

Abstract
Single and combination cytokines offer promise in some patients with advanced cancer. Many spontaneous and experimental cancers naturally express ligands for the lectin-like type-2 transmembrane stimulatory NKG2D immunoreceptor; however, the role this tumor recognition pathway plays in immunotherapy has not been explored to date. Here, we show that natural expression of NKG2D ligands on tumors provides an effective target for some cytokine-stimulated NK cells to recognize and suppress tumor metastases. In particular, interleukin (IL)-2 or IL-12 suppressed tumor metastases largely via NKG2D ligand recognition and perforin-mediated cytotoxicity. By contrast, IL-18 required tumor sensitivity to Fas ligand (FasL) and surprisingly did not depend on the NKG2D–NKG2D ligand pathway. A combination of IL-2 and IL-18 stimulated both perforin and FasL effector mechanisms with very potent effects. Cytokines that stimulated perforin-mediated cytotoxicity appeared relatively more effective against tumor metastases expressing NKG2D ligands. These findings indicate that a rational choice of cytokines can be made given the known sensitivity of tumor cells to perforin, FasL, and tumor necrosis factor–related apoptosis-inducing ligand and the NKG2D ligand status of tumor metastases.
Keywords: tumor, NK cell, Fas ligand, IL-2, IL-18
Introduction
Cytokines have played an important role in new progress in tumor immunology and immunotherapy. The use of IL-2 in patients with metastatic melanoma and renal cell cancer has demonstrated that manipulation of the immune system is capable of mediating the durable regression of established metastatic tumors (1). The mechanism of antitumor efficacy of IL-2 is closely related to its ability to expand and activate NK and T cells that express IL-2 receptors. Other promising cytokines in cancer immunotherapy, including IL-12 (2) and IL-18 (3) or combination IL-2/IL-18 (4), have also been shown to mediate their antitumor activities in mice to a large extent via NK cells. IL-12 plays an essential role in the interaction between the innate and adaptive arms of immunity (5), produced by APC and acting upon T cells and NK cells to generate cytotoxic lymphocytes. IL-12 is also the major cytokine responsible for Th1 cell differentiation, allowing potent production of IFN-γ. IL-18 is a potent immunoregulatory cytokine that was initially described as an IFN-γ–inducing factor (6). IL-18 enhances T and NK cell cytokine production, proliferation, and cytolytic activity (7, 8) and the expression of Fas ligand (FasL) and FasL- or perforin-mediated antitumor activity (9–11). Systemic administration of IL-18 has demonstrated considerable therapeutic activity in several murine tumor models (10, 12).
Recent understanding of the means by which NK cells kill target cells through a complex set of activating and inhibitory receptors recognizing corresponding ligands on tumor cells has paved the way for the design of improved strategies for NK cell–based immunotherapy. One key activating receptor on NK cells for the elimination of tumor cells is NKG2D. NKG2D is present as a homodimeric receptor (13), is expressed on the cell surface of almost all NK cells (14), and is inducible by exposure to IL-15 (15). Several ligands, which bind to NKG2D, are structurally related to MHC class I molecules (16–18). In humans, the polymorphic MHC class I chain–related molecules (MIC)A and MICB can be recognized by NKG2D (19, 20). Unlike conventional MHC class I, those MIC proteins display up-regulated surface expression on stressed cells and are frequently overexpressed by tumors (21). Although MIC molecules have not been found in mice, the retinoic acid early inducible-1 (Rae-1) gene products and a distantly related minor histocompatibility antigen, H60, have been reported as NKG2D ligands in mice (17, 18). Mouse UL16-binding protein-like transcript 1 (Mult1), a third relative of this growing protein family, was identified more recently and shown to bind NKG2D (22, 23).
Natural or induced expression of NKG2D ligands markedly enhances the sensitivity of tumor cells to NK cells in vitro (14, 17, 19, 22, 24, 25). In general, the lysis of tumor cells that naturally express NKG2D ligands is partially inhibited by NKG2D-specific antibodies, indicating that NKG2D is an important receptor in the recognition of target cells by NK cells but not the only one (14, 25). Indeed, some target cells that lack expression of NKG2D ligands are nevertheless sensitive to NK cells (14), in line with the identification of other NK cell stimulatory receptors that participate in tumor cell recognition (26). Expression of NKG2D ligands by tumor cells also results in immune destruction in vivo. Recent studies show that the ectopic expression of NKG2D ligands, Rae-1 and H60, in several tumor cell lines resulted in the rejection of the tumor cells, even when the tumor cells expressed normal levels of MHC class I molecules (27, 28). Immune depletion studies showed that rejection was dependent on NK cells and/or CD8+ T cells depending on the parent tumor cell line and the dose of tumor cells that were transferred (27). These studies together with the in vitro studies leave little doubt that expression of NKG2D ligands confers an effective barrier to tumor formation. Interestingly, our recent study suggested that ectopic expression of the NKG2D ligand, Rae-1β, in a MHC class I–deficient tumor rendered it particularly susceptible to perforin-mediated tumor rejection (29).
Despite our clear knowledge of the therapeutic value of cytokines in promoting NK cell–mediated suppression of tumor growth and metastases, no previous study has elucidated whether direct immune recognition of tumor cells is a requirement for cytokine efficacy. Here, we illustrate using a series of cytokines with distinct means of activating NK cell effector function that the NKG2D–NKG2D ligand recognition pathway is pivotal in the antimetastatic activity of cytokines that promote perforin-mediated cytotoxicity. This study now provides a fundamental basis for some rational selection of cytokines in NK cell–mediated therapy of tumor metastases that either have or lack NKG2D ligand expression.
Materials and Methods
Mice.
Inbred C57BL/6 and BALB/c WT mice were purchased from The Walter and Eliza Hall Institute of Medical Research. The following gene-targeted mice were bred at the Peter MacCallum Cancer Centre: C57BL/6 perforin (pfp)-deficient (B6 pfp−/−); C57BL/6 FasL mutant (B6 gld); C57BL/6 RAG-1-deficient (B6 RAG-1−/−) (from Dr. Corcoran, The Walter and Eliza Hall Institute of Medical Research); BALB/c IFN-γ−/−; BALB/c pfp−/−; BALB/c pfp IFN-γ−/−; BALB/c TNF-related apoptosis-inducing ligand (TRAIL)−/− (from Dr. Peschon, AMGEN, Seattle, WA) (30); and BALB/c pfp TRAIL−/− mice. All mice originally generated on a 129 background have been backcrossed between 10–12 times onto the C57BL/6 or BALB/c background. Mice of 6–12 wk of age were used in all experiments that were performed according to animal experimental ethics committee guidelines.
Isolation of Spleen NK Cells and Cytotoxicity Assay.
NK cells were prepared from the spleen of B6 RAG-1−/− mice as described previously (31). Purity was always >90%. The cytolytic activity of NK cells from various cytokine-treated mice was tested against tumor target cells by a standard 12-h 51Cr release assay as described previously. In some experiments, the assay was performed in the presence of neutralizing hamster anti-mNKG2D mAb (C7) (30 μg/ml) or control hamster Ig (30 μg/ml).
Flow Cytometric Analysis.
Tumor cell lines were assessed for NKG2D ligand expression as follows. To avoid the nonspecific binding of mAbs to FcγR, anti–mouse CD16/32 (2.4G2) mAb was added to the mAb cocktail. After washing the cells, staining was performed in PBS with 5% FCS and 0.02% sodium azide on ice using the PE-conjugated NKG2D tetramer as described previously (32). Additionally, tumor cell lines were screened using anti-pan Rae− and anti-H60 antibodies (33). Anti-pan Rae-1 mAb (clone 186107, rat IgG2a isotype) reacts with Rae-1α, β, γ, δ, and ɛ as described previously (34). The stained cells were analyzed on a FACScan (Becton Dickinson), and the data were processed by the CELLQuest program (Becton Dickinson).
Tumor Cell Lines.
The following standard experimental mouse tumor cell lines were employed in vitro and in vivo. B16F10 melanoma (perforin-sensitive, FasL- and TRAIL-insensitive, H-2b); RMA-S lymphoma (perforin-sensitive, FasL- and TRAIL-insensitive, H-2b); RMA-S-Rae-1β lymphoma (perforin-sensitive, FasL- and TRAIL-insensitive, H-2b); 3LL Lewis lung carcinoma (perforin-sensitive, FasL-sensitive and TRAIL-insensitive, H-2b); Renca renal cell carcinoma (perforin-, and TRAIL-sensitive and FasL-insensitive, H-2d), DA3-m (mock vector alone infected) mammary carcinoma (perforin-, FasL-, and TRAIL-sensitive, H-2d), and DA3-H60 (H60 infected) mammary carcinoma (perforin-, FasL-, and TRAIL-sensitive, H-2d). DA3-H60 cells were prepared and selected by flow cytometry as described previously (31). The maintenance of all tumor cell lines and the sensitivities of lung and liver metastases to various cytotoxic molecules in vitro and in vivo have been described previously (35).
Tumor Models In Vivo.
3LL, Renca, DA3, and DA3-H60 tumor cell lines were inoculated i.v. at a dose indicated and previously shown to result in a maximal number of lung metastases regardless of whether inoculation was in WT, gene-targeted, or antibody-treated mice. The ability of each cytokine treatment schedule was then evaluated for its ability to reduce the expected metastatic tumor burden. For all experimental metastasis models, mice were injected i.v. with tumor cells and killed 14 d later, the lungs removed, and surface metastases counted with the aid of a dissecting microscope. In all metastasis models, the data was recorded as the mean number of metastases ± SE of the mean.
Cytokine Treatment Protocols.
Recombinant mouse IL-12 and IL-2 was provided by Genetics Institute and Chiron Corporation, respectively. Recombinant mouse IL-18 was provided by Glaxo Smith Kline. The preparations of IL-2, IL-12, and IL-18 were diluted in PBS immediately before use. Some groups of mice were treated with one of the following: (a) 500 U IL-12 i.p. on days 3–7 after tumor inoculation; (b) 100,000 U IL-2 i.p. on days 3–7; (c) 2 μg of IL-18 i.p. on days 0–4; (d) 100,000 U IL-2 i.p. on days 4, 6 and 8; (e) 2 μg of IL-18 i.p. on days 4–8; and (f) schedules (d) and (e) together.
NK Cell Depletion and NKG2D Neutralization.
NK cells were specifically depleted in B6 and BALB/c mice using 100 μg i.p. rabbit anti-asialoGM1 (asGM1) antibody (Wako Chemicals) on days 0, 1, and 7 (after tumor inoculation) as described (36). Some groups of B6 or BALB/c mice were treated with either hamster anti–mouse NKG2D mAb (C7 clone; reference 37) (250 μg i.p.) or hamster control Ig mAb (250 μg i.p.) on days 0, 1, 7, and 8 after tumor inoculation. It should also be noted that NKG2D+ NK effector cells were not depleted by anti-NKG2D mAb treatment (not depicted).
Statistical Analysis.
Significant differences in metastases were determined by the unpaired Mann-Whitney U test. P values less than 0.05 were considered significant.
Results
Experimental Tumor Cell Lines Naturally Express NKG2D Ligands.
Initially we screened several experimental tumors from BALB/c and C57BL/6 mice for their expression of NKG2D ligands using the previously described NKG2D tetramer (27), anti-pan Rae-1 mAb (34), and anti-H60 mAb (Fig. 1). Renca renal carcinoma was shown to express the NKG2D ligands, Rae-1 and H60 (Fig. 1 A). Rae-1ɛ expression was additionally demonstrated by flow cytometry, whereas Rae-1β and H60 expression was confirmed by RT-PCR (not depicted). DA3 mammary carcinoma cells that did not naturally express NKG2D ligands were either mock infected (Fig. 1 B) or infected with a retrovirus to express H60 (Fig. 1 C). The C57BL/6 3LL lung carcinoma was shown to strongly express the NKG2D ligands (Fig. 1 D). By contrast, B16F10 melanoma and RMA-S lymphoma cells did not express NKG2D ligands (Fig. 1, E and F). We have reported previously the ectopic expression of Rae-1β in RMA-S tumor cells (29) (Fig. 1 G).
Figure 1.
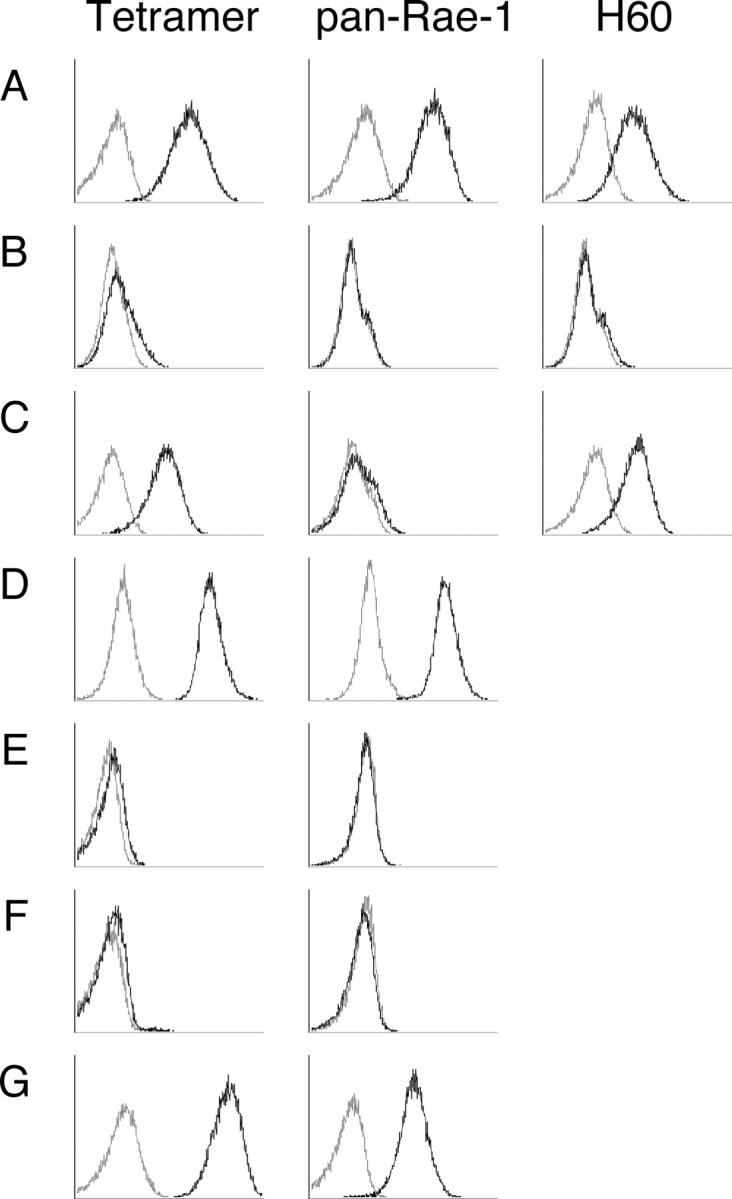
Experimental tumor cell lines naturally express NKG2D ligands. Tumor cell lines were stained with NKG2D tetramer (tetramer), anti-pan Rae-1 mAb and anti-H60 antibody as indicated: (A) Renca; (B) DA3; (C) DA3-H60; (D) 3LL; (E) B16F10; (F) RMA-S; and (G) RMA-S-Rae-1β. Solid black lines, test; gray lines, control (control tetramer, isotype). The data shown are representative of three independent experiments.
Critical Contribution of NKG2D to Perforin-mediated Tumor Suppression by IL-2 or IL-12.
Having established which experimental tumors expressed NKG2D ligands, we next set about defining the role the NKG2D–NKG2D ligand pathway was potentially playing in cytokine-mediated control of tumor growth in vivo. Experiments were performed by challenging mice with a dose of Renca tumor cells that would metastasize equivalently in untreated WT, NK cell–depleted WT mice, or other gene-targeted mice (Fig. 2). Treatment with a course of IL-2 (from day 3 to 7 after tumor inoculation) was effective in WT and RAG-1−/− mice, reducing metastatic load from ∼300 to 100 metastases, but IL-2 was completely ineffective in NK cell–depleted mice (Fig. 2). Interestingly, part of the effectiveness of IL-2 was mediated via the NKG2D–NKG2D ligand pathway since there were a significantly higher number of Renca lung metastases in anti-NKG2D–treated WT mice compared with control Ig-treated WT mice. In a similar fashion, anti-NKG2D–treated RAG-1−/− mice had a significantly higher number of Renca lung metastases compared with control Ig-treated RAG-1−/− mice (not depicted). We next assessed the effector mechanisms used by IL-2–activated NK cells to determine whether they were NKG2D dependent. We had shown previously that IL-2–mediated antimetastatic activity against Renca lung metastases was dependent on perforin and IFN-γ (38). Consistent with these previous observations, IL-2 was less effective in perforin- or IFN-γ–deficient mice and completely inactive in mice gene targeted for both perforin and IFN-γ (Fig. 2). Interestingly, anti-NKG2D mAb only increased metastases in IL-2–treated WT and IFN-γ–deficient mice and not in perforin-deficient mice (Fig. 2). In a similar manner, IL-12 (from day 3 to 7 after tumor inoculation) very significantly reduced the number of Renca lung metastases in WT and RAG-1−/− but not in NK cell–depleted mice (Fig. 3). Once again anti-NKG2D mAb significantly reduced the efficacy of IL-12. IL-12 was less effective in perforin-, TRAIL-, or IFN-γ–deficient mice and completely inactive in mice gene targeted for both perforin and TRAIL, consistent with our previous report (38) (Fig. 3). Anti-NKG2D mAb only increased metastases in IL-12–treated WT and TRAIL-deficient mice and not perforin-deficient mice (Fig. 3). Therefore, two different cytokines that promote NK cell perforin-mediated activity suppress metastases in large part via the NKG2D pathway.
Figure 2.

IL-2 suppresses tumor metastases via NK cell perforin and NKG2D. Groups of 5 to 10 WT, RAG-1−/−, pfp−/−, IFN-γ−/−, or pfp−/−IFN-γ−/− mice were inoculated i.v. with 105 Renca tumor cells on day 0. Some groups of mice, as indicated, received anti-NKG2D mAb or control Ig (250 μg i.p.) on days 0, 1, 7, and 8 or anti-asGM1 (100 μg i.p.) on days 0, 1 and 7 after tumor inoculation. Mice were untreated (dotted bars) or received IL-2 (solid bars) (100,000 U i.p. on days 3, 4, 5, 6, and 7). The lungs were removed from mice on day 14 and the metastatic nodules quantified. Data are recorded as the mean ± SEM with the significance of IL-2 efficacy (*P < 0.05) and significance of anti-NKG2D mAb inhibition (**P < 0.05) recorded as defined by a Mann-Whitney U test.
Figure 3.

IL-12 suppresses tumor metastases via NK cell perforin and NKG2D. Groups of 5 to 10 WT, RAG-1−/−, pfp−/−, IFN-γ−/−, TRAIL−/−, pfp−/−IFN-γ−/−, or pfp−/−TRAIL−/− mice were inoculated i.v. with 105 Renca tumor cells on day 0. Some groups of mice, as indicated, received anti-NKG2D mAb or control Ig (250 μg i.p.) on days 0, 1, 7, and 8 or anti-asGM1 (100 μg i.p.) on days 0, 1, and 7 after tumor inoculation. Mice were untreated (dotted bars) or received IL-12 (solid bars) (500 U i.p. on days 3, 4, 5, 6, and 7). The lungs were removed from mice on day 14 and the metastatic nodules quantified. Data are recorded as the mean ± SEM with the significance of IL-12 efficacy (*P < 0.05) and significance of anti-NKG2D mAb inhibition (**P < 0.05) recorded as defined by a Mann-Whitney U test.
IL-18 Therapy Suppresses Tumor Metastases via NK Cell FasL and Independently of NKG2D.
IL-18 enhances NK cell cytokine production, proliferation, and cytolytic activity and the expression of FasL and FasL- or perforin-mediated antitumor activity (9–11). Given the relatively poor activity of IL-18 alone against Renca tumor metastases, we next assessed the antimetastatic activity of IL-18 alone (day 0–4 after tumor inoculation) against 3LL lung carcinoma (Fig. 4). 3LL tumor cells are sensitive to both perforin- and FasL-dependent pathways (35) and were thus chosen as a tumor target for IL-18 therapy. The activity of IL-18 was moderate but reproducible, and mediated by NK cells rather than T or B cells (Fig. 4). Only a minor increase in 3LL lung metastases was observed in anti-NKG2D mAb– compared with control Ig-treated mice. IL-18 treatment was significantly less effective in FasL mutant gld mice and slightly less so in perforin-deficient mice (Fig. 4). Anti-NKG2D mAb only slightly increased metastases in IL-18–treated WT and gld mice but not perforin-deficient mice. Collectively, these data suggested that the NKG2D pathway was essential for IL-2– and IL-12–mediated antimetastatic activity but not IL-18–mediated antitumor activity.
Figure 4.
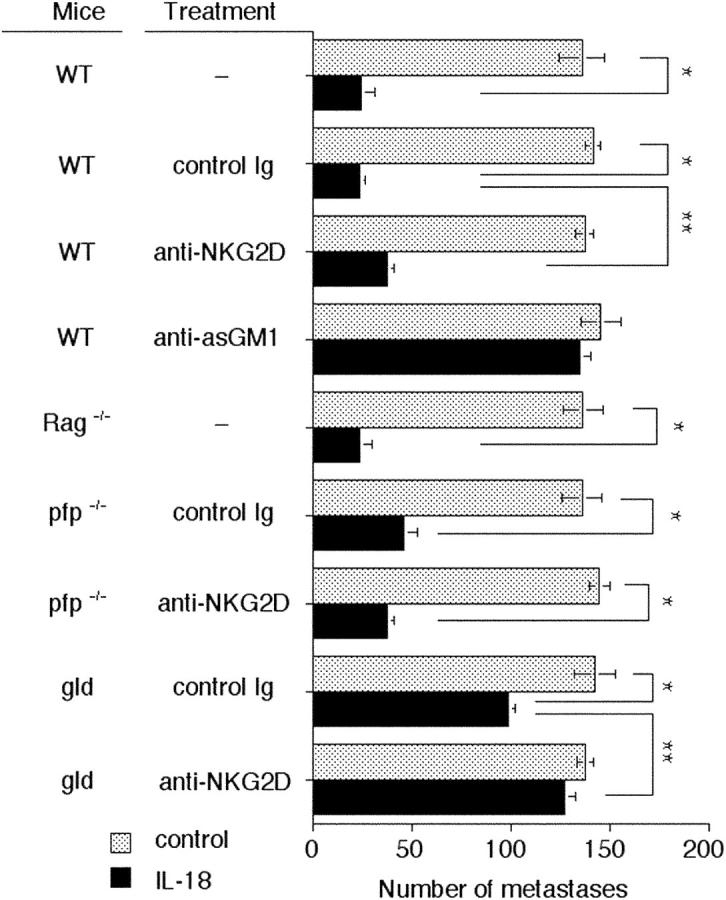
IL-18 suppresses tumor metastases via NK cell FasL and independently of NKG2D. Groups of 5 to 10 WT, RAG-1−/−, pfp−/−, or gld mice were inoculated i.v. with 5 × 105 3LL tumor cells on day 0. Some groups of mice, as indicated, received anti-NKG2D mAb or control Ig (250 μg i.p.) on days 0, 1, 7, and 8 or anti-asGM1 (100 μg i.p.) on days 0, 1, and 7 after tumor inoculation. Mice were untreated (dotted bars) or received IL-18 (solid bars) (2 μg i.p. on days 0, 1, 2, 3, and 4). The lungs were removed from mice on day 14 and the metastatic nodules quantified. Data are recorded as the mean ± SEM, with the significance of IL-18 efficacy (*P < 0.05) and significance of anti-NKG2D mAb inhibition (**P < 0.05) recorded as defined by a Mann-Whitney U test.
Combined IL-2/IL-18 Therapy Suppresses Tumor Metastases via NK Cell FasL and Perforin.
Previous reports had indicated that IL-2 and IL-18 were effective in combination (39), and our data suggested that this combination might engage several distinct effector pathways that were tumor suppressive. Therefore, we examined a single or combined treatment regime with IL-2 and IL-18 against 3LL lung carcinoma. Strikingly, the IL-2/IL-18 combination was extremely effective at suppressing lung metastases compared with treatment with IL-2 or IL-18 alone (Fig. 5 A). Notably, this combination required NK cells but not T and B cells (Fig. 5 A). The extremely potent antimetastatic activity of IL-2/IL-18 was significantly reduced in gld mice and completely inhibited in perforin-deficient mice treated with anti-FasL mAb (Fig. 5 B). These data suggested IL-2/IL-18 may be a particularly effective combination because both FasL and perforin mechanisms are being employed. Once again anti-NKG2D mAb increased metastases in WT and gld mice but not perforin-deficient mice (Fig. 5 B). In summary, these experiments illustrated that cytokine therapies could be tailored to suppress tumor metastases depending on the sensitivity of the tumor to perforin, TRAIL, or FasL and its expression of NKG2D ligands.
Figure 5.
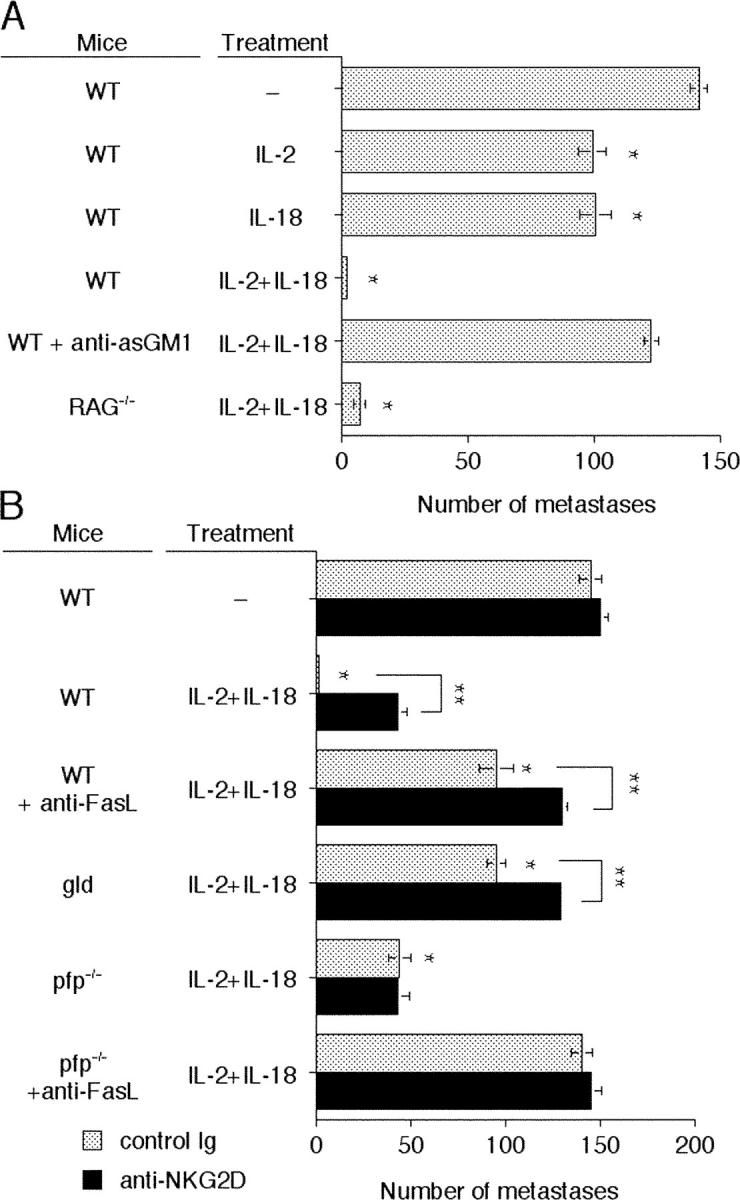
Combination IL-2/IL-18 potently suppresses tumor metastases via perforin and FasL. (A) Groups of 5 to 10 WT or RAG-1−/− mice were inoculated i.v. with 5 × 105 3LL tumor cells on day 0. Some groups of WT mice, as indicated, received anti-asGM1 (100 μg i.p.) on days 0, 1, and 7 after tumor inoculation. (B) Groups of 5 WT, pfp−/−, or gld mice were inoculated i.v. with 5 × 105 3LL tumor cells on day 0. Some groups of mice, as indicated, received anti-NKG2D mAb (solid bars) or control Ig (dotted bars) (250 μg i.p.) on days 0, 1, 7, and 8 or anti-FasL (250 μg i.p.) on days 0, 1 and 7 after tumor inoculation. Mice in A and B were untreated or received IL-2 alone (100,000 U i.p. on days 4, 6, and 8) and IL-18 alone (2 μg i.p. on days 4, 5, 6, 7, and 8) or the combination (IL-2/IL-18). The lungs were removed from mice on day 14 and the metastatic nodules quantified. Data are recorded as the mean ± SEM with the significance of cytokine efficacy (*P < 0.05) and significance of anti-NKG2D mAb inhibition (**P < 0.05) recorded as defined by a Mann-Whitney U test. Regardless of whether in the presence of control Ig or anti-NKG2D mAb, groups of pfp−/− and gld mice or WT and pfp−/− mice treated with anti-FasL, recorded a similar level of metastases (range 137 ± 4 to 145 ± 5 metastases) to WT control mice (not depicted).
Cytokine-activated NK Cells Kill Tumors in an NKG2D-dependent Manner.
To assess the relative contribution of NKG2D to NK cell–mediated cytotoxicity toward 3LL tumor cells following these cytokine protocols, NK cells were isolated from RAG-1−/− mice 24 h after the last cytokine treatment and NK cell–mediated killing examined in a 12-h cytotoxicity assay. Basal NK cell–mediated cytotoxicity was restricted against 3LL, B16F10 (Fig. 6 A), and TAP-2–deficient RMA-S tumor cells (not depicted); however, RMA-S-Rae-1β targets were considerably more sensitive to NK cell–mediated cytotoxicity, and this was inhibited by anti-NKG2D mAb (Fig. 6 A). IL-2 treatment enhanced NK cell–mediated cytotoxicity of 3LL and B16F10 target cells, and the majority of IL-2–enhanced cytotoxicity against 3LL was inhibited by anti-NKG2D mAb (Fig. 6 A). IL-2 also enhanced NK cell–mediated cytotoxicity of RMA-S-Rae-1β (Fig. 6 A) and RMA-S (not depicted) tumor cells, and anti-NKG2D mAb only inhibited lysis of the RMA-S-Rae-1β–expressing target cells (Fig. 6 A). A very similar pattern was observed with NK cells from IL-12–treated mice (Fig. 6 B). Collectively, these data indicated that a large proportion of IL-2– or IL-12–stimulated NK cell cytotoxicity against NKG2D ligand expressing tumor cells was NKG2D dependent. By contrast, the cytotoxicity of NK cells from IL-18–treated mice was not inhibited by anti-NKG2D mAb (Fig. 6 C). Notably, FasL-sensitive 3LL and RMA-S-Rae-1β tumor cells were more sensitive to IL-18–stimulated NK cells than FasL-resistant B16F10 tumor cells. These preliminary data supported the concept that IL-18–stimulated NK cells were capable of FasL-mediated cytotoxicity and that this pathway was not NKG2D dependent. In concert with the potent antimetastatic activity of the IL-2/IL-18 combination, IL-2/IL-18–stimulated NK cells were extremely cytotoxic toward all the tumor target cells and anti-NKG2D mAb partially neutralized lysis of NKG2D ligand expressing 3LL and RMA-S-Rae-1β target cells (Fig. 6 D).
Figure 6.

Some cytokine-activated NK cells kill tumors in an NKG2D-dependent manner. Spleen NK cells were isolated (day 0) from B6 RAG-1−/− mice 24 h after treatment with: PBS (control) and (A) IL-2 (100,000 U i.p. on days –5,–4,–3,–2,–1); (B) IL-12 (500 U i.p. on days –5,–4,–3,–2,–1); (C) IL-18 (2 μg i.p. on days –5, –4, –3, –2, –1); or (D) IL-2/IL-18 (IL-2 100,000 U i.p. on days –5,–3, –1 and IL-18 2 μg i.p. on days –5, –4, –3, –2, –1). Their cytotoxic activities were tested against 3LL, B16F10, RMA-S-Rae1β, and RMA-S (not depicted) tumor cells in the presence of 30 μg/ml of anti-mNKG2D mAb or 30 μg/ml of control hamster Ig by 12 h 51Cr release assay at several effector:target ratios (100:1 to 12.5:1 shown). Data are represented as the mean ± SEM of triplicate samples. Similar results were obtained in two independent experiments.
Tumors Expressing NKG2D Ligands Are More Sensitive to Cytokines That Promote Perforin-mediated Cytotoxicity.
Given that IL-12 and IL-18 cytokine therapies mediated their NK cell–mediated suppression of metastases by very distinct mechanisms, we next assessed whether tumors expressing NKG2D ligands might be relatively more sensitive to IL-12 than IL-18. To compare cytokine activity against matched tumor cells that lack or express a NKG2D ligand, we used DA3 and DA3-H60, the perforin-, FasL-, and TRAIL-sensitive mammary tumors. The growth of DA3-H60 was naturally suppressed in WT mice compared with DA3 tumors (Fig. 7 A). In preliminary experiments, we established that H60 could stimulate natural host NK cell perforin–mediated protection when overexpressed in DA3 tumor cells (Fig. 7 B). Therefore, for cytokine therapy we chose doses of DA3 and DA3-H60 that each generated ∼100 metastases in WT mice (Fig. 7 C). Although IL-18 had an equivalent effect in suppressing DA3 and DA3-H60 tumor metastases in WT mice, IL-12 was far more effective against DA3-H60 than against DA3 tumor (Fig. 7 C). As expected, IL-12–mediated suppression of DA3-H60 metastases was perforin dependent, and IL-18–mediated suppression of DA3-H60 and DA3 tumor metastases was FasL dependent (Fig. 7 C). NK cells were more cytotoxic to DA3-H60 than DA3 tumor cells (Fig. 8 A). The selective effect of IL-12 on DA3-H60 cells was supported by the increased cytotoxicity of spleen NK cells from IL-12–treated mice against DA3-H60 target cells (Fig. 8 A). By contrast, NK cells from IL-18–treated mice were equivalently cytotoxic toward DA3 and DA3-H60 target cells and anti-NKG2D mAb was without effect (Fig. 8 B).
Figure 7.

Tumors expressing NKG2D ligands are more sensitive to cytokines that promote perforin-mediated cytotoxicity. (A) Groups of 5 WT mice were inoculated i.v. on day 0 with increasing doses of DA3 tumor cells (dotted bars) or DA3-H60 tumor cells (solid bars) as indicated. (B) At a dose of 105 tumor cells, metastases were additionally examined in pfp−/− mice or WT mice treated with anti-asGM1 (100 μg i.p.) on days 0, 1, and 7 after tumor inoculation. (C) Groups of 5 WT or pfp−/− mice were inoculated i.v. on day 0 with either 105 DA3 tumor cells or 106 DA3-H60 tumor cells. Some groups of WT mice, as indicated in the legend, received anti-FasL (250 μg i.p.) on days 0, 1, and 7 after tumor inoculation. Mice were untreated (control) or received the following cytokine therapies: IL-12 (solid bars) (500 U i.p. on days 3, 4, 5, 6, and 7) or IL-18 (striped bars) (2 μg i.p. on days 0, 1, 2, 3, and 4). The lungs were removed from mice on day 14 and the metastatic nodules quantified. Data are recorded as the mean ± SEM.
Figure 8.

IL-12–activated NK cells potently kill tumors expressing H60 ligand. Spleen NK cells were isolated (day 0) from BALB/c RAG-1−/− mice 24 h after treatment with: (A) IL-12 (500 U i.p. on days –5, –4, –3, –2, –1); or (B) IL-18 (2 μg i.p. on days –5, –4, –3, –2, –1). Their cytotoxic activities were tested against DA3 and DA3-H60 tumor cells in the presence or absence of 30 μg/ml of anti-mNKG2D mAb or 30 μg/ml of control hamster Ig as indicated by 12 h 51Cr release assay at several effector:target ratios (100:1 to 12.5:1 shown). Data are represented as the mean ± SEM of triplicate samples. Similar results were obtained in two independent experiments.
Discussion
Here we have shown that cytokine therapies that mediate their NK cell activity via FasL or TRAIL do not depend on the NKG2D–NKG2D ligand pathway but require sensitivity of the tumor cells to the appropriate death receptor pathway. By contrast, those cytokines that employ perforin appear more effective against tumor metastases expressing NKG2D ligands. This contention was supported by the ability of IL-18 to equivalently suppress tumor metastases independently of their expression of the NKG2D ligand H60, whereas IL-12 more effectively suppressed the same tumor expressing H60 in a perforin-dependent manner. Our previous analysis had suggested that ectopic expression of NKG2D ligands in the NK cell–sensitive RMA-S and RMA tumor cells triggered natural perforin-mediated immunity (29). This study is the first to illustrate the importance of endogenous NKG2D ligand expression by tumor metastases during cytokine therapy. Herein we have clearly shown in models of NK cell–mediated suppression that the perforin-mediated therapeutic activity of cytokines such as IL-2 and IL-12 is mediated in large part via NKG2D–NKG2D ligand pathway. With knowledge of the relative sensitivity of a tumor to perforin, TRAIL, and FasL and its expression of NKG2D ligands, it should be possible to rationally deliver single and combine cytokine therapies to maximum effect.
Expression of NKG2D ligands may be controlled by various stresses such as heat, retinoids, and carcinogens (40, 41). Expression of MIC-A and MIC-B by DC has also been reported to be induced by IFN-α (42). The signaling events that are responsible for the up-regulation of Rae-1 or H60 expression by tumor cells are not known. Although the tumors employed in our study are unlikely to respond to cytokines IL-2 and IL-12 directly, it remains possible that bystander inflammatory effects of systemic levels of these cytokines may indirectly promote NKG2D ligand expression in the treated mice. This potential mechanism is inherently complicated to examine; however, arguing against such an effect, it appeared that the NKG2D pathway was not important in cytokine control of tumors that did not express detectable NKG2D ligands in vitro. The expression of NKG2D itself on NK cells does not appear to be modulated by these cytokines (unpublished data).
Previous studies have determined the capacity of NKG2D ligands, Rae-1β, Rae-1γ, MULT-1, and H60 in stimulating NK cell and CD8+ T cell–mediated rejection of tumors in vivo (27, 28, 43). This is the first study to describe the potency of H60 in triggering NK cell antitumor function a syngeneic BALB/c background. Clearly, ectopic expression of H60 was sufficient to stimulate NK cell–mediated suppression of DA3 mammary tumor metastases. So far, there is no evidence that this NKG2D ligand induces qualitatively and quantitatively distinct biological effects in responding NK cells, though this remains a possibility to be formally tested. H60 and Rae-1β expression in B16F10 and RMA tumor cells appeared quantitatively equivalent in a previous study; however, this was performed in a C57BL/6 background, where H60 is a minor alloantigen (27). Minimally, the various ligands for NKG2D might be predicted to differ quantitatively in their effects based on the marked differences in their affinity for NKG2D (44). For example, synapse requirements for lymphocyte-mediated cytotoxicity are minimal (45), and triggering granule exocytosis may not require the same interaction time as required to stimulate cytokine secretion. Nonetheless, at present the relevance of NKG2D ligand affinity has not been documented. We note that blockade of anti-NKG2D may influence other activation pathways triggered in cytokine-activated NK cells, since NKG2D has the ability to costimulate multiple NK cell activation receptors (37). Thus, other receptors that trigger perforin-mediated killing may also be affected by inhibition of the NKG2D pathway. The NK cell activation receptors that control the activity of TNF superfamily death ligands remain to be determined.
Clearly, NKG2D ligand expression by tumor cells may not be a barrier to tumor growth since many primary tumors and tumor cell lines naturally express NKG2D ligands (17, 21, 25). It is possible that tumor cells often express insufficient levels of NKG2D ligands to stimulate tumor rejection, either because expression of the ligands is not sufficient early in the development of the tumor or because tumor cells with lower levels of ligand expression are selected by the immune system in vivo as the tumor evolves. Direct experimentation has shown that less tumor rejection occurred when tumors only expressed intermediate levels of Rae1 (27). Ligand-expressing tumors might also evolve mechanisms to evade NKG2D-mediated immunity, as exemplified by human tumors that often produce a soluble version of MICA that reaches high levels in the serum, thereby inhibiting NKG2D in T cells and possibly other immune system cells (46, 47). There is also some indication that circulating soluble MIC in the cancer patients deactivates NK cell immunity by down-modulating important activating and chemokine receptors (48).
Human and mouse ligands for NKG2D are also expressed in cells infected with viruses (49) or bacteria (50). NK cells may also detect infected cells via their expression of NKG2D ligands (22, 51). Indeed viruses encode proteins that prevent NKG2D ligand expression and favor escape (52, 53). The role that the NKG2D–NKG2D ligand pathway plays in pathogen clearance is of particular interest given our observations. Pathogens themselves are recognized via TLRs expressed predominantly on APC-like DCs (54). TLR signaling in some APC induces ligands for the NKG2D receptor (55). All three cytokines, IL-2, IL-12, and IL-18, are products of DCs. Thus, it will be important to establish whether distinct TLR stimulate IL-18 versus IL-2 and IL-12 secretion, since during infection it is possible that these endogenous cytokines have very different roles in modulating NK cell effector function, engaging the NKG2D pathway, and altering the outcome of NK cell–DC interactions.
Unstimulated NK cells mainly express the long isoform of NKG2D (NKG2DL), which associates with DNAX-activating protein of 10 kD (DAP10) and not DAP12 and, therefore, is predicted to activate the phosphatidylinositol 3-kinase–initiated pathway. NK cells from untreated mice lack the NKG2DS isoform and, therefore, presumably lack NKG2D–DAP12 complexes (56). Cytokine-activated (e.g., IL-2) NK cells have been shown previously to express the short isoform of NKG2D (NKG2DS), which associates with both DAP12 and DAP10. Long term culture in the presence of IL-2 results in the down-regulation of expression of both isoforms of NKG2D, especially the short form, indicating that NKG2D in these cells mainly signals through DAP10. Activation of killing and cytokine release occurs when both the DAP10- and DAP12-associated forms are activated; however, the DAP10-associated form of NKG2D might be sufficient to activate killing (56). In this context, the C7 mAb might be blocking the costimulatory function of NKG2D in cytokine-activated NK cells (37). The cytotoxicities of freshly isolated NK cells and NK cells from cytokine-activated mice were both inhibited by the C7 anti-NKG2D mAb suggesting that primary NKG2D pathways were also sensitive to the mAb neutralization. We have shown that the C7 mAb blocks DAP10-deficient NK cell killing of Rae1-γ–transfected cells (57). This killing is presumably due to NKG2D that is coupled to DAP12 and would further support the contention that the C7 mAb blocks primary stimulation by NKG2D. Nevertheless, the relative importance of the different NKG2D isoforms and their adaptors in the antitumor activity of cytokine-activated NK cells remains to be elucidated.
Selection of future cancer patients for IL-18 therapy should consider the FasL sensitivity of their tumors; however, NKG2D ligand status is not likely to influence responsiveness. High dose IL-2 has had spectacular effects in a small fraction of patients with melanoma and renal cell cancer (1). Given our experimental data, it will be critical to retrospectively assess tumor NKG2D ligand expression and secretion in the large number of nonresponders and responders that have taken part in previous clinical trials of IL-2.
Acknowledgments
We thank Mark Shannon for reagent acquisition, Sally Mitchell for genotyping, and Rachel Cameron and Shannon Griffiths for maintaining the gene-targeted mice. We also thank Dr. Lewis Lanier (University of California, San Francisco, San Francisco, CA) for providing the anti-H60 mAb and discussions.
M.J. Smyth is supported by a National Health and Medical Research Council of Australia (NH&MRC) Principal Research Fellowship. The project was supported by a program grant from the NH&MRC and Glaxo Smith Kline. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract number N01-C0-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
The authors have no conflicting financial interests.
Abbreviations used in this paper: asGM1, asialo GM1; FasL, Fas ligand; pfp, perforin; MIC, MHC class I chain–related molecule; TRAIL, TNF-related apoptosis-inducing ligand.
References
- 1.Rosenberg, S.A. 2001. Progress in human tumour immunology and immunotherapy. Nature. 411:380–384. [DOI] [PubMed] [Google Scholar]
- 2.Smyth, M.J., M. Taniguchi, and S.E. Street. 2000. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J. Immunol. 165:2665–2670. [DOI] [PubMed] [Google Scholar]
- 3.Hashimoto, W., F. Tanaka, P.D. Robbins, M. Taniguchi, H. Okamura, M.T. Lotze, and H. Tahara. 2003. Natural killer, but not natural killer T, cells play a necessary role in the promotion of an innate antitumor response induced by IL-18. Int. J. Cancer. 103:508–513. [DOI] [PubMed] [Google Scholar]
- 4.Son, Y.I., R.M. Dallal, R.B. Mailliard, S. Egawa, Z.L. Jonak, and M.T. Lotze. 2001. Interleukin-18 (IL-18) synergizes with IL-2 to enhance cytotoxicity, interferon-gamma production, and expansion of natural killer cells. Cancer Res. 61:884–888. [PubMed] [Google Scholar]
- 5.Trinchieri, G. 1995. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 13:251–276. [DOI] [PubMed] [Google Scholar]
- 6.Okamura, H., H. Tsutsi, T. Komatsu, M. Yutsudo, A. Hakura, T. Tanimoto, K. Torigoe, T. Okura, Y. Nukada, K. Hattori, et al. 1995. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 378:88–91. [DOI] [PubMed] [Google Scholar]
- 7.Dinarello, C.A. 1999. Interleukin-18. Methods. 19:121–132. [DOI] [PubMed] [Google Scholar]
- 8.Okamura, H., H. Tsutsui, S. Kashiwamura, T. Yoshimoto, and K. Nakanishi. 1998. Interleukin-18: a novel cytokine that augments both innate and acquired immunity. Adv. Immunol. 70:281–312. [DOI] [PubMed] [Google Scholar]
- 9.Tsutsui, H., K. Nakanishi, K. Matsui, K. Higashino, H. Okamura, Y. Miyazawa, and K. Kaneda. 1996. IFN-gamma-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J. Immunol. 157:3967–3973. [PubMed] [Google Scholar]
- 10.Hashimoto, W., T. Osaki, H. Okamura, P.D. Robbins, M. Kurimoto, S. Nagata, M.T. Lotze, and H. Tahara. 1999. Differential antitumor effects of administration of recombinant IL-18 or recombinant IL-12 are mediated primarily by Fas-Fas ligand- and perforin-induced tumor apoptosis, respectively. J. Immunol. 163:583–589. [PubMed] [Google Scholar]
- 11.Dao, T., W.Z. Mehal, and I.N. Crispe. 1998. IL-18 augments perforin-dependent cytotoxicity of liver NK-T cells. J. Immunol. 161:2217–2222. [PubMed] [Google Scholar]
- 12.Micallef, M.J., T. Tanimoto, K. Kohno, M. Ikeda, and M. Kurimoto. 1997. Interleukin 18 induces the sequential activation of natural killer cells and cytotoxic T lymphocytes to protect syngeneic mice from transplantation with Meth A sarcoma. Cancer Res. 57:4557–4563. [PubMed] [Google Scholar]
- 13.Houchins, J.P., T. Yabe, C. McSherry, and F.H. Bach. 1991. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 173:1017–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamieson, A.M., A. Diefenbach, C.W. McMahon, N. Xiong, J.R. Carlyle, and D.H. Raulet. 2002. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 17:19–29. [DOI] [PubMed] [Google Scholar]
- 15.Sutherland, C.L., N.J. Chalupny, K. Schooley, T. VandenBos, M. Kubin, and D. Cosman. 2002. UL16-binding proteins, novel MHC class I-related proteins, bind to NKG2D and activate multiple signaling pathways in primary NK cells. J. Immunol. 168:671–679. [DOI] [PubMed] [Google Scholar]
- 16.Diefenbach, A., and D.H. Raulet. 2001. Strategies for target cell recognition by natural killer cells. Immunol. Rev. 181:170–184. [DOI] [PubMed] [Google Scholar]
- 17.Diefenbach, A., A.M. Jamieson, S.D. Liu, N. Shastri, and D.H. Raulet. 2000. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 1:119–126. [DOI] [PubMed] [Google Scholar]
- 18.Cerwenka, A., and L.L. Lanier. 2001. Ligands for natural killer cell receptors: redundancy or specificity. Immunol. Rev. 181:158–169. [DOI] [PubMed] [Google Scholar]
- 19.Bauer, S., V. Groh, J. Wu, A. Steinle, J.H. Phillips, L.L. Lanier, and T. Spies. 1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729. [DOI] [PubMed] [Google Scholar]
- 20.Stephens, H.A. 2001. MICA and MICB genes: can the enigma of their polymorphism be resolved? Trends Immunol. 22:378–385. [DOI] [PubMed] [Google Scholar]
- 21.Groh, V., R. Rhinehart, H. Secrist, S. Bauer, K.H. Grabstein, and T. Spies. 1999. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc. Natl. Acad. Sci. USA. 96:6879–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cosman, D., J. Mullberg, C.L. Sutherland, W. Chin, R. Armitage, W. Fanslow, M. Kubin, and N.J. Chalupny. 2001. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 14:123–133. [DOI] [PubMed] [Google Scholar]
- 23.Carayannopoulos, L.N., O.V. Naidenko, D.H. Fremont, and W.M. Yokoyama. 2002. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 169:4079–4083. [DOI] [PubMed] [Google Scholar]
- 24.Cerwenka, A., A.B. Bakker, T. McClanahan, J. Wagner, J. Wu, J.H. Phillips, and L.L. Lanier. 2000. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 12:721–727. [DOI] [PubMed] [Google Scholar]
- 25.Pende, D., P. Rivera, S. Marcenaro, C.C. Chang, R. Biassoni, R. Conte, M. Kubin, D. Cosman, S. Ferrone, L. Moretta, and A. Moretta. 2002. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 62:6178–6186. [PubMed] [Google Scholar]
- 26.Moretta, A., C. Bottino, M. Vitale, D. Pende, C. Cantoni, M.C. Mingari, R. Biassoni, and L. Moretta. 2001. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 19:197–223. [DOI] [PubMed] [Google Scholar]
- 27.Diefenbach, A., E.R. Jensen, A.M. Jamieson, and D.H. Raulet. 2001. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 413:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerwenka, A., J.L. Baron, and L.L. Lanier. 2001. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA. 98:11521–11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayakawa, Y., J.M. Kelly, J.A. Westwood, P.K. Darcy, A. Diefenbach, D. Raulet, and M.J. Smyth. 2002. Cutting edge: tumor rejection mediated by NKG2D receptor-ligand interaction is dependent upon perforin. J. Immunol. 169:5377–5381. [DOI] [PubMed] [Google Scholar]
- 30.Cretney, E., K. Takeda, H. Yagita, M. Glaccum, J.J. Peschon, and M.J. Smyth. 2002. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 168:1356–1361. [DOI] [PubMed] [Google Scholar]
- 31.Kelly, J.M., P.K. Darcy, J.L. Markby, D.I. Godfrey, K. Takeda, H. Yagita, and M.J. Smyth. 2002. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat. Immunol. 3:83–90. [DOI] [PubMed] [Google Scholar]
- 32.Street, S.E., Y. Hayakawa, Y. Zhan, A.M. Lew, D. MacGregor, A.M. Jamieson, A. Diefenbach, H. Yagita, D.I. Godfrey, and M.J. Smyth. 2004. Innate immune surveillance of spontaneous B cell lymphomas by natural killer cells and γΔ T cells. J. Exp. Med. 199:879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lodoen, M.B., G. Abenes, S. Umamoto, J.P. Houchins, F. Liu, and L.L. Lanier. 2004. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60–NKG2D interactions. J. Exp. Med. 200:1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogasawara, K., J.A. Hamerman, L.R. Ehrlich, H. Bour-Jordan, P. Santamaria, J.A. Bluestone, and L.L. Lanier. 2004. NKG2D Blockade prevents autoimmune diabetes in NOD mice. Immunity. 20:757–767. [DOI] [PubMed] [Google Scholar]
- 35.Smyth, M.J., N.Y. Crowe, D.G. Pellicci, K. Kyparissoudis, J.M. Kelly, K. Takeda, H. Yagita, and D.I. Godfrey. 2002. Sequential production of interferon-gamma by NK1.1(+) T cells and natural killer cells is essential for the antimetastatic effect of alpha-galactosylceramide. Blood. 99:1259–1266. [DOI] [PubMed] [Google Scholar]
- 36.Smyth, M.J., K.Y. Thia, E. Cretney, J.M. Kelly, M.B. Snook, C.A. Forbes, and A.A. Scalzo. 1999. Perforin is a major contributor to NK cell control of tumor metastasis. J. Immunol. 162:6658–6662. [PubMed] [Google Scholar]
- 37.Ho, E.L., L.N. Carayannopoulos, J. Poursine-Laurent, J. Kinder, B. Plougastel, H.R. Smith, and W.M. Yokoyama. 2002. Costimulation of multiple NK cell activation receptors by NKG2D. J. Immunol. 169:3667–3675. [DOI] [PubMed] [Google Scholar]
- 38.Smyth, M.J., E. Cretney, K. Takeda, R.H. Wiltrout, L.M. Sedger, N. Kayagaki, H. Yagita, and K. Okumura. 2001. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 193:661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wigginton, J.M., J.K. Lee, T.A. Wiltrout, W.G. Alvord, J.A. Hixon, J. Subleski, T.C. Back, and R.H. Wiltrout. 2002. Synergistic engagement of an ineffective endogenous anti-tumor immune response and induction of IFN-gamma and Fas-ligand-dependent tumor eradication by combined administration of IL-18 and IL-2. J. Immunol. 169:4467–4474. [DOI] [PubMed] [Google Scholar]
- 40.Raulet, D.H. 2003. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3:781–790. [DOI] [PubMed] [Google Scholar]
- 41.Girardi, M., D.E. Oppenheim, C.R. Steele, J.M. Lewis, E. Glusac, R. Filler, P. Hobby, B. Sutton, R.E. Tigelaar, and A.C. Hayday. 2001. Regulation of cutaneous malignancy by gammadelta T cells. Science. 294:605–609. [DOI] [PubMed] [Google Scholar]
- 42.Jinushi, M., T. Takehara, T. Kanto, T. Tatsumi, V. Groh, T. Spies, T. Miyagi, T. Suzuki, Y. Sasaki, and N. Hayashi. 2003. Critical role of MHC class I-related chain A and B expression on IFN-alpha-stimulated dendritic cells in NK cell activation: impairment in chronic hepatitis C virus infection. J. Immunol. 170:1249–1256. [DOI] [PubMed] [Google Scholar]
- 43.Diefenbach, A., J.K. Hsia, M.Y. Hsiung, and D.H. Raulet. 2003. A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur. J. Immunol. 33:381–391. [DOI] [PubMed] [Google Scholar]
- 44.Carayannopoulos, L.N., O.V. Naidenko, J. Kinder, E.L. Ho, D.H. Fremont, and W.M. Yokoyama. 2002. Ligands for murine NKG2D display heterogeneous binding behavior. Eur. J. Immunol. 32:597–605. [DOI] [PubMed] [Google Scholar]
- 45.Purbhoo, M.A., D.J. Irvine, J.B. Huppa, and M.M. Davis. 2004. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 5:524–530. [DOI] [PubMed] [Google Scholar]
- 46.Groh, V., J. Wu, C. Yee, and T. Spies. 2002. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 419:734–738. [DOI] [PubMed] [Google Scholar]
- 47.Salih, H.R., H. Antropius, F. Gieseke, S.Z. Lutz, L. Kanz, H.G. Rammensee, and A. Steinle. 2003. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood. 102:1389–1396. [DOI] [PubMed] [Google Scholar]
- 48.Doubrovina, E.S., M.M. Doubrovin, E. Vider, R.B. Sisson, R.J. O'Reilly, B. Dupont, and Y.M. Vyas. 2003. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J. Immunol. 171:6891–6899. [DOI] [PubMed] [Google Scholar]
- 49.Groh, V., R. Rhinehart, J. Randolph-Habecker, M.S. Topp, S.R. Riddell, and T. Spies. 2001. Costimulation of CD8 alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2:255–260. [DOI] [PubMed] [Google Scholar]
- 50.Tieng, V., C. Le Bouguenec, L. du Merle, P. Bertheau, P. Desreumaux, A. Janin, D. Charron, and A. Toubert. 2002. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc. Natl. Acad. Sci. USA. 99:2977–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sutherland, C.L., N.J. Chalupny, and D. Cosman. 2001. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol. Rev. 181:185–192. [DOI] [PubMed] [Google Scholar]
- 52.Wu, J., N.J. Chalupny, T.J. Manley, S.R. Riddell, D. Cosman, and T. Spies. 2003. Intracellular retention of the MHC class I-related chain B ligand of NKG2D by the human cytomegalovirus UL16 glycoprotein. J. Immunol. 170:4196–4200. [DOI] [PubMed] [Google Scholar]
- 53.Dunn, C., N.J. Chalupny, C.L. Sutherland, S. Dosch, P.V. Sivakumar, D.C. Johnson, and D. Cosman. 2003. Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J. Exp. Med. 197:1427–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janeway, C.A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. [DOI] [PubMed] [Google Scholar]
- 55.Hamerman, J.A., K. Ogasawara, and L.L. Lanier. 2004. Cutting edge: Toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J. Immunol. 172:2001–2005. [DOI] [PubMed] [Google Scholar]
- 56.Diefenbach, A., E. Tomasello, M. Lucas, A.M. Jamieson, J.K. Hsia, E. Vivier, and D.H. Raulet. 2002. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 3:1142–1149. [DOI] [PubMed] [Google Scholar]
- 57.Gilfillan, S., E.L. Ho, M. Cella, W.M. Yokoyama, and M. Colonna. 2002. NKG2D recruits two distinct adaptors to trigger NK cell activation and costimulation. Nat. Immunol. 3:1150–1155. [DOI] [PubMed] [Google Scholar]