

Abstract
The mechanisms that control neural stem and progenitor cell survival are unknown. In several pathological conditions, death receptor (DR) ligands and inflammatory cytokines exert a deleterious effect on neurons, whereas primitive neural cells migrate and survive in the site of lesion. Here, we show that even in the presence of inflammatory cytokines, DRs are unable to generate death signals in primitive neural cells. Neural stem and progenitor cells did not express caspase 8, the presence of which is required for initiating the caspase cascade. However, exogenous or cytokine-mediated expression of caspase 8 was not sufficient to restore their DR sensitivity. Searching for molecules potentially able to block DR death-inducing signaling complex (DISC), we found that primitive neural cells expressed high levels of the death effector domain-containing protein PED (also known as PEA-15). PED localized in the DISC and prevented caspase 8 recruitment and activation. Moreover, lentiviral-mediated delivery of PED antisense DNA resulted in dramatic down-regulation of the endogenous gene expression and sensitization of primitive neural cells to apoptosis mediated by inflammatory cytokines and DRs. Thus, absence of caspase 8 and high expression of PED constitute two levels of protection from apoptosis induced by DRs and inflammatory cytokines in neural stem and progenitor cells.
Keywords: neural stem cells, apoptosis, inflammatory cytokines, death receptors, death-inducing signaling complex
Introduction
Neural stem cells are primitive cells able to undergo self-renewal and generate the neural progeny through asymmetric divisions (1, 2). These cells are responsible for the development of the nervous system and contribute to adult neurogenesis (3–5). In the adult brain, neural stem and progenitor cells are primarily localized in the subventricular zone and migrate to the olfactory bulb, which is the primary source for the isolation of primitive neural cells from adult individuals (6–8). Primitive neural cells can be grown in vitro as neurospheres, which are clusters of undifferentiated cells containing a minority of neural stem cells and a high number of progenitors able to produce astrocytes, neurons, and oligodendrocytes upon exposure to differentiating agents (9).
Neural stem and progenitor cells apoptosis play a major role in the development of the nervous system. However, the mechanisms that control the survival of undifferentiated neural cells are poorly understood. Two major apoptotic pathways generate death signals in mammalian cells. The intrinsic pathway involves the release of apoptotic mediators from mitochondria with the formation of a high molecular weight complex containing cytochrome c, Apaf-1, and caspase 9, which activates caspase 3 and the execution phase of apoptosis. The extrinsic pathway is activated by death receptors (DRs), a widely expressed subgroup of the TNF receptor superfamily able to transduce apoptotic signals. After ligand binding, the recruitment of the adaptor protein FADD and caspase 8 to DRs leads to the formation of the death-inducing signaling complex (DISC; reference 10), resulting in activation and processing of caspase 8 and downstream effector caspases (11). DR signaling can be reduced or neutralized by the presence of high levels of cFLIP (12) or PED/PEA-15 (13), two DR inhibitory proteins that interact with FADD and caspase 8 through their DED domains, thus preventing caspase 8 activation.
Experimental evidences obtained with knockout mice indicate that neuronal cell ablation during development occurs through the activation of the intrinsic apoptotic pathway, as mice lacking caspase 9 (14, 15) or the caspase adaptor Apaf-1 (16, 17) have abnormal neuronal growth and severe neurological malformations. In contrast, mice lacking caspase 8 do not have neuronal defects (18), suggesting that the extrinsic apoptotic pathway is not exploited during this process. The strict developmental dependence on the intrinsic apoptotic pathway may be related to a possible deficiency of the DR/ligand expression or signaling in embryonic neural stem and progenitor cells.
DRs may contribute to neuronal death in several acute pathological conditions, such as brain ischemia or traumatic head injury. Neutralization of CD95 (Fas/APO-1) ligand (CD95L) and TNF-α protects from stroke-induced neurodegeneration by attenuating reperfusion damage and increasing neuronal viability (19, 20). Similarly, CD95L blockade significantly reduces neuronal and oligodendrocytic apoptosis and promotes functional recovery in spinal cord–lesioned mice (21). Moreover, CD95 up-regulation and caspase 8 activation has been consistently observed in human brain after severe traumatic injury (22). The inflammatory events occurring after acute brain damage are thought to cooperate with DRs for neuronal destruction (23). Glial cells can actively contribute to this process through the release of inflammatory cytokines, such as TNF-α, IL-1β, and IFN-γ, which may prime neurons to DR-mediated apoptosis.
After neural cell death, endogenous progenitors may differentiate and restore the damaged areas. The increased number of neural progenitor cells (NPCs) observed in acute neuronal lesions results from enhanced proliferation and migration to the injured region (24). Therefore, NPCs survive and differentiate in the sites of acute neurodegeneration under the harmful presence of inflammatory mediators and DR ligands, again suggesting that the extrinsic pathway is defective in primitive neural cells.
The possible impairment of DR signaling in both embryonic and adult NPCs prompted us to investigate the integrity of the extrinsic pathway in primitive neural cells and their progeny. Here, we show that NPCs display a double level of protection from DR-induced apoptosis, which neutralizes the combined proapoptotic activities of DR ligands and inflammatory cytokines.
Materials and Methods
Cell Cultures.
Human embryonic tissue was obtained from the brains of 12-wk-old embryos legally aborted, according to the ethical guidelines of the European Network for Transplantation (NECTAR). The ethical committee of “C. Besta” Neurological Institute and of the “L. Mangiagalli” Obstetric-Gynecological Clinic authorized the use of human central nervous system tissue. Human neural adult tissue was obtained following the ethical guidelines of the NECTAR and the Declaration of Helsinki from patients undergoing particularly invasive neurosurgery. NPCs were isolated from human tissues as described previously (25–27). NPCs were cultured in the presence of 20 ng/ml of human recombinant epidermal growth factor and 10 ng/ml of human recombinant basic fibroblast growth factor in a NS-A basal serum-free medium (Euroclone) containing 2 mM l-glutamine, 0.6% glucose, 9.6 μg/ml putrescine, 6.3 ng/ml progesterone, 5.2 ng/ml sodium selenite, 0.025 mg/ml insulin, and 0.1 mg/ml transferrin sodium salt (Sigma-Aldrich). Alternatively, commercial NPCs were purchased from BioWhittaker and cultured according to the manufacturer's protocols. Both commercial and tissue-purified NPCs were used from passage 5 to 15 throughout the study. These cells expressed the progenitor marker nestin (>99%) and were essentially negative for neuronal and glial antigens (<2%), as evaluated by immunofluorescence staining and confocal microscopy analysis. The NT2/D1 cell line was grown in DMEM (GIBCO BRL) supplemented with 10% heat-inactivated FBS (Hyclone Laboratories). NT2/D1 differentiation into postmitotic neurons (referred to as NT2-N cells) was performed as described previously (28). In brief, 2 × 106 cells were plated in a 75-cm2 flask and treated with 10 μM retinoic acid (RA; Sigma-Aldrich) twice a week for 4 wk. After retinoic acid treatment, cells were seeded in DMEM with 10% FBS at a reduced density. After 48 h, day-0 (neuronal progenitor) NT2-N cells were mechanically dislodged and replated on Matrigel (Becton Dickinson) in DMEM with 10% FBS supplemented with 1 μM cytosine arabinoside, 10 μM fluorodeoxyuridine, and 10 μM uridine (Sigma-Aldrich) for 4 wk, until terminal differentiation. The human neuroblastoma SK-N-AS, T lymphoblastoid HuT-78, T lymphoma Jurkat, and monocytic U937 cell lines were cultured in RPMI 1640 medium (GIBCO BRL) supplemented with 10% FBS (Hyclone Laboratories).
Antibodies and Flow Cytometry.
To detect surface DRs expression, 5 × 104 NPCs were washed twice with cold PBS and incubated for 1 h at 4°C with 10 μg/ml of antibodies specific to CD95 (DX2, mouse IgG1, DakoCytomation), TNFR1 (16805.21, mouse IgG1; R&D Systems), TRAIL-R1/DR4 (AF347, goat IgG; R&D Systems), TRAIL-R2/DR5 (AF631, goat IgG, R&D Systems), or control antibody. After two washes, cells were stained for 30 min with fluorescein isothiocyanate–conjugated anti–mouse or anti–goat secondary antibodies (Jackson ImmunoResearch Laboratories). Cytometric staining was analyzed on a FACSCalibur Instrument (Becton Dickinson), and data were analyzed with CELLQuest software (Becton Dickinson).
Cell Death Quantification.
To minimize the effects of growth factors on the survival of NPCs, epidermal growth factor and basic fibroblast growth factor were removed from the culture medium 2 h before DR stimulation. Apoptotic death was assessed by DNA staining and flow cytometry. In brief, cells were incubated overnight at 4°C in a propidium iodide solution (50 μg/ml) with 0.1% sodium citrate and 0.1% Triton X-100, until flow cytometric analysis. The percentage of apoptotic cells was determined by evaluating the number of hypodiploid nuclei. Alternatively, cell death was evaluated by using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) according to the manufacturer's protocol. The assay is based on reduction of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) to a colored formazan product that is measured spectrophotometrically. Cells were plated in 96-well plates in triplicate, stimulated, and incubated at 37°C in a 5% CO2 incubator. Metabolically active cells were detected by adding 20 μl of MTS to each well. After 2 h of incubation, the plates were red on a Multilabel Counter (Victor2; PerkinElmer). Before the apoptotic assays, cell viability was routinely evaluated at single cell level by ethidium bromide–acridine orange staining and fluorescence microscopy analysis (29).
Immunoblot Analysis and RNase Protection Assay.
Cell pellets were washed twice with cold PBS, resuspended in a 50-mM Tris-HCl, pH 7.5, 200 mM NaCl, and 1% NP-40 ice-cold buffer containing proteinase inhibitor cocktail (Sigma-Aldrich), and incubated for 30 min on ice. After centrifugation at 10,000 g for 10 min, lysates were collected as supernatants. For each sample, 20 μg of cell extracts was resolved on a 12% SDS-polyacrylamide gel using a mini-gel apparatus (Bio-Rad Laboratories) and transferred to Hybond-C extra nitrocellulose (Amersham Biosciences). Membrane was blocked for 1 h with 5% nonfat dry milk in TBS containing 0.05% Tween-20 and incubated for 2 h with specific antibodies. The following antibodies were used for immunoblotting: anti–caspase 8 (5F7, mouse IgG2b; Upstate Biotechnology); anti–caspase 3 (rabbit polyclonal IgG; Upstate Biotechnology); anti-FADD (mouse IgG1; Transduction Laboratories); anti-CD95 (C-20, rabbit polyclonal IgG; Santa Cruz Biotechnology, Inc.); anti-FLIP (NF6, mouse IgG1; Qbiogene); anti-PED/PEA-15 serum as described previously (30); anti–caspase 9 (goat IgG; R&D Systems); and anti–β-actin (Ab-1, mouse IgM; Oncogene Research Products). Washed filters were incubated for 45 min with horseradish peroxidase–conjugated anti–rabbit or anti–mouse secondary antibodies (Amersham Biosciences) and visualized by using an enhanced chemiluminescence detection system (SuperSignal West Pico chemiluminescent substrate; Pierce Chemical Co.). Total RNA was isolated from cells using RNeasy kit (QIAGEN). mRNA levels were evaluated using Riboquant Multi-Probe RNase Protection Assay System (hAPO-1c and hAPO-3c; BD Biosciences) according to the manufacturer's protocols.
Immunofluorescence Microscopy.
Cells were grown on polylysine-coated glass coverslips for immunofluorescence microscopy. After a fixing step in 2% paraformaldehyde-PBS for 20 min at 37°C, cells were permeabilized in 0.2% Triton X-100 PBS for 3 min and washed three times for 5 min with PBS. Slides were incubated for 1 h at 37°C with anti–caspase 8 (N-19, goat polyclonal IgG; Santa Cruz Biotechnology, Inc.) and antineuron-specific β-III tubulin–specific antibodies (mouse IgG1; Serotec Inc.). Nuclei were counterstained with propidium iodide (Sigma-Aldrich). After two washes in PBS, slides were incubated with secondary antibodies for 45 min at 37°C. Secondary antibodies, including FITC-conjugated goat anti–mouse IgG and Cy5-conjugated donkey anti–goat IgG, (Jackson ImmunoResearch Laboratories) were used at 2.5 μg/ml. Images were collected with a laser scanning microscope (IX81; Olympus).
Transduction of NPCs with Lentiviral Vectors.
Gene transfer was performed by using pRRLsin.cPPT.hCMV.GFP.Wpre and pRRLsin.cPPT.hPGK.GFP.Wpre, new variants of third-generation lentiviral vectors described previously (31). To simultaneously transduce both reporter and target gene, a new lentiviral vector, Tween, was generated by engineering pRRLsin.cPPT. hCMV.GFP.Wpre. In this vector, the hCMV.GFP cassette was substituted with the hCMV.hPGK.GFP. A multiple cloning site was inserted downstream of hCMV. Caspase 8 cDNA was subcloned in the XhoI site of Tween vector. PED/PEA-15 antisense was obtained by PCR amplification of the human PED/PEA-15 cDNA using the following primers: 5′-CCCGCTAGCGCTCAATGTAGGAGAGGTTG-3′ and 5′-CCCCTCGAGGCCAGAGCGCGCGGGGCAGTGTG-3′ containing the NheI and XhoI cloning sites, respectively (32). The amplified fragment was subcloned in the XbaI site of the Tween vector.
Lentiviral supernatants were produced by calcium phosphate transient cotransfection of a three-plasmid expression system in the packaging human embryonic kidney cell line 293T. The calcium-phosphate DNA precipitate was removed after 14–16 h by replacing the medium. Viral supernatant was collected 48 h after transfection, filtered through 0.45 μm–pore nitrocellulose filters, and frozen in liquid nitrogen. On the same day of transfection, NPCs were plated in a six-well plate in presence of viral supernatant. 4 μg/ml of polybrene was added to the viral supernatant to improve the infection efficiency (31). Cells were centrifuged for 45 min at 1,800 revolutions/min and incubated for 75 min in a 5% CO2 incubator. After the infection cycles, NPCs were washed twice and replated in fresh medium. Infection efficiency was evaluated after 48 h by flow cytometry.
DISC Analysis by Immunoprecipitation.
NPCs were pretreated with 200 U/ml of human recombinant TNF-α, 500 U/ml IFN-γ, and 100 U/ml IL-1β (PeproTech) for 60 h and stimulated with 1 μg/ml of CD95 agonistic antibody (CH11, mouse IgM; Upstate Biotechnology) for 90 min at 37°C in a 5% CO2 incubator. In unstimulated controls, 100 ng of CH11 was added after lysis in NP-40 lysis buffer to immunoprecipitate nonstimulated DRs. 15 μg of total proteins were immunoprecipitated with goat anti–mouse IgM antibody (Abcam Ltd.) bound to protein A/G–Sepharose beads (Pierce Chemical Co.) overnight at 4°C. Immunoprecipitates were washed three times with lysis buffer, mixed with 20 μl of 2 × Laemmli sample buffer, boiled for 10 min, and analyzed by Western blotting for the presence of CD95, Caspase 8, FADD, and PED/PEA-15.
Results
NPCs Are Resistant to DR-induced Apoptosis and Do Not Express Caspase 8.
To investigate the role of the extrinsic cell death pathway, we evaluated DR expression and apoptosis sensitivity in adult and embryonic NPCs. Flow cytometric analysis showed that CD95 and TRAIL-R2/DR5 were highly expressed in the majority of NPCs, whereas the levels of TRAIL-R1/DR4 and TNFR1 were lower (Fig. 1 A). This expression pattern was compatible with the ability of DRs to transduce apoptotic signals. Therefore, we measure the viability of undifferentiated NPCs exposed to anti-CD95 agonistic antibody, TNF-α and TRAIL. Unlike the control cell line U937, NPCs were completely resistant to DR stimulation (Fig. 1 B), whereas NPCs exposed to staurosporine exhibited massive apoptosis, suggesting the functional integrity of the intrinsic death pathway and common downstream components of both pathways (33).
Figure 1.
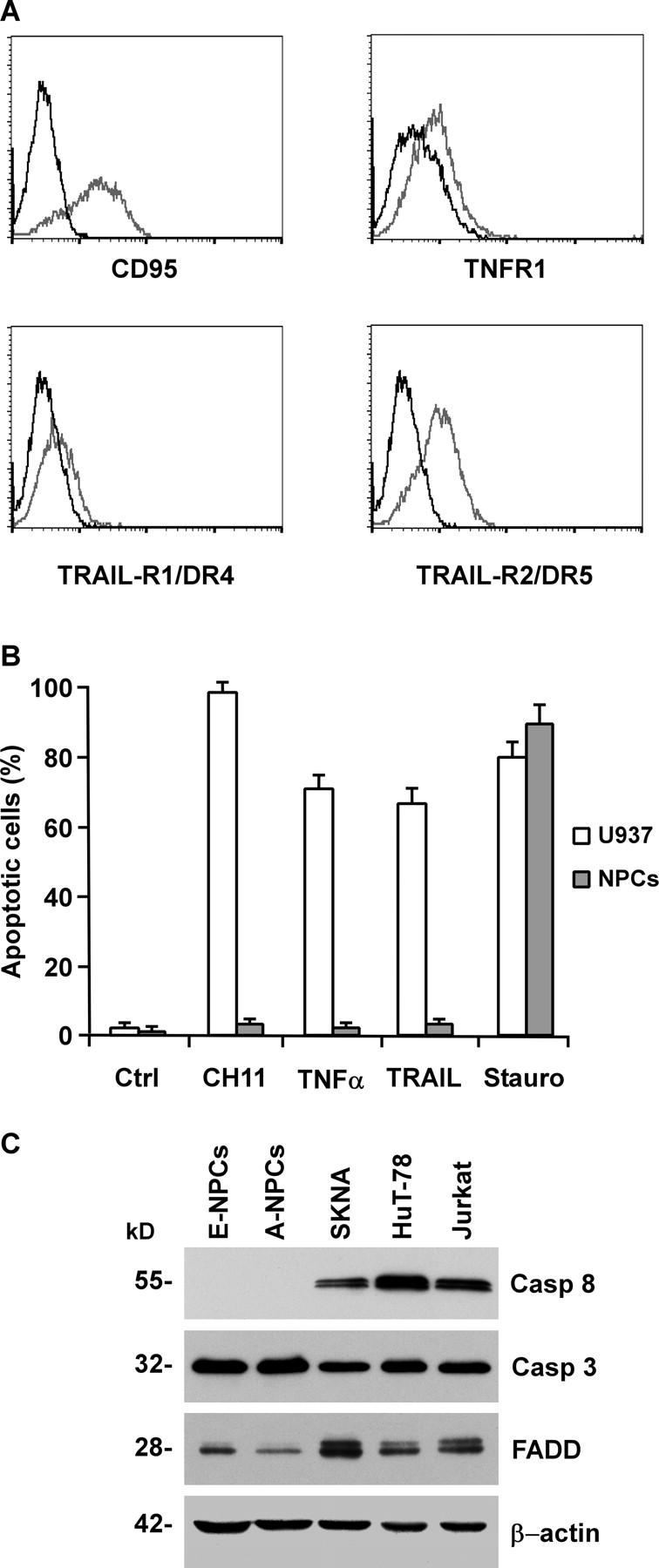
Expression of DRs and sensitivity to apoptosis in tissue-isolated embryonic and adult NPCs. (A) Flow cytometry analysis of DRs in NPCs. Cells were stained with antibodies specific to CD95, TNFR1, TRAIL-R1/DR4 and TRAIL-R2/DR5 (gray), or control IgGs (black). (B) Percentage of apoptotic cells in NPCs untreated (Ctrl) or exposed for 24 h to 500 ng/ml anti-CD95 (CH11), 500 U/ml TNF-α, 500 ng/ml TRAIL, or 0.25 μM staurosporine (Stauro). The human monocytic cells U937 were used as a positive control. The results are the mean ± SD of four independent experiments. (C) Immunoblot analysis of caspase 8 (Casp 8), caspase 3 (Casp 3), and FADD on embryonic (E-NPCs) and adult NPCs (A-NPCs) as compared with control cell lines. Detection of β-actin in the same membrane blot served as loading control. One representative experiment out of four is shown.
This observation led us to evaluate the expression and function of the upstream mediators of the extrinsic pathway, whose possible impairment could account for NPC resistance to DR-induced cell death. Western blot analysis of major DISC components showed that FADD was expressed in both embryonic and adult NPCs, whereas caspase 8 was not detectable (Fig. 1 C), suggesting that the absence of caspase 8 does not allow the activation of the caspase cascade after DR triggering. In contrast, in line with the assumed integrity of the downstream apoptotic machinery, the executioner caspase 3 was expressed at high levels as compared with those observed in DR-sensitive neural or hematopoietic cells (Fig. 1 C).
Caspase 8 Expression during Neuronal Differentiation.
Next, we analyzed the expression of caspase 8 in NPCs undergoing serum-induced differentiation. RNase protection assay and immunoblot analysis revealed barely detectable levels of caspase 8 mRNA and protein that increased during NPC differentiation (Fig. 2, A and B). Serum-induced NPC differentiation generates a population mainly composed of astrocytes, whereas a low percentage (∼15%) of differentiated cells displays a neuronal phenotype. Because mature astrocytes do not express caspase 8 (34), it is likely that the limited increase in caspase 8 levels may reflect the low proportion of neuronal cells generated by this system. Therefore, the same molecular analysis was performed on differentiating NT2/D1 cells. This cell line was derived from a human germ cell tumor and consists of progenitors cells that gradually differentiate into a virtually pure population of neuronlike cells upon exposure to retinoic acid (35, 36). Like NPCs, undifferentiated NT2-N cells expressed extremely low levels of caspase 8 mRNA and protein. However, caspase 8 levels dramatically increased during neuronal differentiation, indicating that the presence of caspase 8 in neurons is a developmentally regulated process (Fig. 2, A and B). Accordingly, three-color fluorescence and confocal analysis of differentiating NPCs showed that caspase 8 was expressed only in cells positive for the neuron-specific marker βIII-tubulin (Fig. 2 C), indicating that primitive neural cells acquire the expression of caspase 8 during neuronal differentiation.
Figure 2.
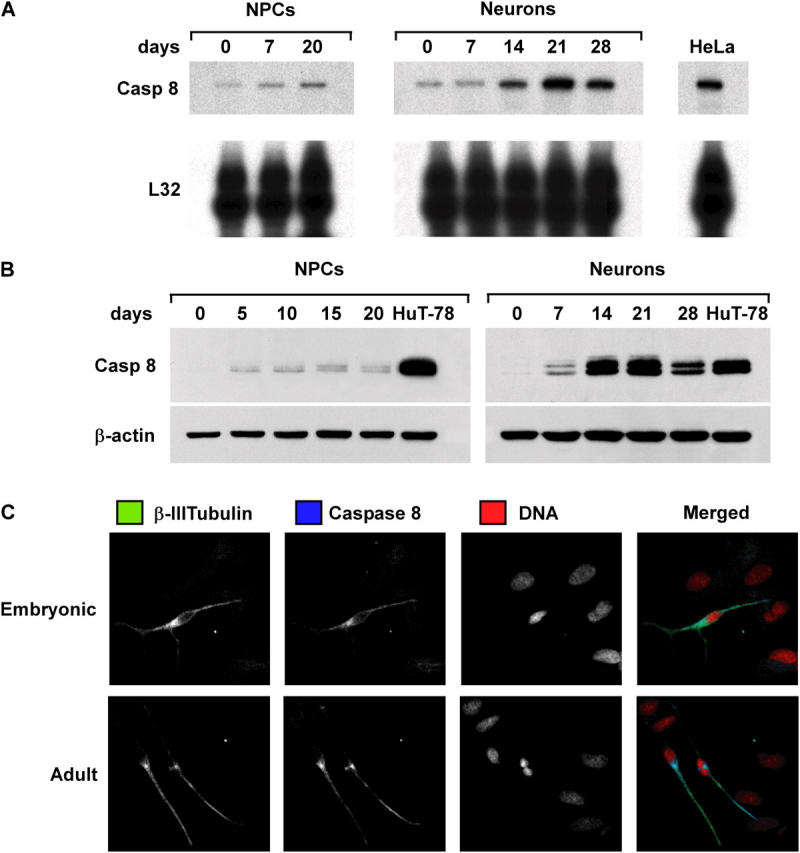
Expression of caspase 8 during NPC and NT2/D1 differentiation. (A) RNase protection assay for caspase 8 mRNA in NPCs and NT2-neurons at different days of differentiation. L32 levels were used as loading control, and RNA of HeLa cells were used as reference control. (B) Immunoblot analysis of caspase 8 expression in cells treated as in A. Cell lysates of HuT-78 cells were used as control. (C) Cells from differentiated embryonic and adult NPCs were analyzed by confocal microscopy to detect β-III tubulin (green), caspase 8 (blue), and DNA (red). One representative experiment out of four performed with commercial and tissue-isolated NPCs is shown.
Inflammatory Cytokines Do Not Sensitize NPCs to CD95-mediated Apoptosis.
During serum-induced differentiation, NPCs exposed to inflammatory cytokines give rise to reactive astrocytes, which are sensitive to CD95 stimulation (37 and unpublished data). Given the ability of inflammatory molecules to up-regulate caspase 8 in different cellular systems, we exposed neurons and NPCs to TNF-α, IFN-γ, IL-1β, or to the combinations of the three inflammatory cytokines and analyzed the expression of CD95 DISC components and susceptibility to apoptosis. RNase protection and immunoblot analysis showed that exposure of NT2-derived neurons to each single cytokine was sufficient to significantly up-regulate CD95 and caspase 8 expression (Fig. 3, A and B), suggesting that the presence of inflammatory cytokines may prime neurons to DR-induced apoptosis. Differently from neurons, exposure of NPCs to single inflammatory cytokines resulted in limited up-regulation of caspase 8 and CD95. However, simultaneous exposure of NPCs to TNF-α, IFN-γ, and IL-1β potently up-regulated both CD95 and caspase 8, suggesting a synergistic effect of different inflammatory stimuli (Fig. 3, A and B). Although the majority of NT2 neurons underwent apoptosis after CD95 stimulation in the presence of inflammatory cytokines, despite the presence of cytokine-induced caspase 8, NPCs remained completely refractory to CD95 stimulation (Fig. 3 C), thus suggesting that the absence of caspase 8 is not the only antiapoptotic feature responsible for DR refractoriness of NPCs.
Figure 3.

Effect of inflammatory cytokines on CD95 signaling in NPCs and differentiated neurons. (A) RNase protection assay for caspase 8 and CD95 mRNAs on 28-d NT2-neurons and NPCs untreated (Control) or treated with 200 U/ml TNF-α, 500 U/ml IFN-γ, 100 U/ml IL-1β, or a mixture of the three cytokines (Mix). L32 levels were used as loading control, and RNA of HeLa cells were used as reference control. (B) Immunoblot analysis of caspase 8 and CD95 expression on NT2-neurons and NPCs treated as in A. Cell lysates of HuT-78 and Jurkat cells were used as control. Detection of β-actin in the same membrane blot served as loading control. One representative experiment out of three performed with commercial and tissue-isolated embryonic NPCs is shown. (C) Percentage of apoptotic cells from NT2-neurons and NPCs exposed to anti-CD95 and to the cytokine mix as indicated. The results represent the mean of four independent experiments, using tissue-isolated embryonic and adult NPCs.
Exogenous Caspase 8 Expression Does Not Prime NPCs to DR-induced Apoptosis.
To confirm that the absence of NPC sensitivity to DR-induced apoptosis was not exclusively due to the absence of caspase 8, NPCs were transduced with a lentiviral vector carrying caspase 8a cDNA and the GFP as a reporter. After infection, NPCs transduced with control vector or caspase 8 were analyzed by flow cytometry. As shown in Fig. 4 A, the lentiviral infection yielded an essentially pure population of GFP positive cells. The expression of caspase 8 was further monitored by immunoblot analysis, which revealed that the exogenous expression of caspase 8 was comparable to that induced in NPCs by the combined treatment with TNF-α, IFN-γ, and IL-1β (Fig. 4 B). In line with the experiments concerning cytokine induction of caspase 8, exogenous caspase 8 expression was not sufficient to sensitize NPCs to CD95-mediated apoptosis, whereas the cells remained equally responsive to staurosporine (Fig. 4 C). To investigate whether the inability to transduce apoptotic signals by DR-stimulated NPCs, expressing endogenous or exogenous caspase 8, resulted from a defect at the DISC level, we directly analyzed caspase 8 processing after CD95 stimulation. Immunoblot analysis of NPCs transduced with caspase 8 or stimulated with TNF-α, IFN-γ, and IL-1β showed no caspase 8 processing, whereas the control cell line HuT-78 displays massive caspase 8 degradation with formation of the canonical fragments of 43-41 and 20 kD (Fig. 4 D). These data suggest that in addition to the absence of caspase 8, NPCs have a second mechanism operating at the DISC level responsible for early impairment of the apoptosis signaling generated by DRs.
Figure 4.
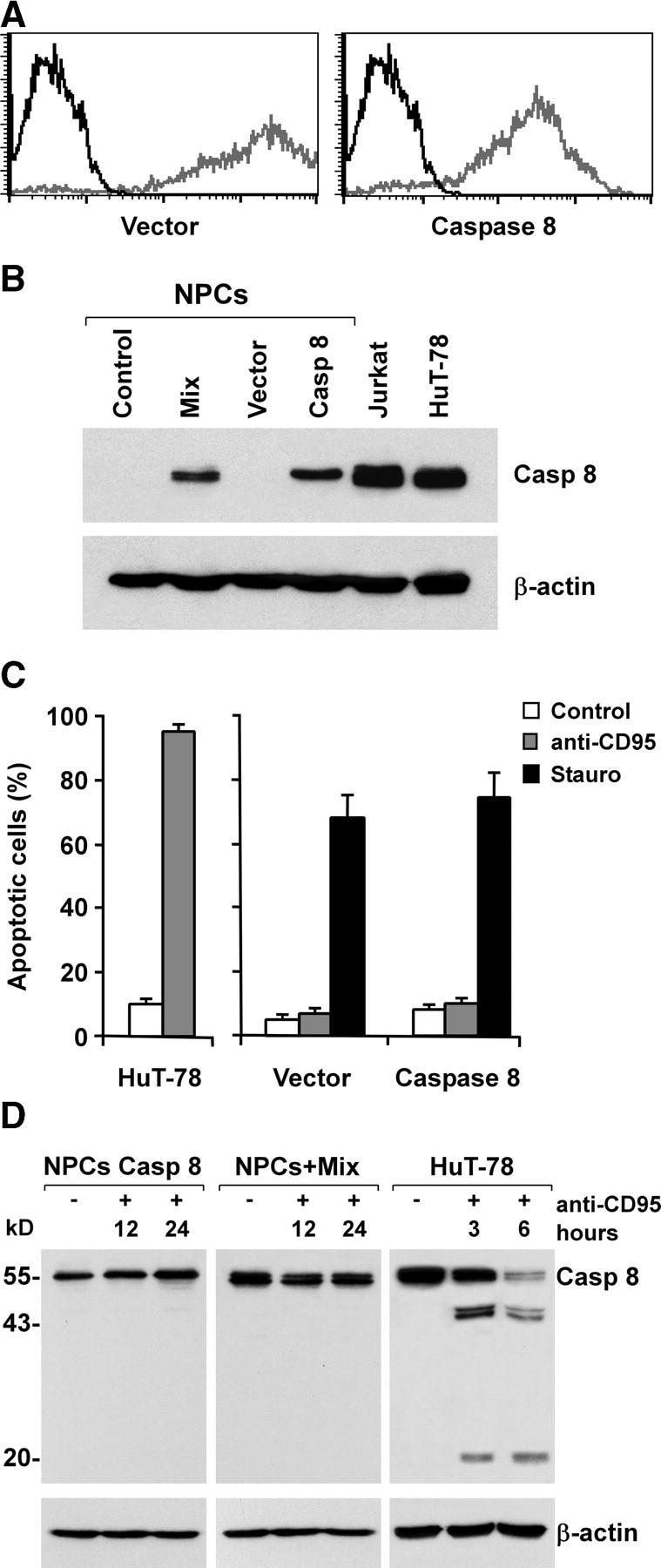
Absence of sensitization to CD95-induced apoptosis by exogenous caspase 8 expression in adult NPCs. (A) Flow cytometry profiles of GFP-positive NPCs transduced with empty Tween (Vector) or Tween/Caspase 8 (Caspase 8) vector (gray) as compared with nontransduced NPCs (black). (B) Immunoblot analysis of caspase 8 and β-actin in NPCs transduced as in A or treated with TNF-α, IFN-γ, and IL-1β (Mix). Jurkat and HuT-78 cell lines were used as positive controls. (C) Percentage of apoptosis in NPCs transduced as in A and treated with anti-CD95 or staurosporine. HuT-78 cells were used as positive control. The results are the mean ± SD of four independent experiments. (D) Immunoblot analysis of caspase 8 and β-actin in NPCs cultured with or without inflammatory cytokines and exposed to anti-CD95 for 12 or 24 h. In the control cell line HuT-78, the proteolytic products of caspase 8 were detectable shortly after CD95 stimulation.
PED/PEA-15 Mediates NPC Resistance to Inflammatory Cytokines and DR Stimulation.
The lack of caspase 8 processing after CD95 stimulation in caspase 8–expressing NPCs may result from the inhibitory action of antiapoptotic factors that act at the DISC level. Immunoblot analysis of those antiapoptotic proteins that block the caspase activation cascade through inhibition of caspase 8 activation showed that cFLIP was expressed at low levels in NPCs, whereas PED/PEA-15 was strongly expressed (Fig. 5, A and B) and not influenced by the presence of exogenous caspase 8. Next, we investigated whether inflammatory cytokines could modulate PED/PEA-15 expression in undifferentiated NPCs or in neurons. Exposure of NPCs to IFNγ or IL-1β considerably increased PED/PEA-15 expression (Fig. 5 C). In contrast, treatment with inflammatory cytokines resulted in dramatic downmodulation of PED/PEA-15 expression in neurons (Fig. 5 D). The opposite effect generated by inflammatory cytokines on PED/PEA-15 expression in NPCs and in differentiated neurons was in line with their inability to restore NPC sensitivity to DR-mediated apoptosis, despite the significant induction of caspase 8. To determined whether PED/PEA-15 up-regulation precedes caspase 8 induction, we evaluated the levels of the two proteins in NPCs at different time points after stimulation with inflammatory cytokines. In line with their inability to sensitize NPCs to DR-mediated apoptosis, inflammatory cytokines induced a significant and stable increase in PED/PEA-15 expression in 12 h, whereas caspase 8 reached considerable levels 24–48 h after stimulation (Fig. 5 E).
Figure 5.

Expression of DISC inhibitory proteins in tissue-isolated embryonic and adult NPCs. Immunoblot analysis of cFLIP (A) and PED/PEA-15 (B) on NPCs, JY, and 293T cell lines, the latter transfected with Tween/cFLIP (293 FLIP) or Tween/MYC-PED/PEA-15 (293 MYC-PED), were used as positive control. Immunoblot analysis of PED/PEA-15 in NPCs (C) and NT2-neurons (D) untreated (Control) or treated with TNF-α, IFN-γ, and IL-1β alone or in combination (Mix). One representative experiment out of three is shown. (E) Immunoblot analysis of PED/PEA-15 and caspase 8 in NPCs at different time points after stimulation with inflammatory cytokines. Detection of β-actin in the same membrane blot served as loading control. (F) Immunoprecipitation (IP) of the CD95 DISC in control (−) or CD95-stimulated (+) NPCs treated with TNF-α, IFN-γ, and IL-1β. CD95 was immunoprecipitated and blot probed for CD95, caspase 8, FADD, and PED/PEA-15. One representative experiment out of four is shown.
PED/PEA-15 contains a DED domain and inhibits DR-mediated apoptosis by competing with caspase 8 for FADD binding. To demonstrate the antiapoptotic activity of PED/PEA-15 in NPCs, the CD95 DISC was analyzed in cells pretreated with TNF-α, IFN-γ, and IL-1β to induce caspase 8 expression. Immunoblot analysis after CD95 immunoprecipitation showed a massive recruitment of PED/PEA-15 and FADD into the DISC of NPCs after CD95 stimulation, whereas caspase 8 was scarcely present (Fig. 5 F). To formally prove that PED/PEA-15 mediates DR resistance in NPCs, PED/PEA-15 antisense was cloned in the Tween lentiviral vector and transduced in NPCs. Flow cytometry analysis of NPCs transduced with PED/PEA-15 antisense or control lentiviral vector showed that the lentiviral infection produced a virtually pure population of GFP positive cells (Fig. 6 A). The expression of PED/PEA-15 was subsequently monitored by immunoblot analysis in normal culture conditions or after exposure to TNF-α, IFN-γ, and IL-1β. Transduction of NPCs with PED/PEA-15 antisense considerably reduced PED/PEA-15 expression, both in basal conditions and after treatment with inflammatory cytokines (Fig. 6 B). Accordingly, NPCs transduced with PED/PEA-15 antisense acquired CD95 sensitivity and underwent caspase activation upon exposure to inflammatory cytokines and anti-CD95 antibody, as shown by caspase degradation (Fig. 6 C) and susceptibility to apoptosis (Fig. 6 D). Thus, absence of caspase 8 and PED/PEA-15 overexpression generate a double level of protection from DR-mediated apoptosis in NPCs.
Figure 6.

PED/PEA-15–mediated protection of adult NPCs from CD95-induced apoptosis. (A) Flow cytometry analysis of GFP positive NPCs transduced with empty Tween (Vector) or Tween/PED/PEA-15 antisense (PED/AS) vectors (gray), as compared with nontransduced NPCs (black). (B) Immunoblot analysis of PED/PEA-15 expression in NPCs transduced as in A, untreated or treated TNF-α, IFN-γ, and IL-1β (+Mix). (C) Caspase expression and degradation in NPCs transduced with empty Tween (Vector) or PED/AS and exposed to anti-CD95 where indicated. (D) Percentage of apoptosis in untransduced (Mock) and empty Tween (Vector) or PED/AS transduced NPCs exposed to anti-CD95 for 24 or 48 h. All samples in C and D were preincubated with TNF-α, IFN-γ, and IL-1β 48–60 h before anti-CD95 stimulation. Results shown are representative of four independent experiments with tissue-isolated embryonic and adult NPCs.
Discussion
For several years, one of the main dogmas in neuroscience was based on the assumption that the nervous system's ability to recover from any kind of injury was due to neuronal plasticity, but not regeneration. Recently, the discovery of in-brain self-renewing populations of cells, potentially capable of differentiating in functional mature neurons and glial cells, has widened up the horizons in the therapy of neurodegenerative disorders (3, 38, 39). However, a profound understanding of the biological behavior of primitive neural cells is required to allow their manipulation and therapeutic utilization.
Here, we have demonstrated that embryonic and adult NPCs express functionally inactive DRs, whose binding by their specific ligands does not generate apoptotic signals because of the absence of caspase 8. Although the extrinsic apoptotic pathway controls the developmental production and homeostasis of hematopoietic cells (40), the intrinsic pathway is exploited during the development of the nervous system to shape the brain through the activation of programmed cell death in selected neural populations (41). The absence of caspase 8 in primitive neural cells may contribute to the developmental dependence on the intrinsic pathway of neural cells, which accumulate abnormally in embryos of mice lacking caspase 9 or its adaptor Apaf-1.
The refractoriness to the activation of the extrinsic pathway in primitive neural cells may have considerable implications in adult neurogenesis. In several pathological conditions, DR ligands exert a deleterious effect on neurons, whereas NPCs migrate and survive in the site of lesion. Brain injury creates in degenerating areas an inflammatory environment that enhances neuronal death (23). The ability of NPCs to survive in the damaged areas of the brain apparently conflicts with the up-regulation of CD95 and caspase 8 observed after their exposure to the inflammatory cytokines that are commonly produced during acute brain lesions (23). However, exogenous or cytokine-induced expression of caspase 8 is not sufficient to prime primitive neural cells to DR-mediated apoptosis, even in stimulatory conditions resulting in massive neuronal death. After DR stimulation in sensitive cells, caspase 8 undergoes an intramolecular refolding responsible for its proteolytic activation as death signal transducer. Primitive neural cells do not activate caspase 8 after DR triggering. Refractoriness to DR stimulation in cells expressing all the different components of the apoptotic machinery may result from the inability to recruit FADD or caspase 8 into the DISC. Primitive neural cells express high levels of PED/PEA-15, a DED domain containing protein that binds the DED domain of FADD or caspase 8, thereby preventing the recruitment of caspase 8 to the DISC and subsequent activation of the apoptotic cascade. The antiapoptotic role of PED/PEA-15 is particularly clear in NPCs exposed to inflammatory cytokines, where the rapid increase in PED/PEA-15 expression neutralizes the up-regulation of CD95 and caspase 8. Although the ability of these cytokines to modulate the expression of PED/PEA-15 has not been explored before, this behavior seems to typically characterize undifferentiated neural cells because the same cytokines down-regulate PED/PEA-15 protein levels in NT2/D1-derived neurons. Thus, it is likely that the modulation of PED/PEA-15 by inflammatory cytokines differentially regulate the survival of primitive neural cells and their progeny. The molecular basis underlying the opposite effects of inflammatory cytokines on PED/PEA-15 expression in primitive neural cells and neurons should be a matter of investigation. A possible explanation may reside in the different targets of the signals generated by inflammatory cytokines in undifferentiated and differentiated cells. Because Akt-mediated phosphorylation of PED/PEA-15 increases its protein stability (42), the different activation of this kinase in mature and immature cells represents a possible mechanism responsible for the opposite effect on PED/PEA-15 expression.
The inflammatory products generated in the lesioned areas of the brain during acute pathological conditions may impair neuronal cell survival, even without the contribution of DR triggering. However, high PED/PEA-15 expression in neural stem and progenitor cells may not exclusively protect these cells from DR ligands, as PED/PEA-15 levels comparable or lower than those observed in primitive neural cells have been shown to inhibit apoptosis induced by growth factor deprivation, exposure to H2O2, and anisomycin (43). Therefore, constitutive and cytokine-induced PED/PEA-15 may theoretically contribute to promote the survival of primitive neural cells during oxidative stress, inflammation, or growth factor deprivation. The ability of PED/PEA-15 to circumvent the extrinsic death signals makes it a suitable therapeutic target to enhance the prosurvival response of injured differentiated neurons. Likewise, the capacity of the PI3K–Akt pathway to sustain the levels of PED/PEA-15 may provide a further explanation to the neuroprotective potential of PI3K–Akt-inducing signals (42).
In conclusion, this work demonstrates that primitive neural cells display a double level of protection from the deadly activity of DR ligands, which are unable to deliver apoptosis signals in these cells even in enhancing situations, such as the inflammatory response typical of many neurodegenerative conditions. Thus, our results may explain the ability of endogenous neural stem cells to survive in acute brain injury. Although in vivo studies are required to confirm that the high expression of PED/PEA-15 and the absence of caspase 8 observed in vitro are relevant features of NPCs, these findings may provide useful information for therapeutic approaches exploiting the endogenous neurogenesis or based on delivery of neural stem or progenitor cells to replace damaged neurons.
Acknowledgments
This paper is dedicated to Stefano Pagano.
The authors thank Dr. L. Naldini for providing lentiviral vectors, Drs. F. Condorelli and A. Pagliuca for suggestions and critically reading the manuscript, and P. Di Matteo and G. Loreto for technical assistance.
This work was supported by funds from Associazione Italiana per la Ricerca sul Cancro, Ministero dell'Istruzione e dell'Università, and Ministero della Salute.
The authors have no conflicting financial interests.
Abbreviations used in this paper: DISC, death-inducing signaling complex; DR, death receptor; NPC, neural progenitor cell.
References
- 1.Chenn, A., and S.K. McConnell. 1995. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell. 82:631–641. [DOI] [PubMed] [Google Scholar]
- 2.Lu, B., L. Jan, and Y.N. Jan. 2000. Control of cell divisions in the nervous system: symmetry and asymmetry. Annu. Rev. Neurosci. 23:531–556. [DOI] [PubMed] [Google Scholar]
- 3.Gage, F.H. 2000. Mammalian neural stem cells. Science. 287:1433–1438. [DOI] [PubMed] [Google Scholar]
- 4.Temple, S. 2001. The development of neural stem cells. Nature. 414:112–117. [DOI] [PubMed] [Google Scholar]
- 5.Weissman, I.L., D.J. Anderson, and F. Gage. 2001. Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 17:387–403. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Verdugo, J.M., F. Doetsch, H. Wichterle, D.A. Lim, and A. Alvarez-Buylla. 1998. Architecture and cell types of the adult subventricular zone: in search of the stem cells. J. Neurobiol. 36:234–248. [DOI] [PubMed] [Google Scholar]
- 7.Lois, C., and A. Alvarez-Buylla. 1993. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl. Acad. Sci. USA. 90:2074–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss, S., B.A. Reynolds, A.L. Vescovi, C. Morshead, C.G. Craig, and D. van der Kooy. 1996. Is there a neural stem cell in the mammalian forebrain? Trends Neurosci. 19:387–393. [DOI] [PubMed] [Google Scholar]
- 9.Caldwell, M.A., X. He, N. Wilkie, S. Pollack, G. Marshall, K.A. Wafford, and C.N. Svendsen. 2001. Growth factors regulate the survival and fate of cells derived from human neurospheres. Nat. Biotechnol. 19:475–479. [DOI] [PubMed] [Google Scholar]
- 10.Peter, M.E., and P.H. Krammer. 2003. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10:26–35. [DOI] [PubMed] [Google Scholar]
- 11.Green, D.R. 2000. Apoptotic pathways: paper wraps stone blunts scissors. Cell. 102:1–4. [DOI] [PubMed] [Google Scholar]
- 12.Irmler, M., M. Thome, M. Hahne, P. Schneider, K. Hofmann, V. Steiner, J.L. Bodmer, M. Schroter, K. Burns, C. Mattmann, et al. 1997. Inhibition of death receptor signals by cellular FLIP. Nature. 388:190–195. [DOI] [PubMed] [Google Scholar]
- 13.Xiao, C., B.F. Yang, N. Asadi, F. Beguinot, and C. Hao. 2002. Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-FLIP and PED/PEA-15 in glioma cells. J. Biol. Chem. 277:25020–25025. [DOI] [PubMed] [Google Scholar]
- 14.Kuida, K., T.F. Haydar, C.Y. Kuan, Y. Gu, C. Taya, H. Karasuyama, M.S. Su, P. Rakic, and R.A. Flavell. 1998. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 94:325–337. [DOI] [PubMed] [Google Scholar]
- 15.Hakem, R., A. Hakem, G.S. Duncan, J.T. Henderson, M. Woo, M.S. Soengas, A. Elia, J.L. de la Pompa, D. Kagi, W. Khoo, et al. 1998. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 94:339–352. [DOI] [PubMed] [Google Scholar]
- 16.Cecconi, F., G. Alvarez-Bolado, B.I. Meyer, K.A. Roth, and P. Gruss. 1998. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 94:727–737. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida, H., Y.Y. Kong, R. Yoshida, A.J. Elia, A. Hakem, R. Hakem, J.M. Penninger, and T.W. Mak. 1998. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 94:739–750. [DOI] [PubMed] [Google Scholar]
- 18.Varfolomeev, E.E., M. Schuchmann, V. Luria, N. Chiannilkulchai, J.S. Beckmann, I.L. Mett, D. Rebrikov, V.M. Brodianski, O.C. Kemper, O. Kollet, et al. 1998. Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 9:267–276. [DOI] [PubMed] [Google Scholar]
- 19.Martin-Villalba, A., I. Herr, I. Jeremias, M. Hahne, R. Brandt, J. Vogel, J. Schenkel, T. Herdegen, and K.M. Debatin. 1999. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J. Neurosci. 19:3809–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin-Villalba, A., M. Hahne, S. Kleber, J. Vogel, W. Falk, J. Schenkel, and P.H. Krammer. 2001. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 8:679–686. [DOI] [PubMed] [Google Scholar]
- 21.Demjen, D., S. Klussmann, S. Kleber, C. Zuliani, B. Stieltjes, C. Metzger, U.A. Hirt, H. Walczak, W. Falk, M. Essig, et al. 2004. Neutralization of CD95 ligand promotes regeneration and functional recovery after spinal cord injury. Nat. Med. 10:389–395. [DOI] [PubMed] [Google Scholar]
- 22.Zhang, X., S.H. Graham, P.M. Kochanek, D.W. Marion, P.D. Nathaniel, S.C. Watkins, and R.S. Clark. 2003. Caspase-8 expression and proteolysis in human brain after severe head injury. FASEB J. 17:1367–1369. [DOI] [PubMed] [Google Scholar]
- 23.Wang, C.X., and A. Shuaib. 2002. Involvement of inflammatory cytokines in central nervous system injury. Prog. Neurobiol. 67:161–172. [DOI] [PubMed] [Google Scholar]
- 24.Kokaia, Z., and O. Lindvall. 2003. Neurogenesis after ischaemic brain insults. Curr. Opin. Neurobiol. 13:127–132. [DOI] [PubMed] [Google Scholar]
- 25.Galli, R., S.F. Pagano, A. Gritti, and A.L. Vescovi. 2000. Regulation of neuronal differentiation in human CNS stem cell progeny by leukemia inhibitory factor. Dev. Neurosci. 22:86–95. [DOI] [PubMed] [Google Scholar]
- 26.Pagano, S.F., F. Impagnatiello, M. Girelli, L. Cova, E. Grioni, M. Onofri, M. Cavallaro, S. Etteri, F. Vitello, S. Giombini, et al. 2000. Isolation and characterization of neural stem cells from the adult human olfactory bulb. Stem Cells. 18:295–300. [DOI] [PubMed] [Google Scholar]
- 27.Vescovi, A.L., E.A. Parati, A. Gritti, P. Poulin, M. Ferrario, E. Wanke, P. Frolichsthal-Schoeller, L. Cova, M. Arcellana-Panlilio, A. Colombo, and R. Galli. 1999. Isolation and cloning of multipotential stem cells from the embryonic human CNS and establishment of transplantable human neural stem cell lines by epigenetic stimulation. Exp. Neurol. 156:71–83. [DOI] [PubMed] [Google Scholar]
- 28.Pleasure, S.J., C. Page, and V.M. Lee. 1992. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J. Neurosci. 12:1802–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Maria, R., L. Lenti, F. Malisan, F. d'Agostino, B. Tomassini, A. Zeuner, M.R. Rippo, and R. Testi. 1997. Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science. 277:1652–1655. [DOI] [PubMed] [Google Scholar]
- 30.Condorelli, G., G. Vigliotta, C. Iavarone, M. Caruso, C.G. Tocchetti, F. Andreozzi, A. Cafieri, M.F. Tecce, P. Formisano, L. Beguinot, and F. Beguinot. 1998. PED/PEA-15 gene controls glucose transport and is overexpressed in type 2 diabetes mellitus. EMBO J. 17:3858–3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Follenzi, A., L.E. Ailles, S. Bakovic, M. Geuna, and L. Naldini. 2000. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat. Genet. 25:217–222. [DOI] [PubMed] [Google Scholar]
- 32.Hao, C., F. Beguinot, G. Condorelli, A. Trencia, E.G. Van Meir, V.W. Yong, I.F. Parney, W.H. Roa, and K.C. Petruk. 2001. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 61:1162–1170. [PubMed] [Google Scholar]
- 33.Sleeper, E., C. Tamm, J. Frisen, B. Zhivotovsky, S. Orrenius, and S. Ceccatelli. 2002. Cell death in adult neural stem cells. Cell Death Differ. 9:1377–1378. [DOI] [PubMed] [Google Scholar]
- 34.Wosik, K., B. Becher, A. Ezman, J. Nalbantoglu, and J.P. Antel. 2001. Caspase 8 expression and signaling in Fas injury-resistant human fetal astrocytes. Glia. 33:217–224. [DOI] [PubMed] [Google Scholar]
- 35.Andrews, P.W. 1984. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 103:285–293. [DOI] [PubMed] [Google Scholar]
- 36.Pierce, T., H.J. Worman, and J. Holy. 1999. Neuronal differentiation of NT2/D1 teratocarcinoma cells is accompanied by a loss of lamin A/C expression and an increase in lamin B1 expression. Exp. Neurol. 157:241–250. [DOI] [PubMed] [Google Scholar]
- 37.Falsig, J., M. Latta, and M. Leist. 2004. Defined inflammatory states in astrocyte cultures: correlation with susceptibility towards CD95-driven apoptosis. J. Neurochem. 88:181–193. [DOI] [PubMed] [Google Scholar]
- 38.Reynolds, B.A., and S. Weiss. 1992. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 255:1707–1710. [DOI] [PubMed] [Google Scholar]
- 39.Rossi, F., and E. Cattaneo. 2002. Opinion: neural stem cell therapy for neurological diseases: dreams and reality. Nat. Rev. Neurosci. 3:401–409. [DOI] [PubMed] [Google Scholar]
- 40.De Maria, R., A. Zeuner, A. Eramo, C. Domenichelli, D. Bonci, F. Grignani, S.M. Srinivasula, E.S. Alnemri, U. Testa, and C. Peschle. 1999. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature. 401:489–493. [DOI] [PubMed] [Google Scholar]
- 41.Troy, C.M., and G.S. Salvesen. 2002. Caspases on the brain. J. Neurosci. Res. 69:145–150. [DOI] [PubMed] [Google Scholar]
- 42.Trencia, A., A. Perfetti, A. Cassese, G. Vigliotta, C. Miele, F. Oriente, S. Santopietro, F. Giacco, G. Condorelli, P. Formisano, and F. Beguinot. 2003. Protein kinase B/Akt binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic action. Mol. Cell. Biol. 23:4511–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Condorelli, G., A. Trencia, G. Vigliotta, A. Perfetti, U. Goglia, A. Cassese, A.M. Musti, C. Miele, S. Santopietro, P. Formisano, and F. Beguinot. 2002. Multiple members of the mitogen-activated protein kinase family are necessary for PED/PEA-15 anti-apoptotic function. J. Biol. Chem. 277:11013–11018. [DOI] [PubMed] [Google Scholar]