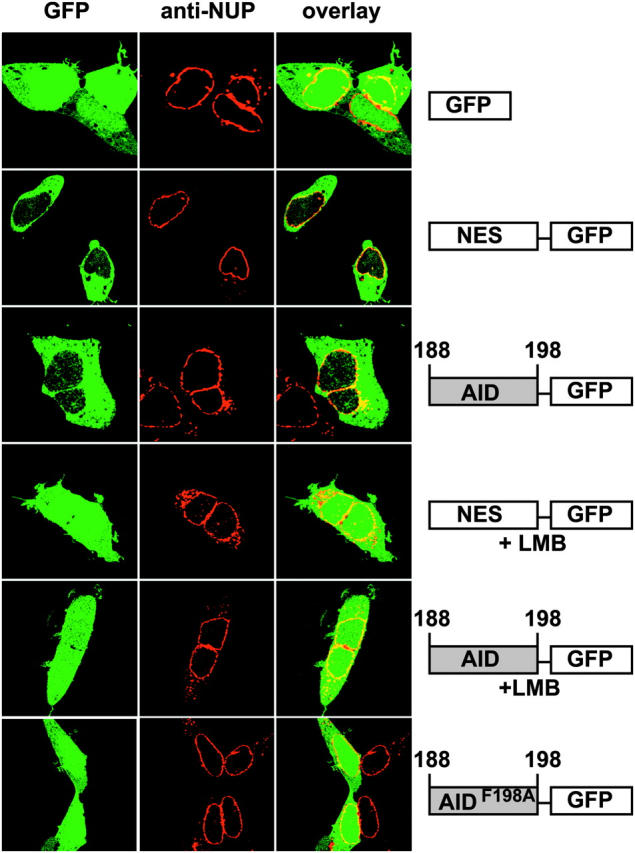
AID residues 188–198 comprise a functional NES. Diagrams show maps (not to scale) of transfected indicator constructs. +LMB indicates that transfected cells were treated with 10 nM LMB for 1 h. Representative confocal micrographs show 293 cells expressing green GFP and red nuclear membranes stained with fluorescent anti-NUP antibodies.
The CRM1 export pathway is found in all cell types, but B cells may have specific factors that control the nucleo-cytoplasmic movement of AID. To examine the regulation of AID in B cells undergoing CSR, AID-GFP or AIDF198A-GFP was expressed in LPS plus IL-4–stimulated AID−/− B cells by retrovirus infection. 48 h after infection, AID-GFP was localized in the cytoplasm and excluded from the nucleus (Fig. 3; reference 36). In contrast, AIDF198A-GFP, with a defective NES, was found in both the nuclear and cytoplasmic compartments, a result consistent with free diffusion of a 52-kD protein that is neither actively imported into nor exported from the B cell nucleus (Fig. 3). We conclude that the AID NES controls the subcellular localization of AID in B cells.
Figure 3.

Nuclear export controls AID localization in B cells. (A) Retroviral vector used to infect B cells. (B) AID-GFP or AIDF198A-GFP expressed in LPS and IL-4–stimulated AID−/− B cells after retroviral infection. Diagrams (not to scale) of indicator constructs (right). Representative confocal micrographs show green GFP and red nuclear membranes stained with fluorescent anti-NUP antibodies.
The AID NES Limits SHM in 3T3-NTZ Cells.
To determine whether the NES affects AID activity, we measured mutation in E. coli (25). Indicator uracil-DNA glycosylase-deficient E. coli carry an inactive kanamycin (Kan) gene (kanL94P, TTGL94→CCAP94) that can be reverted to Kan resistance (KanR) by C to T mutation. Thus, Kan-resistant colonies are a measure of the cytidine deamination activity of AID. AIDF198A and wild-type AID were expressed at comparable levels in E. coli as determined by Western blotting (Fig. 4 A) and showed a slightly higher level of mutator activity (Fig. 4 B; AIDF198A = 1.7 × AID, P = 0.027). We conclude that AIDF198A is marginally more active than wild-type AID.
Figure 4.

SHM by NES mutant AID in E. coli. (A) AID, AIDF198A, and AIDL62A (a catalytically inactive control; reference 25) protein expression in E. coli measured by Western blotting with rabbit anti–AID antiserum. (B) SHM measured by Kan-resistance assay for AID-mediated cytidine deamination in UDG (uracil DNA glycosylase)-deficient E. coli. Circles represent the mutation frequency of individual starting colonies and horizontal bars represent mean values. Results from two independent experiments are shown. Mean frequencies of KanR colonies are indicated underneath each group.
To determine whether nuclear export in eukaryotic cells regulates SHM, we used 3T3-NTZ indicator cells, which carry an actively transcribed GFP gene with a premature stop codon whose reversion can be assayed by flow cytometry (12, 45). AID expression in 3T3-NTZ initially induces GFP fluorescence, but prolonged AID expression inactivates GFP by accumulation of deleterious mutations (12). Although AIDF198A was expressed at 3.6-fold lower levels than AID by Western blotting (Fig. 5 B), AIDF198A-infected 3T3-NTZ cells initially showed greater SHM than AID-infected cells (Fig. 5 C). At later time points the percentage of GFP expression by AIDF198A-infected 3T3-NTZ cells leveled off and then decreased relative to wild-type AID (Fig. 5 C). To determine whether this pattern of GFP expression was due to increased SHM, we sequenced the GFP gene from AID and AIDF198A-expressing 3T3-NTZ cells 3 d after infection. Relative to AIDF198A, AID showed a small increase in the number of mutated GFP genes (35 vs. 51%, respectively, P = 0.08) and a significant increase in the number of mutations per GFP gene (Fig. 5 D). The average number of mutations per mutated clone in AIDF198A was 4.4 compared with 1.5 for AID (Fig. 5 D, P = 0.003), with a maximum of 3 mutations per clone in AID and a maximum of 16 mutations per clone in AIDF198A (Fig. 5 D). In addition, 10 out of 77 GFP clones sequenced from AIDF198A showed deletions ranging from 11–45 bp, but there were no deletions in 79 clones from AID-expressing cells (Fig. 5 E). Despite the increase in rate of mutation, AIDF198A showed the same hotspot preferences as AID (Fig. 5 E). We conclude that nuclear export limits AID-mediated SHM in 3T3-NTZ cells.
Figure 5.

SHM by NES-mutant AID in transfected fibroblasts. (A) Diagrammatic representation of retroviral vector used to infect 3T3-NTZ cells. (B) AID and AIDF198A protein expression in 3T3-NTZ cells after 3 d of culture measured by Western blotting of 15 μg whole cell extracts. Anti-PK Western blot served as a loading control. (C) Percent of GFP expressing 3T3-NTZ cells measured by flow cytometry (y axis) and plotted over time in days (x axis). Results show mean of triplicate cultures from one of n = 5 representative experiments. (D) Proportion of GFP sequences cloned from AID or AIDF198A-expressing 3T3-NTZ cells 3 d after infection carrying a different number of mutations. Segment sizes in the pie charts are proportional to the number of sequences carrying the number of mutations indicated in the periphery of the charts. The total number of independent sequences analyzed is indicated in the center of each chart. Deletions, duplications, and insertions were excluded from this analysis. (E) Comparison of GFP gene sequences cloned from AID or AIDF198A-expressing 3T3-NTZ cells after 3 d of culture. Single nucleotide substitutions are indicated above AID or below AIDF198A, the authentic GFP sequence. Bold indicates deletions and boxes indicate WRC (R, purine; W, A or T) hotspots (reference 70) are underlined.
Switch Region Hypermutation and CSR in B cells.
To determine whether nuclear export also regulates SHM and CSR in B cells, we measured the rate of CSR and hypermutation of the switch μ region in B cells expressing AIDF198A. AID−/− B cells infected with retroviruses encoding either AID or AIDF198 and a GFP indicator were stimulated with LPS plus IL-4 to undergo CSR and switch μ mutation (Fig. 6 A). 72 h after infection, CSR was measured by surface IgG1 expression and mutation was evaluated by cloning and sequencing a region 5′ of the Igμ switch repeats from IgG1-expressing B cells. AID−/− B cells showed 3.5-fold lower levels of AIDF198A protein than wild-type AID by Western blotting (Fig. 6 B), and lower levels of CSR (Fig. 6 C; 60% of AID in n = 3 experiments). AIDF198A also showed a decrease in the number of clones that contained mutations in the switch μ region relative to AID (Fig. 6 D; 17 vs. 32%, respectively, P = 0.006). However, within each switch μ region that was mutated, the average number of mutations was similar in that AIDF198A had 1.7 compared with 1.4 for AID (Fig. 6 D; P = 0.3), with a similar distribution, spectrum, and hotspot preference (Fig. 6 E).
Figure 6.

SHM by NES mutant AID in B cells. (A) Representation of retroviral vectors used to infect B cells. (B) AID and AIDF198A protein expression in AID−/− B cells after 3 d of culture measured by Western blotting of whole cell extracts. Anti-PK Western blot served as a loading control. The results are representative of three independent experiments. (C) CSR in AID−/− spleen B cells expressing AID or AIDF198A. Dot plots show IgG1 surface expression on AID or AIDF198A-expressing AID−/− B cells that had been stimulated with LPS plus IL-4 for 3 d. Numbers indicate the percentage of GFP+ cells that were IgG1+. The results are representative of three independent experiments. (D) Proportion of GFP sequences cloned from AID or AIDF198A-expressing, surface IgG1+ AID−/− B cells 3 d after retrovirus infection carrying a different number of mutations. Segment sizes in the pie charts are proportional to the number of sequences carrying the number of mutations indicated in the periphery of the charts. The total number of independent sequences analyzed is indicated in the center of each chart. Deletions, duplications, and insertions were excluded from this analysis. (E) Comparison of Sμ sequences cloned from AID or AIDF198A-expressing, surface IgG1+ AID−/− B cells 3 d after retrovirus infection. Single nucleotide substitutions are indicated above (AID) or below (AIDF198A) the authentic Sμ sequence. Hollow boxes indicate deletions. WRC (R, purine; W, A or T) hotspots (reference 70) are underlined.
Discussion
AID initiates SHM and CSR by deaminating cytidine residues in ssDNA, a reaction that has the potential to produce significant collateral genomic damage. Nevertheless, most B cells expressing AID do not suffer widespread mutation or chromosome instability (46–48). Our experiments show that AID contains an NES that controls its subcellular localization and limits nonphysiologic target gene mutation in fibroblasts. However, the NES does not appear to impact on Ig switch region mutation in B lymphocytes.
We have not addressed the question of how AID gets into the nucleus. In this study we found no direct evidence for a constitutive NLS in AID. By itself, AID is small enough to passively diffuse into the nucleus. We used a PK-GFP indicator, which is too large to enter the nucleus by diffusion, to discriminate between active transport and diffusion (40, 41). AID was unable to transport the PK-GFP to the nucleus even when nuclear export was blocked by LMB. We also failed to find nuclear accumulation of AID in cells expressing an amino terminal–tagged NES mutant AID (GFP-AIDF198A; Fig. 1). Instead, GFP-AIDF198A was found in the cytoplasm and nucleus of fibroblasts and B cells undergoing CSR, a result consistent with passive diffusion.
Although we see no evidence of active import, the amino terminus of AID contains an amino acid sequence that resembles a bipartite NLS, and it has been reported that this region is required for the nuclear accumulation of AID (44). AID is homologous to APOBEC-1, an mRNA editing enzyme (49), and an analogous basic region required for nuclear accumulation is also found in the amino terminus of APOBEC-1 (50–52). This region of APOBEC-1 is involved in nuclear accumulation, but it does not appear to function as an autonomous NLS because it cannot direct heterologous proteins such as PK or GST to the nucleus (50–52). Mutagenesis studies on this region of APOBEC-1 reveal that the arginine and lysine residues, normally the critical elements of a bipartite NLS, can be mutated without loss of nuclear accumulation (50, 52). Furthermore, the carboxyl terminal domain (residues 97–172) of APOBEC-1 is also critical for nuclear accumulation APOBEC-1 (51). All of these characteristics indicate that if APOBEC-1 has an NLS, it certainly is an atypical one. A recent study by Ito et al. (44) also suggested that the same amino terminal region of AID plays a critical role in localizing AID to the nucleus. However, the authors used an AID-GFP, which differed from ours in the linker sequence between AID and GFP, and because of its size, did not discriminate between active nuclear transport and passive diffusion with nuclear retention (44). If the studies of APOBEC-1 serve as any indication, then the regulation of AID nuclear transport might be a complex process.
In fibroblasts, absence of the AID nuclear export was associated with increased SHM of a nonantibody substrate, but the hotspot target preference and the nature of the mutations did not change. The increase in mutation was due to an increase in the frequency of mutated genes and an increase in the absolute number of mutations per altered sequence. In addition, we found increased numbers of deletions of the target gene. We interpret this as due to a greater frequency of AID-induced lesions that increase the probability of a double strand DNA break.
We did not find increased CSR or mutation in the 5′ of switch region in B cells stimulated with LPS and IL-4 when we blocked nuclear export. Thus, increasing the nuclear concentration of AID is sufficient to enhance mutation of a nonantibody gene in fibroblasts but not on a normal target of AID, the switch μ region in B cells.
The NES in AID overlaps a region that is essential for CSR but not required for SHM in humans and mice (residues 189–198; references 37 and 53). Consistent with this observation, AIDF198A shows decreased CSR (60% of wild-type; Fig. 6) and a more dramatic defect is seen with deletion of the three carboxyl terminal amino acid residues (10% of wild-type; reference 44), underlining the importance of the carboxyl terminus of AID in CSR. AIDΔ189–198 differs from AIDF198A in that it shows a 10-fold increase in catalytic activity in E. coli when compared with wild-type AID, and it has lost the ability to mediate CSR entirely. Despite nuclear trapping by loss of the NES and the 10-fold increase in catalytic activity of AIDΔ189–198, this and related mutants do not show enhanced Ig switch region or Ig V region SHM (37, 53). The function of the last 10 amino acids in CSR has not been determined, but this region of the molecule does not appear to be essential for targeting to the antibody switch regions or in limiting SHM because B cells expressing AIDΔ189–198 show normal switch μ lesion formation. It has been proposed that the carboxyl terminus functions in the synapsis phase of the CSR reaction (37).
Transcription targets SHM to a 1.5–2 kb region starting at the 5′ boundary of the variable region exon (54, 55), but the sequence of the exon does affect the rate of mutation (56, 57). Furthermore, there is no promoter specificity for SHM because non-Ig promoters are sufficient for SHM when combined with Ig enhancers (58–61). Transcription also targets CSR to specific switch regions by intronic promoters 5′ of switch DNA (62, 63), and CSR is absent after deletion of these promoters or their regulatory elements (64–67). How transcription targets AID to antibody genes is still to be determined, but the finding that in E. coli transcription facilitates SHM by exposing ssDNA suggests that one link between mutation and transcription might be that RNA polymerase uncovers the ssDNA template for AID (25). Thus, highly transcribed genes loaded with many molecules of polymerase would display more ssDNA making them better substrates for AID. This idea is supported by experiments in transfected cell lines overexpressing AID where non-Ig genes are targeted for mutation by a mechanism that is directly dependent on the rate of target gene transcription (11, 12, 68, 69). Our experiments show that increasing the nuclear concentration of AID in these cells also increases the amount of SHM and that regulated nuclear export of AID might be one of the molecular devices that restricts AID activity on nonantibody genes in fibroblasts. In contrast, in B cells, increasing nuclear AID does not increase Ig gene mutation and therefore mutation must be limited either by target gene availability, i.e., the rate of Ig gene transcription, or yet to be identified AID-interacting factors.
Acknowledgments
We are indebted to members of the Nussenzweig laboratory; E. Besmer, F. Papavasiliou, and T. Honjo for discussions, AID−/− mice, and 3T3-NZT cells; K. Velinzon for cell sorting; and G. Blobel and J. Enninga for anti-NUP antibodies and help with confocal microscopy.
This work was supported in part by grants from the United States National Institutes of Health (NIH) and the Leukemia Society to M.C. Nussenzweig. M.C. Nussenzweig is a Howard Hughes Medical Institute Investigator, K.M. McBride is supported by a fellowship from the NIH, V. Barreto is a fellow of the Fundação para a Ciência e Tecnologia (Portugal), and A.R. Ramiro is a fellow of the Ministerio de Educacion, Cultura y Deporte (Spain).
Abbreviations used in this paper: AID, activation-induced deaminase; CSR, class switch recombination; GFP, green fluorescent protein; Kan, kanamycin; LMB, leptomycin B; NES, nuclear export signal; NLS, nuclear localization signal; PK, pyruvate kinase; SHM, somatic hypermutation; ssDNA, single-stranded DNA.
References
- 1.McKean, D., K. Huppi, M. Bell, L. Staudt, W. Gerhard, and M. Weigert. 1984. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA. 81:3180–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honjo, T., and T. Kataoka. 1978. Organization of immunoglobulin heavy chain genes and allelic deletion model. Proc. Natl. Acad. Sci. USA. 75:2140–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stavnezer, J. 1996. Immunoglobulin class switching. Curr. Opin. Immunol. 8:199–205. [DOI] [PubMed] [Google Scholar]
- 4.Iwasato, T., A. Shimizu, T. Honjo, and H. Yamagishi. 1990. Circular DNA is excised by immunoglobulin class switch recombination. Cell. 62:143–149. [DOI] [PubMed] [Google Scholar]
- 5.Matsuoka, M., K. Yoshida, T. Maeda, S. Usuda, and H. Sakano. 1990. Switch circular DNA formed in cytokine-treated mouse splenocytes: evidence for intramolecular DNA deletion in immunoglobulin class switching. Cell. 62:135–142. [DOI] [PubMed] [Google Scholar]
- 6.von Schwedler, U., H.M. Jack, and M. Wabl. 1990. Circular DNA is a product of the immunoglobulin class switch rearrangement. Nature. 345:452–456. [DOI] [PubMed] [Google Scholar]
- 7.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 8.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 9.Muramatsu, M., V.S. Sankaranand, S. Anant, M. Sugai, K. Kinoshita, N.O. Davidson, and T. Honjo. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476. [DOI] [PubMed] [Google Scholar]
- 10.Martin, A., P.D. Bardwell, C.J. Woo, M. Fan, M.J. Shulman, and M.D. Scharff. 2002. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature. 415:802–806. [DOI] [PubMed] [Google Scholar]
- 11.Okazaki, I.M., K. Kinoshita, M. Muramatsu, K. Yoshikawa, and T. Honjo. 2002. The AID enzyme induces class switch recombination in fibroblasts. Nature. 416:340–345. [DOI] [PubMed] [Google Scholar]
- 12.Yoshikawa, K., I.M. Okazaki, T. Eto, K. Kinoshita, M. Muramatsu, H. Nagaoka, and T. Honjo. 2002. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 296:2033–2036. [DOI] [PubMed] [Google Scholar]
- 13.Petersen-Mahrt, S.K., R.S. Harris, and M.S. Neuberger. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–103. [DOI] [PubMed] [Google Scholar]
- 14.Bertocci, B., L. Quint, F. Delbos, C. Garcia, C.A. Reynaud, and J.C. Weill. 1998. Probing immunoglobulin gene hypermutation with microsatellites suggests a nonreplicative short patch DNA synthesis process. Immunity. 9:257–265. [DOI] [PubMed] [Google Scholar]
- 15.Kim, N., G. Bozek, J.C. Lo, and U. Storb. 1999. Different mismatch repair deficiencies all have the same effects on somatic hypermutation: intact primary mechanism accompanied by secondary modifications. J. Exp. Med. 190:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rada, C., M.R. Ehrenstein, M.S. Neuberger, and C. Milstein. 1998. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 9:135–141. [DOI] [PubMed] [Google Scholar]
- 17.Schrader, C.E., W. Edelmann, R. Kucherlapati, and J. Stavnezer. 1999. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 190:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiesendanger, M., B. Kneitz, W. Edelmann, and M.D. Scharff. 2000. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 191:579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Noia, J., and M.S. Neuberger. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 419:43–48. [DOI] [PubMed] [Google Scholar]
- 20.Rada, C., G.T. Williams, H. Nilsen, D.E. Barnes, T. Lindahl, and M.S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755. [DOI] [PubMed] [Google Scholar]
- 21.Faili, A., S. Aoufouchi, E. Flatter, Q. Gueranger, C.A. Reynaud, and J.C. Weill. 2002. Induction of somatic hypermutation in immunoglobulin genes is dependent on DNA polymerase iota. Nature. 419:944–947. [DOI] [PubMed] [Google Scholar]
- 22.Rogozin, I.B., Y.I. Pavlov, K. Bebenek, T. Matsuda, and T.A. Kunkel. 2001. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat. Immunol. 2:530–536. [DOI] [PubMed] [Google Scholar]
- 23.Zan, H., A. Komori, Z. Li, A. Cerutti, A. Schaffer, M.F. Flajnik, M. Diaz, and P. Casali. 2001. The translesion DNA polymerase zeta plays a major role in Ig and bcl-6 somatic hypermutation. Immunity. 14:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng, X., D.B. Winter, C. Kasmer, K.H. Kraemer, A.R. Lehmann, and P.J. Gearhart. 2001. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2:537–541. [DOI] [PubMed] [Google Scholar]
- 25.Ramiro, A.R., P. Stavropoulos, M. Jankovic, and M.C. Nussenzweig. 2003. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4:452–456. [DOI] [PubMed] [Google Scholar]
- 26.Bransteitter, R., P. Pham, M.D. Scharff, and M.F. Goodman. 2003. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA. 100:4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaudhuri, J., M. Tian, C. Khuong, K. Chua, E. Pinaud, and F.W. Alt. 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 422:726–730. [DOI] [PubMed] [Google Scholar]
- 28.Dickerson, S.K., E. Market, E. Besmer, and F.N. Papavasiliou. 2003. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 197:1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doi, T., K. Kinoshita, M. Ikegawa, M. Muramatsu, and T. Honjo. 2003. Inaugural article: de novo protein synthesis is required for the activation-induced cytidine deaminase function in class-switch recombination. Proc. Natl. Acad. Sci. USA. 100:2634–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen, S., R. Casellas, B. Reina-San-Martin, H.T. Chen, M.J. Difilippantonio, P.C. Wilson, L. Hanitsch, A. Celeste, M. Muramatsu, D.R. Pilch, et al. 2001. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 414:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nambu, Y., M. Sugai, H. Gonda, C.-G. Lee, T. Katakai, Y. Agata, Y. Yokota, and A. Shimizu. 2003. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 302:2137–2140. [DOI] [PubMed] [Google Scholar]
- 32.Davis, L.I. 1995. The nuclear pore complex. Annu. Rev. Biochem. 64:865–896. [DOI] [PubMed] [Google Scholar]
- 33.Rout, M.P., and J.D. Aitchison. 2001. The nuclear pore complex as a transport machine. J. Biol. Chem. 276:16593–16596. [DOI] [PubMed] [Google Scholar]
- 34.Pemberton, L.F., G. Blobel, and J.S. Rosenblum. 1998. Transport routes through the nuclear pore complex. Curr. Opin. Cell Biol. 10:392–399. [DOI] [PubMed] [Google Scholar]
- 35.Mattaj, I.W., and L. Englmeier. 1998. Nucleocytoplasmic transport: the soluble phase. Annu. Rev. Biochem. 67:265–306. [DOI] [PubMed] [Google Scholar]
- 36.Rada, C., J.M. Jarvis, and C. Milstein. 2002. AID-GFP chimeric protein increases hypermutation of Ig genes with no evidence of nuclear localization. Proc. Natl. Acad. Sci. USA. 99:7003–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barreto, V., B. Reina-San-Martin, A.R. Ramiro, K.M. McBride, and M.C. Nussenzweig. 2003. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol. Cell. 12:501–508. [DOI] [PubMed] [Google Scholar]
- 38.Reina-San-Martin, B., S. Difilippantonio, L. Hanitsch, R.F. Masilamani, A. Nussenzweig, and M.C. Nussenzweig. 2003. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 197:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis, L.I., and G. Blobel. 1986. Identification and characterization of a nuclear pore complex protein. Cell. 45:699–709. [DOI] [PubMed] [Google Scholar]
- 40.Michael, W.M., M. Choi, and G. Dreyfuss. 1995. A nuclear export signal in hnRNP A1: a signal-mediated, temperature-dependent nuclear protein export pathway. Cell. 83:415–422. [DOI] [PubMed] [Google Scholar]
- 41.Paine, P.L., L.C. Moore, and S.B. Horowitz. 1975. Nuclear envelope permeability. Nature. 254:109–114. [DOI] [PubMed] [Google Scholar]
- 42.Wolff, B., J.J. Sanglier, and Y. Wang. 1997. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 4:139–147. [DOI] [PubMed] [Google Scholar]
- 43.McBride, K.M., C. McDonald, and N.C. Reich. 2000. Nuclear export signal located within the DNA-binding domain of the STAT1 transcription factor. EMBO J. 19:6196–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ito, S., H. Nagaoka, R. Shinkura, N. Begum, M. Muramatsu, M. Nakata, and T. Honjo. 2004. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc. Natl. Acad. Sci. USA. 101:1975–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bachl, J., C. Carlson, V. Gray-Schopfer, M. Dessing, and C. Olsson. 2001. Increased transcription levels induce higher mutation rates in a hypermutating cell line. J. Immunol. 166:5051–5057. [DOI] [PubMed] [Google Scholar]
- 46.Shen, H.M., A. Peters, B. Baron, X. Zhu, and U. Storb. 1998. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science. 280:1750–1752. [DOI] [PubMed] [Google Scholar]
- 47.Pasqualucci, L., P. Neumeister, T. Goossens, G. Nanjangud, R.S.K. Chaganti, R. Küppers, and R. Dalla-Favera. 2001. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 412:341–346. [DOI] [PubMed] [Google Scholar]
- 48.Müschen, M., D. Re, B. Jungnickel, V. Diehl, K. Rajewsky, and R. Küppers. 2000. Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. J. Exp. Med. 192:1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teng, B., C.F. Burant, and N.O. Davidson. 1993. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 260:1816–1819. [DOI] [PubMed] [Google Scholar]
- 50.Yang, Y., M.P. Sowden, and H.C. Smith. 2001. Intracellular trafficking determinants in APOBEC-1, the catalytic subunit for cytidine to uridine editing of apolipoprotein B mRNA. Exp. Cell Res. 267:153–164. [DOI] [PubMed] [Google Scholar]
- 51.Yang, Y., and H.C. Smith. 1997. Multiple protein domains determine the cell type-specific nuclear distribution of the catalytic subunit required for apolipoprotein B mRNA editing. Proc. Natl. Acad. Sci. USA. 94:13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chester, A., A. Somasekaram, M. Tzimina, A. Jarmuz, J. Gisbourne, R. O'Keefe, J. Scott, and N. Navaratnam. 2003. The apolipoprotein B mRNA editing complex performs a multifunctional cycle and suppresses nonsense-mediated decay. EMBO J. 22:3971–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ta, V.T., H. Nagaoka, N. Catalan, A. Durandy, A. Fischer, K. Imai, S. Nonoyama, J. Tashiro, M. Ikegawa, S. Ito, et al. 2003. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat. Immunol. 4:843–848. [DOI] [PubMed] [Google Scholar]
- 54.Lebecque, S.G., and P.J. Gearhart. 1990. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5′ boundary is near the promoter, and 3′ boundary is approximately 1 kb from V(D)J gene. J. Exp. Med. 172:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Both, G.W., L. Taylor, J.W. Pollard, and E.J. Steele. 1990. Distribution of mutations around rearranged heavy-chain antibody variable-region genes. Mol. Cell. Biol. 10:5187–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yelamos, J., N. Klix, B. Goyenechea, F. Lozano, Y.L. Chui, A. Gonzalez Fernandez, R. Pannell, M.S. Neuberger, and C. Milstein. 1995. Targeting of non-Ig sequences in place of the V segment by somatic hypermutation. Nature. 376:225–229. [DOI] [PubMed] [Google Scholar]
- 57.Azuma, T., N. Motoyama, L.E. Fields, and D.Y. Loh. 1993. Mutations of the chloramphenicol acetyl transferase transgene driven by the immunoglobulin promoter and intron enhancer. Int. Immunol. 5:121–130. [DOI] [PubMed] [Google Scholar]
- 58.Goyenechea, B., N. Klix, J. Yelamos, G.T. Williams, A. Riddell, M.S. Neuberger, and C. Milstein. 1997. Cells strongly expressing Ig(kappa) transgenes show clonal recruitment of hypermutation: a role for both MAR and the enhancers. EMBO J. 16:3987–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peters, A., and U. Storb. 1996. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 4:57–65. [DOI] [PubMed] [Google Scholar]
- 60.Betz, A.G., C. Milstein, A. Gonzalez-Fernandez, R. Pannell, T. Larson, and M.S. Neuberger. 1994. Elements regulating somatic hypermutation of an immunoglobulin kappa gene: critical role for the intron enhancer/matrix attachment region. Cell. 77:239–248. [DOI] [PubMed] [Google Scholar]
- 61.Fukita, Y., H. Jacobs, and K. Rajewsky. 1998. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity. 9:105–114. [DOI] [PubMed] [Google Scholar]
- 62.Stavnezer-Nordgren, J., and S. Sirlin. 1986. Specificity of immunoglobulin heavy chain switch correlates with activity of germline heavy chain genes prior to switching. EMBO J. 5:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yancopoulos, G.D., R.A. DePinho, K.A. Zimmerman, S.G. Lutzker, N. Rosenberg, and F.W. Alt. 1986. Secondary genomic rearrangement events in pre-B cells: VHDJH replacement by a LINE-1 sequence and directed class switching. EMBO J. 5:3259–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gu, H., Y.R. Zou, and K. Rajewsky. 1993. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 73:1155–1164. [DOI] [PubMed] [Google Scholar]
- 65.Jung, S., K. Rajewsky, and A. Radbruch. 1993. Shutdown of class switch recombination by deletion of a switch region control element. Science. 259:984–987. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, J., A. Bottaro, S. Li, V. Stewart, and F.W. Alt. 1993. A selective defect in IgG2b switching as a result of targeted mutation of the I gamma 2b promoter and exon. EMBO J. 12:3529–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinaud, E., A.A. Khamlichi, C. Le Morvan, M. Drouet, V. Nalesso, M. Le Bert, and M. Cogne. 2001. Localization of the 3′ IgH locus elements that effect long-distance regulation of class switch recombination. Immunity. 15:187–199. [DOI] [PubMed] [Google Scholar]
- 68.Bachl, J., C. Carlson, V. Gray-Schopfer, M. Dessing, and C. Olsson. 2001. Increased transcription levels induce higher mutation rates in a hypermutating cell line. J. Immunol. 166:5051–5057. [DOI] [PubMed] [Google Scholar]
- 69.Lee, C.G., K. Kinoshita, A. Arudchandran, S.M. Cerritelli, R.J. Crouch, and T. Honjo. 2001. Quantitative regulation of class switch recombination by switch region transcription. J. Exp. Med. 194:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rogozin, I.B., and N.A. Kolchanov. 1992. Somatic hypermutagenesis in immunoglobulin genes. II. Influence of neighbouring base sequences on mutagenesis. Biochim. Biophys. Acta. 1171:11–18. [DOI] [PubMed] [Google Scholar]