

Abstract
Interleukin (IL)-10 is a regulator of inflammatory responses and is secreted by a variety of different cell types including T cells. T regulatory cells have been shown to suppress immune responses by IL-10–dependent, but also IL-10–independent, mechanisms. Herein, we address the role of T cell–derived IL-10 in mice with an inactivation of the IL-10 gene restricted to T cells generated by Cre/loxP-mediated targeting of the IL-10 gene. Splenocytes from this T cell–specific mutant secrete increased amounts of proinflammatory cytokines after activation in vitro compared with show enhanced contact hypersensitivity reactions, and succumb to severe immunopathology upon infection with Toxoplasma gondii. Despite intact IL-10 genes in other cell types, the dysregulation of T cell responses observed in the T cell–specific IL-10 mutant closely resembles the phenotype in complete IL-10 deficiency. However, in contrast to complete IL-10 deficiency, sensitivity to endotoxic shock and irritant responses of the skin are not enhanced in the T cell–specific IL-10 mutant. Our data highlight the importance of T cell–derived IL-10 in the regulation of T cell responses and demonstrate that endotoxic shock and the irritant response of the skin are controlled by IL-10 from other cell types.
Keywords: cytokines, gene targeting, inflammation, inflammatory bowel disease, allergic contact dermatitis
Introduction
IL-10 is a regulator of inflammatory responses that limits damage to host tissue by effectors of the immune system in the course of responses to microbial pathogens (1). By inhibiting the activation of macrophages, IL-10 controls responses of the innate immune system. Furthermore, effects of IL-10 on antigen-presenting cells as well as direct suppressive effects on T cells modulate T cell responses by inhibiting Th1 cell differentiation and action (1). IL-10–deficient (IL-10 − / −) mice mount exaggerated Th1 cell responses and spontaneously develop inflammatory bowel disease (IBD) as a result of an unbalanced response to environmental antigens (2). They also exhibit increased susceptibility to immunopathology upon parasitic and bacterial infection (3–6) or LPS exposure (7), as well as enhanced contact hypersensitivity reactions and irritant responses of the skin (8).
Originally identified as a Th2 cell–derived factor (9), IL-10 was later found to be secreted by a variety of haemopoietic cell types including activated macrophages (10, 11), dendritic cells (12–15), B cells (16–18), and mast cells (19). In addition, keratinocytes can be induced to secrete IL-10 by contact allergens (20) or UV irradiation (21). Human intestinal epithelial cells were reported to constitutively secrete the cytokine (22).
Over the past few years, a number of different T cell subsets were identified as important regulators of immune responses (23, 24). The lack of the best characterized subset of regulatory T cells, the CD4+ CD25+ cells, in mice deficient for the transcription factor foxp3 results in severe inflammatory multiorgan disease (25–27). In IL-10–deficient mice, functionally active CD4+ CD25+ regulatory T cells are present (28) and in comparison to foxp3-deficient mice, IL-10 − / − animals develop a less severe disease with a later onset. IL-10–dependent, but also IL-10–independent, suppressive effects mediated by regulatory T cells have been described in a variety of experimental systems (28–32). Thus, the relevance of T cell–derived IL-10 in the regulation of immune responses remains unclear.
To determine the role of T cell–derived IL-10 in vivo, we generated mice with a selective deficiency for IL-10 only in T cells by Cre/loxP-mediated gene targeting and compared T cell and innate immune responses of these animals to responses of mice with complete IL-10 deficiency. We found that although T cell responses were similarly enhanced in mice with T cell–specific or complete IL-10 deficiency in comparison to control animals, innate responses to LPS or irritation of the skin were deregulated under the condition of complete, but not in T cell–specific, IL-10 deficiency.
Materials and Methods
Gene Targeting.
The targeting vector was constructed by cloning two genomic fragments of the murine IL-10 gene (long and short arm of homology) into plasmid peasy flox (provided by M. Alimzhanov, Harvard Medical School, Boston, MA) after a loxP site along with an additional PstI restriction site had been inserted into the long arm of homology. The linearized construct was transfected into Ola129-derived embryonic stem cells (IB-10). Homologous recombinants were analyzed for single copy integration of the targeting vector by Southern blot analysis and were transfected with ppgk-Cre for Cre-mediated deletion of the neomycin selection marker in vitro. Clones carrying the IL-10 FL mutation were injected into CB20 blastocysts, which were then implanted into foster mothers. Chimeric offspring were mated to C57BL/6 mice and the F1 generation was screened for germline transmission of the mutation. Mice were kept in a specific pathogen-free facility. IL-10 FL mice were crossed to C57BL/6 deleter Cre mice (33) and to CD4-Cre mice (34) on a mixed C57BL/6-DBA/2-129v/J background. All animal experimentation was performed in accordance with institutional guidelines.
Genotyping.
Tail DNA samples were digested with PstI. Southern blots were hybridized with probe 3 for detection of IL-10 alleles and probe 10 for detection of Cre transgenes using the Gene Images CDP-Star random prime labeling and detection modules (Amersham Biosciences) according to the manufacturer's instructions. For radioactive hybridization, the Laderman labeling kit (TaKaRa) was used to label probe DNA and hybridization was performed in QuikHyb Hybridization Solution (Stratagene). Primers for amplification of probes were probe3up (5′ aca gac tag aca gga gac tag gta a), probe3do (5′ c agc aga gga cag cct aga aag atg), CreC1 (5′ caa ttt act gac cgt aca c), and CreDo (5′ gtt ttg cac gtt cac cgg cat caa).
Cell Sorting.
Spleen cell suspensions were subjected to erythrocyte lysis and Ficoll-Paque (Amersham Biosciences) gradient centrifugation for depletion of residual erythrocytes and dead cells. T cells, B cells, and dendritic cells were enriched immunomagnetically using CD3, CD19, and CD11c microbeads (Miltenyi Biotec), respectively, and then were sorted from the enriched suspensions after immunostaining for CD3, CD19, and CD11c (BD Biosciences) on a FACStar (BD Biosciences). Macrophages were isolated from peritoneal lavage fluid by FACS sorting of F4/80-stained (Serotec) cells.
Splenocyte Stimulation and Cytokine Quantification.
96-well plates were antibody coated by incubation for 90 min at 37°C with a solution of 10 μg/ml anti-CD3 (clone 145-2C11; BD Biosciences) and 5 μg/ml anti-CD28 (clone 37.51; BD Biosciences) antibodies in PBS. Splenocyte suspensions were depleted of erythrocytes and dead cells by Ficoll-Paque gradient centrifugation (Amersham Biosciences) before addition to coated 96 wells in a density of 106 cells/ml. Culture supernatants were harvested after 48 h. Measurement of cytokine concentrations in culture supernatants was performed by Cytometric Bead Array (BD Biosciences) according to the manufacturer's instructions.
Toxoplasma Infection.
IL-10 FL/FL CD4-Cre+ and Cre− control mice were infected orally with the low virulence DX strain of Toxoplasma gondii as described previously (35). Animals were killed on days 7 and 10 after injection and liver tissue was snap-frozen in liquid nitrogen or fixed and paraffin embedded. Immunohistochemistry was performed on 10-μm cryostat sections. CD4, CD8, CD45, B220, F4/80, and T. gondii antigens were detected as described previously (35).
Contact Hypersensitivity Reaction and Irritant Response.
Contact hypersensitivity was induced by painting the shaved abdomen with 100 μl of 5% (wt/vol) 2,4-dinitrochlorobenzene (DNCB; Sigma-Aldrich) in a 4:1 mixture of acetone and olive oil for sensitization. The mice were challenged 6 d later by application of 10 μl of 1% (wt/vol) DNCB in olive oil to each side of one ear. Ear thickness was determined before challenge and daily thereafter for 4 d using an engineer's caliper (Mitutoyo). Percent increase of ear thickness was compared for the different experimental groups using the student's t test for independent samples. The irritant response was elicited by applying 20 μl of 1% (vol/vol) Croton oil (Sigma-Aldrich) in acetone to one ear, 10 μl to each side of the ear. Ear thickness was measured before as well as 24 and 48 h after irritant application and evaluated as described above.
LPS Sensitivity.
The response to LPS (LPS from Escherichia coli serotype 055:B5; Sigma-Aldrich) was tested by a single i.p. injection of 20 μg/g body weight in 200 μl sterile saline. Survival was monitored for 5 d thereafter. In additional animals, blood samples were taken 6 h after LPS injection and assayed for serum TNF-α, IFN-γ, and IL-12 p70 concentrations by a cytometric bead array kit (BD Biosciences).
Results
Conditional Mutagenesis of the IL-10 Gene.
We generated mice with an IL-10 allele mutated by the insertion of two loxP sites flanking parts of the promoter region, transcription initiation site, and the first exon (Fig. 1, floxed allele). IL-10 FL/FL mice develop normally, are fertile, and do not show any sign of IBD macroscopically or histologically (n = 5; not depicted), indicating that the insertion of the loxP sites does not significantly interfere with regulation of the IL-10 gene. Crossing of IL-10 FL/FL mice to a Cre transgenic strain expressing Cre recombinase ubiquitously in early stages of ontogeny (33) yielded mice carrying deleted alleles in all cell types (IL-10 D/D). As expected, these animals exhibited IBD, enhanced contact hypersensitivity and irritant reactions, as well as increased sensitivity to LPS (see below). IL-10 D/D mice thus reproduce the phenotype of conventional IL-10–deficient mice (2), which indicates that as intended, Cre-mediated deletion of the loxP-flanked fragment inactivates the IL-10 gene.
Figure 1.

Conditional targeting of the IL-10 gene. (A) Scheme showing the WT IL-10 genomic locus (WT) and the mutant IL-10 alleles after homologous recombination of the targeting vector (Neo), after Cre-mediated deletion of the neomycin resistance gene (FL), and after Cre-mediated deletion of the loxP-flanked promoter region and exon1 (D). The location of the probe for Southern blot analysis as well as restriction enzyme sites (P, PstI) are indicated. PstI fragments are in kilobases (kb). Filled boxes represent exons. LoxP sites are depicted as arrowheads. (B) Southern blot analysis of DNA from tail biopsies from a WT, an IL-10 FL/FL, and an IL-10 D/D mouse.
T Cell–specific Inactivation of the IL-10 Gene.
IL-10 FL mice were crossed to a transgenic line expressing Cre under the control of the murine CD4 enhancer/promoter/silencer, which results in high efficiency Cre-mediated deletion commencing in CD4+ CD8+ (double positive) progenitors of CD4 and CD8 lymphocytes (CD4-Cre transgenic mice; references 34 and 36). The efficiency and T cell specificity of Cre-mediated deletion was verified by Southern blot analysis of DNA extracted from FACS-sorted cell populations. Complete deletion of the loxP-flanked fragment was found in CD3+ T cells from the spleens of IL-10 FL/FL CD4-Cre+ mice (Fig. 2 A). In contrast, no deletion was observed in CD19+ splenic B cells (Fig. 2 A), in peritoneal macrophages (Fig. 2 B), or in CD11c+ splenic dendritic cells (Fig. 2 C). In addition, DNA extracted from the brain, heart, lung, thymus, liver, muscle, kidney, and tail tissue was analyzed for deletion of the loxP-flanked fragment by Southern blot hybridization (Fig. 2 D). The degree to which deletion occurred corresponded with the number of resident and blood-borne T cells expected to be contained in these tissues. Thus, inactivation of the IL-10 gene in IL-10 FL/FL CD4-Cre+ mice is efficient in T cells and was not detected in other cell types by Southern blot analysis. Peritoneal macrophages from IL-10 FL/FL CD4-Cre+ mice secreted similar amounts of IL-10, as did macrophages from control animals after stimulation with LPS in vitro (not depicted).
Figure 2.

T cell–specific deletion of the IL-10 gene in IL-10 FL/FL CD4-Cre+ mice. Southern blot analysis of DNA extracted from different FACS-sorted cell populations and tissues after PstI digest. (A) CD3+ T and CD19+ B cells sorted to a purity of 98 and 96%, respectively, from splenocyte suspension of a 3-mo-old mouse. The result is representative of five animals. (B) F4/80+ macrophages from peritoneal lavage fluid (purity 97%, representative of four animals) and splenic T cells. (C) Splenic CD11c+ dendritic cells sorted to a purity of 95% (representative of three animals) and splenic T cells. (D) Various tissues and splenic T cells from an 8-wk-old mouse.
Enhanced Secretion of Proinflammatory Cytokines by Splenocytes from T Cell–specific IL-10 Mutants.
To compare cytokine production in mice with T cell–specific or complete IL-10 deficiency, we stimulated IL-10 FL/FL CD4-Cre+ and Cre− as well as IL-10 D/D and IL-10 D/WT spleen cells with anti-CD3 and anti-CD28 antibodies in vitro. The IL-10 FL/FL CD4-Cre+ animals analyzed all displayed rectal prolapse by the time of the experiment, indicating with high probability the presence of intestinal inflammation (see below). TNF-α levels of culture supernatants from stimulated IL-10 FL/FL CD4-Cre+ splenocytes were on average three times higher than those of control cultures from Cre− animals (Fig. 3 A). Similar results were obtained for the TNF-α–induced chemokine monocyte chemoattractant protein 1 (MCP-1) and IL-6. IFN-γ concentrations were similar in stimulated Cre+ and Cre− cultures. IL-12 levels of five stimulated Cre+ cultures were in the same range as those in Cre− cultures. Four cultures reached concentrations that were three- to sevenfold higher. IL-10 was secreted in very low or undetectable amounts not different for Cre− versus Cre+ cells (not depicted). These results demonstrate that T cell responses in IL-10 FL/FL CD4-Cre+ mice are accompanied by increased secretion of inflammatory mediators. Four additional IL-10 FL/FL CD4-Cre+ animals analyzed did not show rectal prolapse and were thus probably devoid of IBD. Interestingly, secretion of cytokines released by splenocytes from these four animals after T cell stimulation was similar to that of splenocytes from Cre− controls or was only moderately increased (not depicted).
Figure 3.

Cytokine and chemokine secretion by splenocytes from mice with complete or T cell–specific IL-10 deficiency after stimulation by plate-bound anti-CD3 and anti-CD28 antibodies in vitro. (A) Concentrations of IFN-γ, TNF-α, IL-12, IL-6, and MCP-1 in 48-h culture supernatants of splenocytes from IL-10 FL/FL CD4-Cre+ animals (n = 9; □, unstimulated; ▪, stimulated), IL-10 FL/FL Cre− controls (n = 8; ○, unstimulated; •, stimulated), and (B) in supernatants of splenocyte cultures from IL-10 D/D animals (n = 5; □, unstimulated; ▪, stimulated) and IL-10 WT/D controls (n = 5; ○, unstimulated; •, stimulated).
The increased production of proinflammatory mediators by cells from T cell–specific mutant mice in vitro was similar to the results obtained for IL-10 D/D mice (Fig. 3 B). In accordance with published experiments with IL-10−/− mice (2), secretion of IFN-γ, TNF-α, IL-6, and MCP-1 by stimulated splenocytes from IL-10 D/D mice was clearly increased in comparison with cell suspensions from control animals.
T Cell–specific IL-10 Mutants Spontaneously Develop IBD.
IL-10 FL/FL CD4-Cre+ and IL-10 D/D were kept under standard specific pathogen-free conditions. A fraction of IL-10 FL/FL CD4-Cre+ mice developed rectal prolapse, which is an indicator of severe intestinal inflammation in mice. Histologically, the most prominent inflammatory changes were found in the cecum and ascending colon (Fig. 4 A), where multifocal severe inflammation affected all strata of the colonic wall, in some animals including the serosa with consecutive peritonitis. Multifocal mucosal necroses resulted in numerous ulcerations. The mucosa showed massive hyperplasia and infiltration with inflammatory cells, ectatic lymphatic vessels, and crypt abscesses. In addition to these focal changes, there was diffuse infiltration of the colonic mucosa with neutrophils, lymphocytes, plasma cells, and macrophages. Lympho-plasmocytic inflammation with focally hyperplastic mucosa was also found in duodenum and rectum, although it was less prominent in these locations. All animals that displayed the histologic changes described above (n = 9) also had macroscopically evident prolapse of the rectal mucosa. In IL-10 FL/FL CD4-Cre+ mice without prolapse (n = 6), we did not find signs of IBD histologically. The earliest occurrence of rectal prolapse was at the age of 4 wk. The incidence of bowel disease in the T cell–specific IL-10 mutants increased with age, reaching 33% at 6 mo (Fig. 4 B).
Figure 4.

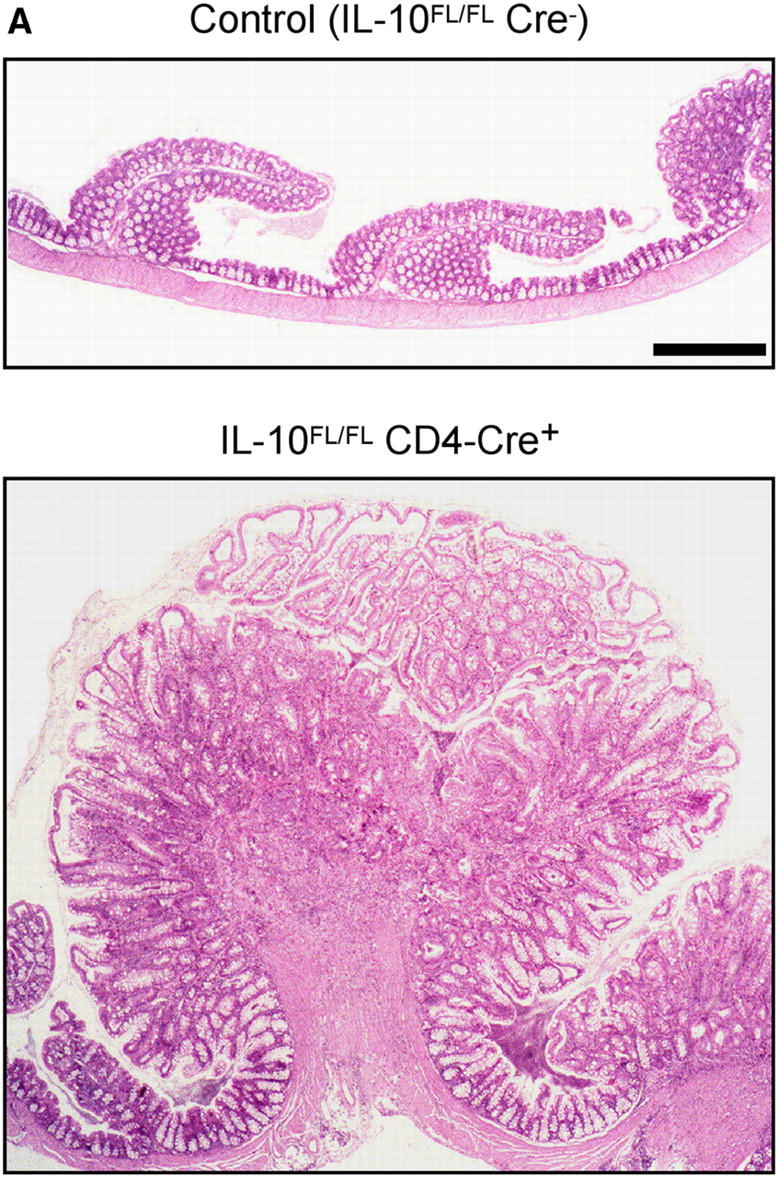
(A) Histology of colonic mucosa. Note massive hyperplasia and edema of the mucosa in the T cell–specific IL-10 mutant. Paraffin sections, hematoxylin and eosin stain. Bar, 40 μm. (B) Cumulative incidence of rectal prolapse indicating severe intestinal inflammation in IL-10 D/D mice (•, n = 90) and IL-10 FL/FL CD4-Cre+ mice (□, n = 142).
Intestinal inflammation in the T cell–specific IL-10 mutant mice closely resembled IBD in IL-10 − / − mice (2) or IL-10 D/D animals with complete IL-10 deficiency with respect to incidence, localization, and severity. The incidence of IBD in IL-10 D/D mice was 41% at the age of 6 mo (Fig. 4 B).
In RNA samples from the cecum of WT as well as healthy or diseased T cell–specific mutant mice, but not from IL-10D/D mice, we detected IL-10 mRNA by RT-PCR (not depicted). These data suggest that there is IL-10 production from non–T cells in the T cell–specific mutant animals. However, IL-10 secretion from cell types other than T cells does not prevent intestinal inflammation.
All animals were kept in a facility that was specific pathogen-free except for a high prevalence of infection with Helicobacter ganmani (37) as detected by PCR and sequencing. Although some Helicobacter species are known to exacerbate IBD in susceptible animals (38), this has not been described for H. ganmani. Thus, we can only speculate that H. ganmani infection may enhance intestinal inflammation in our mutant mice.
T Cell–specific IL-10 Mutants Succumb to Infection with T. gondii.
To further characterize immune reactions in the T cell–specific IL-10 mutant animals, we infected IL-10 FL/FL CD4-Cre+ mice orally with a low virulence strain of T. gondii. All seven mutants succumbed by 11 d after infection, whereas none of the eight Cre− control animals developed disease (Fig. 5 A). Death was associated with severe hepatitis characterized by disseminated focal inflammatory infiltrates (Fig. 5 B, top), whereas only mild hepatitis was detected in the controls (n = 5). In the mutants, numerous CD4+ T cells were detected in the liver by day 7 after infection (Fig. 5 B, bottom), with numbers increasing until day 10 (not depicted), whereas the scarce infiltrates in the livers of control mice contained only low numbers of CD4+ T cells at both time points. There were no differences in the numbers of infiltrating CD8+ T cells or macrophages between mutants and controls (not depicted). Because the parasite load was low in both experimental groups, there was no evidence for insufficient parasite control in IL-10 FL/FL CD4-Cre+ mice.
Figure 5.

Infection of T cell–specific IL-10 mutants with T. gondii. (A) Survival of IL-10 FL/FL CD4-Cre+ mice (n = 7) and IL-10 FL/FL Cre− control animals (n = 8). Two and three mutants were killed because of severe, overt disease on days 7 and 10 after infection, respectively. Two and three control mice, which never showed symptoms or signs of disease, were killed in parallel on days 7 and 10, respectively. Three controls were killed 10 wk after infection. (B) Cryostat liver sections. On the top, hematoxylin and eosin stain is shown at a magnification of 25. Note the more intense infiltration of liver from T cell–specific mutants with inflammatory cells 7 d after infection. On the bottom, immunostaining for CD4 (Hemalum counterstain) is shown at a magnification of 100. Note the increased number of CD4+ cells in mutant liver compared with the controls.
IL-10 − / − mice develop CD4+ T cell–dependent lethal immunopathology upon infection with T. gondii (5). Our findings indicate that T cell–derived IL-10 normally prevents lethal T. gondii–induced immunopathology in mice.
T Cell–specific IL-10 Mutants Display Enhanced Contact Hypersensitivity Reactions.
To analyze cutaneous T cell responses in animals with T cell–specific IL-10 deficiency, we elicited contact hypersensitivity in IL-10 FL/FL CD4-Cre+ mice and IL-10 FL/FL Cre− littermates. Data presented in Fig. 6 A show the ear swelling response after treatment of ears with the contact allergen DNCB. The increase in ear thickness was on average about twofold higher in IL-10 FL/FL CD4-Cre+ compared with IL-10 FL/FL Cre− mice at all time points measured. The difference between mutants and controls was highly significant (P < 0.01 for 24, 48, and 96 h; P = 0.02 for 72 h; the standard deviation observed was most likely due to mixed genetic backgrounds). Thus, the T cell–mediated contact hypersensitivity reaction is subject to regulation by T cell–derived IL-10. An enhanced ear swelling response, similar to the results obtained for the T cell–specific IL-10 mutant, was observed for IL-10 D/D mice (Fig. 6 B), confirming published experiments with IL-10 − / − mice (8).
Figure 6.

Contact hypersensitivity reactions of (A) IL-10 FL/FL CD4-Cre+ (n = 28) and IL-10 FL/FL Cre− control mice (n = 26), and (B) IL-10 D/D (n = 28) and IL-10 D/WT control mice (n = 21). The increase in ear thickness of sensitized mice was measured daily after challenge with DNCB.
In Contrast to Complete IL-10 Deficiency, T Cell–specific IL-10 Mutants Display a Normal Cutaneous Irritant Response.
The irritant response of the skin is a function of the local innate immune system. To find out whether T cell–derived IL-10 also regulates this type of inflammatory response, we treated IL-10 FL/FL CD4-Cre+ mice and IL-10 FL/FL Cre− littermates with the irritant croton oil. The ear swelling response in the T cell–specific IL-10 mutant mice was not increased 24 and 48 h after croton oil application compared with the control animals (Fig. 7 A), demonstrating that cutaneous innate inflammatory responses are not subject to regulation by IL-10–producing T cells. In contrast, IL-10 D/D animals displayed significantly enhanced irritant responses compared with IL-10–competent control mice (Fig. 7 B), reproducing earlier findings in IL-10−/− mice (8). Thus, the cutaneous irritant response is subject to regulation by IL-10 secreted by cell types other than T cells.
Figure 7.

Irritant response of (A) IL-10 FL/FL CD4-Cre+ (n = 36) and IL-10 FL/FL Cre− control mice (n = 35), and (B) IL-10 D/D (n = 17) and IL-10 D/WT control mice (n = 16). The percent increase in ear thickness was measured 24 and 48 h after application of croton oil.
In Contrast to Complete IL-10 Deficiency, T Cell–specific IL-10 Mutants Do Not Display Increased Sensitivity to LPS.
To further analyze responses of the innate immune system, IL-10 mutants were injected i.p. with LPS. None of the 10 IL-10 FL/FL CD4-Cre+ or the 10 Cre− littermates died after LPS exposure (Fig. 8 A). Additional animals were bled 6 h after LPS injection and serum concentrations of IFN-γ, TNFα, and IL-12 were determined (Fig. 8 B). For IL-10 FL/FL CD4-Cre+ mice, serum levels of these three cytokines were in the same low range as those obtained for IL-10–competent control animals, which produced little or no IFN-γ, TNFα, or IL-12. In contrast, 8 of 10 IL-10 D/D, but only 1 of 10 IL-10 D/WT, littermates died within 3 d after LPS injection (Fig. 8 A), consistent with a 20-fold increased LPS sensitivity reported for IL-10 −/− mice as compared with WT controls (7). As expected, drastically increased concentrations of IFN-γ, TNFα, or IL-12 were measured in the LPS-exposed IL-10 D/D mice as compared with IL-10–competent control animals (Fig. 8 B). Taken together, these data show that T cell–derived IL-10 is not involved in regulating the response to systemic LPS exposure, but that this reaction is controlled by IL-10 from non–T cells.
Figure 8.

Response of mice with complete or T cell–specific IL-10 deficiency to LPS. 20 μg of LPS/g body weight were injected i.p. (A) Survival of IL-10 FL/FL CD4-Cre+ mice (□, n = 10) and IL-10 FL/FL Cre− control littermates (▪, n = 10), and IL-10 D/D animals (○, n = 10) and IL-10 D/WT control littermates (•, n = 10). (B) Additional animals were bled 6 h after LPS injection and serum TNF-α, IFN-γ, and IL-12 levels were determined (symbols as in A).
Discussion
Within the population of T cells, the capacity to secrete IL-10 is well documented, in particular for Th2 cells and some T cell subsets with regulatory function. T regulatory cells have been shown to suppress immune responses by IL-10–dependent or IL-10–independent mechanisms in various experimental systems. IL-10–competent, but not IL-10–deficient, CD4+ CD25+ CD45RBlo cells suppressed development of IBD, resulting after adoptive transfer of CD4+ CD45RBhi cells into lymphocyte-deficient (SCID) mice (29). Another subset of regulatory T cells, the T regulatory 1 cells, suppress immune responses through secretion of large amounts of IL-10 (13, 32). In addition, transfer of antigen-pulsed CD40-deficient myeloid dendritic cells induced regulatory T cells, which suppressed T cell responses by an IL-10–dependent mechanism (39). However, suppression of T cell proliferation in response to polyclonal stimulation by CD4+ CD25+ T regulatory cells in vitro requires cell contact, but no IL-10 secretion (28). Likewise, CD4+ CD25+ T cell–mediated suppression of autoimmune gastritis induced by transfer of CD4+ CD25− T cells into lymphocyte-deficient (RAG-2−/−) recipients was independent of IL-10 secretion (40, 41). Regulation of the priming of potentially colitogenic T cells, however, by CD4+ CD25+ T cells occurs primarily independent of IL-10 (30). In vivo control of Leishmania major was shown to be regulated by CD4+ CD25+ T cells through both IL-10–dependent and IL-10–independent mechanisms (31). In addition to T cells, a large number of other cell types are able to secrete IL-10. For example, IL-10–producing regulatory B cells have been shown to prevent autoimmunity (16, 17). Thus, the relevance of immune regulation mediated by T cell–derived IL-10 in vivo remains unclear.
Our results show that T cell–specific IL-10 deficiency results in a dysregulation of T cell–mediated immune responses, which is very similar to the phenotype observed earlier in mice with complete IL-10 deficiency (2, 5, 8). Like IL-10 − / − animals, our T cell–specific IL-10 mutants are susceptible to induction of IBD by environmental bacteria and exhibit severe immunopathology upon infection with T. gondii as well as enhanced contact hypersensitivity reactions. These results indicate that regulation of inflammatory responses by IL-10–producing T cells cannot be substituted for by compensatory IL-10 production from other cell types.
Intestinal inflammation in IL-10 − / − mice is due to T cell–dependent immunopathology as a result of an unbalanced Th1 cell response to luminal bacterial antigens (2). Macrophages are a major source of IL-10 in the gut and secrete the cytokine in late phases of activation, for example by abundant bacterial LPS to contain secretion of proinflammatory mediators in an autocrine fashion (10). Our results, however, show that IL-10–mediated autocrine-negative feedback of macrophages is insufficient to prevent unbalanced immune responses in the intestine. Likewise, IL-10 production by other intestinal cell types does not suffice to compensate T cell–specific IL-10 deficiency.
Acute infection of IL-10 − / − mice with an avirulent strain of T. gondii results in a lethal CD4+ T cell–mediated immune response characterized by overproduction of TNF-α, IFN-γ, and IL-12 (5). Production of IL-10 by macrophages in Toxoplasma infection is well documented (42, 43) and was considered a critical mechanism responsible for controlling the Th1 cell response to T. gondii (5). However, our data show that in the absence of T cell–derived IL-10, the lethal immune response occurs with similar kinetics as in IL-10 − / − mice. This result demonstrates the pivotal role IL-10–producing T cells play in the regulation of T cell responses against microbial pathogens.
Contact hypersensitivity is a T cell–mediated immune response that requires prior sensitization. IL-10 − / − mice show enhanced contact hypersensitivity reactions (8). Keratinocytes, macrophages, dendritic cells, as well as T cells are potential sources of IL-10 in the skin. The enhanced contact hypersensitivity reactions in our T cell–specific IL-10 mutant demonstrate that T cell–derived IL-10 is an important regulator of this type of T cell response.
In addition to unbalanced T cell responses resulting in IBD, susceptibility to immunopathology upon infection, and enhanced contact hypersensitivity reactions, IL-10 − / − mice also show an enhanced inflammatory response of the skin to nonspecific irritation. This response is a T cell–independent function of the innate immune system of the skin. Interestingly, our T cell–specific IL-10 mutants did not exhibit enhanced cutaneous irritant responses. Furthermore, the T cell–specific IL-10 mutant mice did not exhibit exaggerated innate responses to systemic LPS exposure as determined by survival as well as production of TNF-α, IFN-γ, and IL-12 after i.p. injection. Under defined experimental conditions, IL-10–secreting T cells were shown to regulate intestinal innate inflammatory responses to Helicobacter hepaticus in lymphocyte-deficient mice (44). However, the enhanced sensitivity to endotoxic shock and cutaneous irritation observed in complete IL-10 deficiency are due to the lack of IL-10 secretion from non–T cells like keratinocytes, histiocytes, monocytes, or dendritic cells. Future experiments will address this question by cell type–specific deletion of the IL-10 gene in these cell types.
Dysregulation of T cell responses in our T cell–specific IL-10 mutant may reflect the absence or functional impairment of regulatory T cells, which depend on IL-10 for their differentiation and/or their suppressive function. The phenotype our mutant develops is unlikely to be a result of the lack of the CD4+ CD25+ subset of T regulatory cells, as absence of these cells in mice deficient for the transcription factor foxp3 leads to a more severe disease with multiorgan inflammation shortly after birth (25–27). In addition, CD4+ CD25+ cells with suppressive activity are present in IL-10 − / − animals (28). Furthermore, foxp3 mRNA was detectable in CD4+ T cells sorted from suspensions of purified intestinal intraepithelial lymphocytes from all T cell–specific IL-10 mutants analyzed (n = 4) by real-time RT-PCR (not depicted). As CD4+ CD25+ T regulatory cells have been shown to mediate IL-10–dependent as well as IL-10–independent effects (28–32), the phenotype of the T cell–specific IL-10 mutant may result from a partial loss of function of CD4+ CD25+ T regulatory cells. Analysis of suppression of T cell proliferation in vitro, which represents a convenient assay for regulatory function, cannot clarify this potential defect of the CD4+ CD25+ cells, as in vitro suppression of T cell responses by these cells is known to be independent of IL-10 (28).
Our data show that T cell–derived IL-10 is dispensable for the regulation of cutaneous innate irritant responses, but crucial for the regulation of T cell responses. We will expand the experiments presented here by crossing the conditional IL-10–deficient mutation onto other Cre recombinase–expressing mouse lines. By inactivating the IL-10 gene selectively in keratinocytes and macrophages, we hope to identify the source of the IL-10–controlling cutaneous innate responses. In addition, it will be of interest to analyze mice with cell type–specific deficiency for IL-10 in macrophages, dendritic cells, intestinal epithelial cells, or B cells for development of IBD or deregulation of T cell responses. A further extension of our experiments will be the cell type–specific inactivation of the IL-10 receptor to identify the relevant IL-10–responsive cells.
Acknowledgments
The authors wish to thank Christopher B. Wilson for providing the CD4-Cre transgenic line. We thank Fiona Powrie for critically reading the manuscript and Wiebke Hansen and Mariela Bollati for performing the RT-PCR for foxp3 and IL-10. We are also grateful to Angela Egert, Anke Leinhaas, Christoph Göttlinger, Tobias Häring, and the members of the laboratory for skin histopathology of the University of Cologne for technical support.
This work was supported by grants from the Deutsche Forschungsgemeinschaft through the SFB 621 to W. Müller, through the SFB 589 and the Köln Fortune Program/Faculty of Medicine, University of Cologne to A. Roers, from the Center for Molecular Medicine, University of Cologne (CMMC) to W. Müller and K. Rajewsky, and the Juvenile Diabetes Foundation International to K. Rajewsky.
The authors have no conflicting financial interests.
Abbreviations used in this paper: DNCB, 2,4-dinitrochlorobenzene; IBD, inflammatory bowel disease; MCP-1, monocyte chemoattractant protein 1.
References
- 1.Moore, K.W., R. de Waal Malefyt, R.L. Coffman, and A. O'Garra. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683–765. [DOI] [PubMed] [Google Scholar]
- 2.Kühn, R., J. Löhler, D. Rennick, K. Rajewsky, and W. Müller. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 75:263–274. [DOI] [PubMed] [Google Scholar]
- 3.Kullberg, M.C., J.M. Ward, P.L. Gorelick, P. Caspar, S. Hieny, A. Cheever, D. Jankovic, and A. Sher. 1998. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 66:5157–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deckert, M., S. Soltek, G. Geginat, S. Lutjen, M. Montesinos-Rongen, H. Hof, and D. Schlüter. 2001. Endogenous interleukin-10 is required for prevention of a hyperinflammatory intracerebral immune response in Listeria monocytogenes meningoencephalitis. Infect. Immun. 69:4561–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gazzinelli, R.T., M. Wysocka, S. Hieny, T. Scharton-Kersten, A. Cheever, R. Kühn, W. Müller, G. Trinchieri, and A. Sher. 1996. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157:798–805. [PubMed] [Google Scholar]
- 6.Namangala, B., W. Noel, P. De Baetselier, L. Brys, and A. Beschin. 2001. Relative contribution of interferon-gamma and interleukin-10 to resistance to murine African trypanosomosis. J. Infect. Dis. 183:1794–1800. [DOI] [PubMed] [Google Scholar]
- 7.Berg, D.J., R. Kuhn, K. Rajewsky, W. Muller, S. Menon, N. Davidson, G. Grunig, and D. Rennick. 1995. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J. Clin. Invest. 96:2339–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berg, D.J., M.W. Leach, R. Kühn, K. Rajewsky, W. Müller, N.J. Davidson, and D. Rennick. 1995. Interleukin 10 but not interleukin 4 is a natural suppressant of cutaneous inflammatory responses. J. Exp. Med. 182:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiorentino, D.F., M.W. Bond, and T.R. Mosmann. 1989. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 170:2081–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Waal Malefyt, R., J. Abrams, B. Bennett, C.G. Figdor, and J.E. de Vries. 1991. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 174:1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maeda, H., H. Kuwahara, Y. Ichimura, M. Ohtsuki, S. Kurakata, and A. Shiraishi. 1995. TGF-beta enhances macrophage ability to produce IL-10 in normal and tumor-bearing mice. J. Immunol. 155:4926–4932. [PubMed] [Google Scholar]
- 12.Stumbles, P.A., J.A. Thomas, C.L. Pimm, P.T. Lee, T.J. Venaille, S. Proksch, and P.G. Holt. 1998. Resting respiratory tract dendritic cells preferentially stimulate T helper cell type 2 (Th2) responses and require obligatory cytokine signals for induction of Th1 immunity. J. Exp. Med. 188:2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGuirk, P., C. McCann, and K.H.G. Mills. 2002. Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin-10 production by dendritc cells: a novel strategy for evasion of protective T helper type 1 responses by Bordetella pertussis. J. Exp. Med. 195:221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwasaki, A., and B.L. Kelsall. 1999. Freshly isolated Peyer's patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J. Exp. Med. 190:229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khanna, A., A.E. Morell, C. Zhong, T. Takayama, L. Lu, and A.W. Thomson. 2000. Effects of liver-derived dendritic cell progenitors on Th1- and Th2-like cytokine responses in vitro and in vivo. J. Immunol. 164:1346–1354. [DOI] [PubMed] [Google Scholar]
- 16.Mizoguchi, A., E. Mizoguchi, H. Takedatsu, R.S. Blumberg, and A.K. Bhan. 2002. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 16:219–230. [DOI] [PubMed] [Google Scholar]
- 17.Fillatreau, S., C.H. Sweenie, M.J. McGeachy, D. Gray, and S.M. Anderton. 2002. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 3:944–950. [DOI] [PubMed] [Google Scholar]
- 18.O'Garra, A., R. Chang, N. Go, R. Hastings, G. Haughton, and M. Howard. 1992. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur. J. Immunol. 22:711–717. [DOI] [PubMed] [Google Scholar]
- 19.Masuda, A., Y. Yoshikai, K. Aiba, and T. Matsuguchi. 2002. Th2 cytokine production from mast cells is directly induced by lipopolysaccharide and distinctly regulated by c-Jun N-terminal kinase and p38 pathways. J. Immunol. 169:3801–3810. [DOI] [PubMed] [Google Scholar]
- 20.Enk, A.H., and S.I. Katz. 1992. Identification and induction of keratinocyte-derived IL-10. J. Immunol. 149:92–95. [PubMed] [Google Scholar]
- 21.Rivas, J.M., and S.E. Ullrich. 1992. Systemic suppression of delayed-type hypersensitivity by supernatants from UV-irradiated keratinocytes. An essential role for keratinocyte-derived IL-10. J. Immunol. 149:3865–3871. [PubMed] [Google Scholar]
- 22.Autschbach, F., J. Braunstein, B. Helmke, I. Zuna, G. Schurmann, Z.I. Niemir, R. Wallich, H.F. Otto, and S.C. Meuer. 1998. In situ expression of interleukin-10 in noninflamed human gut and in inflammatory bowel disease. Am. J. Pathol. 153:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maloy, K.J., and F. Powrie. 2001. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2:816–822. [DOI] [PubMed] [Google Scholar]
- 24.Shevach, E.M. 2000. Regulatory T cells in autoimmunity. Annu. Rev. Immunol. 18:423–449. [DOI] [PubMed] [Google Scholar]
- 25.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+ CD25+ regulatory cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 26.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 27.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Surfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 28.Thornton, A.E., and E.M. Shevach. 1998. CD4+ CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin-2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asseman, C., S. Mauze, M.W. Leach, R.L. Coffman, and F. Powrie. 1999. An essential role for interleukin-10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 190:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asseman, C., S. Read, and F. Powrie. 2003. Colitogenic Th1 cells are present in the antigen-experienced T cell pool in normal mice: control by CD4(+) regulatory T cells and IL-10. J. Immunol. 171:971–978. [DOI] [PubMed] [Google Scholar]
- 31.Belkaid, Y., C.A. Piccirillo, S. Mendez, E.M. Shevach, and D.L. Sacks. 2002. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 420:502–507. [DOI] [PubMed] [Google Scholar]
- 32.Groux, H., A. O'Garra, M. Bigler, M. Rouleau, S. Antonenko, J.E. de Vries, and M.G. Roncarolo. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 389:737–742. [DOI] [PubMed] [Google Scholar]
- 33.Schwenk, F., U. Baron, and K. Rajewsky. 1995. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23:5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee, P.P., D.R. Fitzpatrick, C. Beard, H.K. Jessup, S. Lehar, K.W. Makar, M. Perez-Melgosa, M.T. Sweetser, M.S. Schlissel, S. Nguyen, S.R. Cherry, J.H. Tsai, et al. 2001. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 15:763–774. [DOI] [PubMed] [Google Scholar]
- 35.Schlüter, D., J. Löhler, M. Deckert, H. Hof, and G. Schwendemann. 1991. Toxoplasma encephalitis of immunocompetent and nude mice: immunohistochemical characterisation of Toxoplasma antigen, infiltrates and major histocompatibility complex gene products. J. Neuroimmunol. 31:185–198. [DOI] [PubMed] [Google Scholar]
- 36.Wolfer, A., T. Bakker, A. Wilson, M. Nicolas, V. Ioannidis, D.R. Littman, P.P. Lee, C.B. Wilson, W. Held, H.R. MacDonald, and F. Radtke. 2001. Inactivation of Notch 1 in immature thymocytes does not perturb CD4 or CD8 T cell development. Nat. Immunol. 2:235–241. [DOI] [PubMed] [Google Scholar]
- 37.Robertson, B.R., J.L. O'Rourke, P. Vandamme, S.L. On, and A. Lee. 2001. Helicobacter ganmani sp. nov., a urease-negative anaerobe isolated from the intestines of laboratory mice. Int. J. Syst. Evol. Microbiol. 51:1881–1889. [DOI] [PubMed] [Google Scholar]
- 38.Kullberg, M.C., A.G. Rothfuchs, D. Jankovic, P. Caspar, T.A. Wynn, P.L. Gorelick, A.W. Cheever, and A. Sher. 2001. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infect. Immun. 69:4232–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin, E., B. O'Sullivan, P. Low, and R. Thomas. 2003. Antigen-specific suppression of a primed immune response by dendritic cells mediated by regulatory T cells secreting interleukin-10. Immunity. 18:155–167. [DOI] [PubMed] [Google Scholar]
- 40.Shevach, E.M., R.S. McHugh, C.A. Piccirillo, and A.M. Thornton. 2001. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol. Rev. 182:58–67. [DOI] [PubMed] [Google Scholar]
- 41.Suri-Payer, E., and H. Cantor. 2000. Differential cytokine requirements for regulation of autoimmune gastritis and colitis by CD4+CD25+ T cells. J. Autoimmun. 16:115–123. [DOI] [PubMed] [Google Scholar]
- 42.Khan, I.A., T. Matsuura, and L.H. Kasper. 1995. IL-10 mediates immunosuppression following primary infection with Toxoplasma gondii in mice. Parasite Immunol. 17:185–195. [DOI] [PubMed] [Google Scholar]
- 43.Grunvald, E., M. Chiaramonte, S. Hieny, M. Wysocka, G. Trinchieri, S.N. Vogel, R.T. Gazzinelli, and A. Sher. 1996. Biochemical characterization and protein kinase C dependency of monokine-inducing activities of Toxoplasma gondii. Infect. Immun. 64:2010–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maloy, K.J., L. Salaun, R. Cahill, G. Dougan, N.J. Saunders, and F. Powrie. 2003. CD4+ CD25+ TR cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 197:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]