

Abstract
It has been reported that the differentiation of CD4+CD25+ regulatory T cells (T reg cells) can be induced by agonist peptide/major histocompatibility complex ligands in the thymus. Exploiting a transgenic mouse line wherein expression of a particular T cell epitope can be controlled temporally and quantitatively, we found that diversion of differentiating thymocytes into the FoxP3 T reg cell pathway by this agonist ligand was essentially nonexistent. However, CD4+CD25+ thymocytes were much less sensitive than their CD4+CD25− companions, by two to three orders of magnitude, to agonist-induced clonal deletion, such that their proportion increased, giving the false impression of induced differentiation. To account for these and prior observations, one can propose that differentiation along the CD4+CD25+ pathway is induced by cues other than recognition of self-agonist cues, which are poorly read by thymocytes, whose T cell receptors are conducive to selection toward the conventional CD4+CD25− lineage. Thus, selective survival, rather than induced differentiation, may explain the apparent enrichment observed here and in previous studies.
Keywords: Foxp3, thymocytes, clonal deletion, inducible expression, transgenic
Introduction
Random rearrangement of TCR gene segments to encode functional TCR chains has obvious benefits, but comes with a price; although it enables vertebrates to generate an enormously diverse T cell repertoire, offering protection against a wide variety of pathogens, it also produces thymocytes displaying TCRs that recognize self-antigens and that have the potential to cause autoimmunity. Clonal deletion of differentiating thymocytes bearing self-reactive TCRs is an important mechanism for maintaining T cell tolerance (1–3). However, this process does not remove all self-reactive cells from the repertoire, as mature, self-reactive T cells can be isolated from healthy individuals. Clearly, there must be additional mechanisms that play a role in maintaining tolerance.
Over the past few years, a variety of T cell subsets that can inhibit T cell responses in vitro and in vivo and can prevent or ameliorate disease in several animal models of autoimmunity have been described (4–6). Given the potential clinical benefit of these so-called regulatory T cell (T reg cell) subsets, it is of clear interest to elucidate the molecular mechanisms via which they differentiate and exert their function. Amongst the best studied is the CD4+CD25+ population, which can be found in the thymus and periphery of naive mice (7–10) and has been isolated from peripheral blood, tonsils, and thymus of healthy humans (11–16). The regulatory properties of murine CD4+ CD25+ T reg cells were originally apparent from these cells' ability to suppress autoimmune responses caused by purified CD4+CD25− T cells upon adoptive transfer into nude mice (7). Recently, it has been found that differentiation and function of CD4+CD25+ T reg cells is critically dependent on expression of the forkhead/winged helix transcription factor Foxp3 (17–19). Perhaps the most compelling argument for the importance of CD4+CD25+ T reg cells in maintenance of tolerance came from the phenotype of the Foxp3 mutant scurfy mice and Foxp3-null mice, which lack CD4+CD25+ T reg cells and die of a lymphoproliferative wasting disease (17, 20). Furthermore, adoptive transfer of CD4+CD25+ T reg cells into neonatal Foxp3-null or scurfy mice protected them, at least temporarily, from disease (17, 21). The genetic mutation in immune disregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX) patients, who succumb to several autoimmune/inflammatory diseases, including inflammatory bowel disease, insulin-dependent diabetes mellitus, and thyroiditis (for review see reference 22), was shown to map to the Foxp3 gene (23, 24). There is no direct evidence linking pathology in these patients to a lack of CD4+CD25+ T reg cells, but the similarities in the disease profile between immune disregulation, polyendocrinopathy, enteropathy, and X-linked inheritance patients and Foxp3-deficient mice suggest that CD4+CD25+ T reg cells play a critical role in T cell tolerance in humans as well.
It has been hypothesized that CD4+CD25+ T reg cells are generated in the thymus after high-affinity interaction of their TCR with peptide/MHC ligands, at a level of affinity or avidity at the brink of that resulting in clonal deletion (25). Consistent with this hypothesis, MHC class II–restricted, TCR-transgenic thymocytes differentiated into CD4+CD25+ T reg cells when agonistic T cell epitopes bound to MHC class II molecules were expressed on radio-resistant cells in the thymus (26–29).
After these reports of induction of CD4+CD25+ T reg cells by agonist ligands, it seemed of interest to ask whether the dose of the agonist peptide/MHC ligand influences the efficiency of generation of CD4+CD25+ T reg cells. Might there be a window of affinity between the induction of CD4+CD25+ T reg cells and clonal deletion? Therefore, we generated a mouse line wherein expression of a T cell epitope from moth cytochrome c (MCC) was under the control of a tetracycline (tet)-regulatable gene expression system (30, 31) and, as a convenient read-out, introduced a further transgene encoding a TCR specific for the MCC peptide (32).
The numbers of AND CD4+CD25+ T reg cells generated in the thymus of mice expressing graded amounts of the agonist peptide/MHC ligand were quantified, and these cells were compared for their phenotypical and functional properties. We arrived at an unexpected conclusion.
Materials and Methods
Constructs.
An invariant chain (Ii) cDNA in which the class II–associated invariant chain peptide (CLIP)–encoding region was substituted by MCC93–103 (DLIAYLKQATK) was constructed by N. Nakano (Research Institute for Biological Sciences, Chiba, Japan). It was constructed via a two-step overlap PCR strategy using WT Ii cDNA as template, flanking oligos containing EcoRI restriction sites, and the following overlapping oligos: MCC96–103Ii99–103, sense: 5′-GCTTACCTGAAACAGGCTACCAAGCGTCCAATGTCCATG-3′; and MCC100–93Ii87–83, antisense: 5′-CTGTTTCAGGTAAGCGATCAAATCCACAGGTTTGGCAGA-3′. This Ii–MCC fragment was cloned into the unique EcoRI site of pKCR3 (33). A PvuI–ApaI fragment containing the SV40 early promoter and part of the rabbit β-globin intron was replaced by a PvuI–ApaI fragment from TetO-Eα (34), containing seven tet operator (TetO) sequences, a CMV minimal promoter, and part of the rabbit β-globin intron, resulting in pTIM.
Mice.
A PvuI–XhoI fragment derived from pTIM was injected into (C57BL × SJL)F2 fertilized eggs. Transgene positive founders were crossed with previously described CII-tTA mice (34). Thymic RNA from double transgenic and single transgenic offspring was tested by semi-quantitative RT-PCR and S1 nuclease protection assays for expression of the tet-regulatable invariant chain with MCC (TIM) transgene. Mice from one of the lines showing complete dependence on the CII-tTA transactivator for TIM expression were backcrossed for three to five generations onto the B10.BR background and used in all described experiments. AND-transgenic mice, carrying a TCR-recognizing MCC88–103 bound to I-Ek, were a gift from S. Hedrick (University of California at San Diego, La Jolla, CA; reference 32). Invariant chain-deficient mice have been described previously (35). Tet treatment was performed by supplementing the drinking water with the indicated concentration of tet-HCl (Sigma-Aldrich) plus 2 g/l equal (Merisant US). Bottles were changed twice a week during the course of treatment. To construct radiation chimeras, donor bone marrow isolated from AND and TA × TIM × AND donors was magnetically depleted of mature T cells (biotin anti-CD4 and anti-CD8 mAbs with streptavidin-conjugated magnetic beads). 2–3-mo-old, γ-irradiated recipients (1,000 rad) were reconstituted with 2 × 106 depleted BM cells and kept on antibiotic treatment (Sulfatrim) for 6 and 8 wk before analysis. All mice were bred and maintained under sterile barrier conditions at the Harvard Center for Animal Resources and Comparative Medicine (protocol no. 2954) in accordance with National Institutes of Health guidelines.
Flow Cytometry.
Thymocyte and LN suspensions were stained in PBS-CMF, 3% heat-inactivated horse serum, 10 mM Hepes, and 0.03% NaN3 and analyzed or sorted by four- or six-color flow cytometry on a MoFlo cell sorter (DakoCytomation). Data were analyzed using Summit software (DakoCytomation). The following antibodies were used for analysis: affinity-purified goat anti–mouse GITR/TNRFSF18 polyclonal antibody (R&D Systems); affinity-purified goat anti–human IgG (H + L) and affinity-purified, FITC-conjugated donkey anti–goat IgG (H + L) F(ab′)2 fragment (Jackson ImmunoResearch Laboratories); FITC-labeled anti-mVβ3 (KJ25, purified and labeled in the laboratory); biotinylated anti-mVα11 (RR8.1), biotinylated anti-mVβ3 (KJ25), PE–anti-mCD25 (PC61), and allophycocyanin–anti-mCD4 (RM4-5; BD Biosciences); PE-Cy7–anti-mCD8α (5H10), PE-Cy7–anti-mCD4 (RM4-5), allophycocyanin–anti-mCD8α (CT-CD8a), and streptavidin-PE–Texas red (Caltag); and allophycocyanin–anti-mCD25 (PC61; eBioscience). Dead cells were excluded from analysis by addition of Hoechst before acquisition.
RNA Isolation and RNA Transcript Quantification.
Total RNA was isolated from thymic lobes by the LiCl/urea method as described previously (36). Sorted thymocyte subpopulations were resuspended in TRIzol (Invitrogen), and RNA was prepared according to the manufacturer's instructions. Residual DNA was removed using a DNA-free kit (Ambion). Randomly primed cDNA was prepared using M-MLV reverse transcriptase (Invitrogen) following standard procedures. Real-time PCR (TaqMan) was performed using the following oligos and probes: TIM sense 5′-GGATCCTGAGAACTTCAGGCTC-3′; TIM antisense 5′-TTGGT-CATCCATGGCTCTAGC-3′; TIM probe 5′-FAMAACGTG-CTGGTTGTTGTGCTGTCTCATC-TAMRA-3′; Foxp3 primers and probe as described in reference 18; HPRT sense 5′-GACCGGTCCCGTCATGC-3′; HPRT antisense 5′-CAGTCCATGAGGAATAAACACTTTTTC-3′; HPRT probe 5′-VICCC-GCAGTCCCAGCGTCGTGATT-TAMRA-3′. Samples were analyzed on ABI PRISM 7700 (Applied Biosystems) or Mx3000p (Stratagene) real-time PCR instruments. S1 protection assays were performed as described previously (36).
Proliferation Assays.
CD4+CD25+ T reg cell thymocytes and CD4+CD25− peripheral responder T cells were sorted from TAND mice and AND mice, respectively. 2 × 104 responder cells were cultured in round bottom 96-well plates with graded amounts of CD4+CD25+ T reg cell thymocytes in the presence of 5 × 104 irradiated B10.BR spleen cells and 10 μM of the MCC88–103 peptide. Cells were cultured for a 72-h period, and 1 μCi of [3H]thymidine was added 16 h before harvesting and counting.
Results
A Transgenic Mouse Line with Regulated Self-Antigen Expression.
We used a tet-regulatable gene expression system to generate mice with quantitatively controlled expression of the MCC-derived T cell epitope (30, 31). This is a double transgenic system in which expression of a reporter gene is dependent on binding of a tet-sensitive transactivator to regulatory sequences upstream of the reporter gene. The transactivator normally activates reporter constructs, but is inhibited from binding in the presence of tet in a dose-dependent fashion (Fig. 1 A).
Figure 1.

Tet-controlled expression of a T cell epitope. (A) Schematic depiction of the tet-responsive double transgenic system. A transactivator (TA) consisting of the tet-repressor (TetR) fused to the VP16-activating domain is expressed via the Eα promoter. Binding of the transactivator to tet operator (TetO) regulatory sequences upstream of the minimal CMV promoter is necessary to drive expression of a modified invariant chain protein and can be blocked by tet. The modified invariant chain construct (TIM) contains a MCC–derived, I-Ek–restricted minimal T cell epitope instead of the CLIP region. (B) Predominant expression of the TIM transgene in the thymus. RNA isolated from the indicated organs was analyzed via S1 nuclease protection assays for transcription of the TIM gene. The DNA probe used in this assay was complementary to the first 174 bases of the recombinant invariant chain cDNA and 54 bases from the upstream rabbit β-globin exon sequence, allowing discrimination of TIM transcripts from the endogenous invariant chain (Ii) transcripts on the basis of a difference in size of the protected fragments. (C) Immunohistochemical analysis of the thymus from invariant chain-deficient TA × TIM mice. Adjacent frozen tissue sections were stained with the anti–MHC class II mAb M5/114 and the antimurine invariant chain mAb IN1.1. Note that, due to the absence of endogenous invariant chain, the IN1.1 antibody reveals expression of the TIM transgene. (D) Differentiation of AND thymocytes in BM chimeras. Profiles show the CD8 versus CD4 distribution and expression of the clonotypic Vα11 and Vβ3 TCR chains on mature CD4+ T cells in the indicated chimeras. Note that little, if any, negative selection of AND thymocytes occurs when only BM-derived cells are positive for the TA and TIM genes.
T cell epitopes embedded in the CLIP region of the MHC class II–associated invariant chain can be presented efficiently to T cell clones (37, 38). Therefore, we generated a reporter gene in which the CLIP region of a murine invariant chain cDNA was replaced by the Ek-restricted minimal T cell epitope of MCC93–103 (39). This construct was used to generate transgenic mice, referred to as TIM.
Transgenic animals were crossed with a previously reported mouse line expressing a transactivator under the control of the Eα promoter (CII-tTA, referred to as TA; reference 34). The TA line's transactivator is predominantly expressed in radio-resistant stromal cells of the thymus, in cells that promote positive selection of CD4+ lymphocytes by an MHC II reporter (34). Because expression of the reporter is dependent on the presence of the transactivator, we expected that the TIM transgene in TA × TIM double transgenic mice would have a similar expression pattern. Indeed, S1-nuclease protection assays on RNA isolated from lymphoid and nonlymphoid organs of untreated TA × TIM mice revealed TIM RNA only in the thymus (Fig. 1 B). The TIM protein was detected by immunostaining of thymus cryostat sections from TA × TIM mice on an invariant chain-deficient background (35), staining serial sections with anti–MHC class II mAbs for localization (Fig. 1 C). The TIM protein was expressed in a subset of cells in both the cortex and medulla, at levels similar to those of standard invariant chain protein.
The results of Witherden et al. (34) indicate that expression should be found predominantly in radio-resistant cells of the thymic stroma. To verify this point, a set of reciprocal bone marrow chimeras were constructed with the MCC93–103 reactive AND TCR-transgenic line (32), where the TA × TIM transgenes were present in either the donor bone marrow or the host radio-resistant stroma (Fig. 1 D). In AND→WT chimeras, a large population of mature CD4+ T cells was present, most expressing the clonotypic Vα11 and Vβ3 TCR chains. A similar population was found in TA × TIM × AND→WT chimeras, indicating that little if any TIM protein was expressed by BM-derived cells. In contrast, clonotype positive cells were largely deleted when radio-resistant thymic stromal cells expressed TIM. Thus, the TIM protein is primarily expressed in both cortical and medullary stromal cells in the thymus of TA × TIM mice.
Next, we established that graded expression of the TIM transgene could be achieved by providing transgenic breeders with graded amounts of tet in the drinking water. TA × TIM offspring were kept under the same treatment conditions for 5–7 wk, and transcripts from the TIM transgene were quantitated by real-time PCR (Fig. 2 A). Administering increasing doses of tet resulted in a progressive reduction in TIM RNA expression. Although there was some mouse-to-mouse variability, careful titration of tet permitted modulation of TIM expression over four orders of magnitude, a dynamic range matching that reported previously for tissue culture and select organs in transgenic mice (31, 40). We were unable to detect any TIM transcripts in the thymus of control TIM animals lacking the TA transgene, regardless of tet treatment, indicating that the TIM transgene is completely dependent on a transactivator for its expression.
Figure 2.
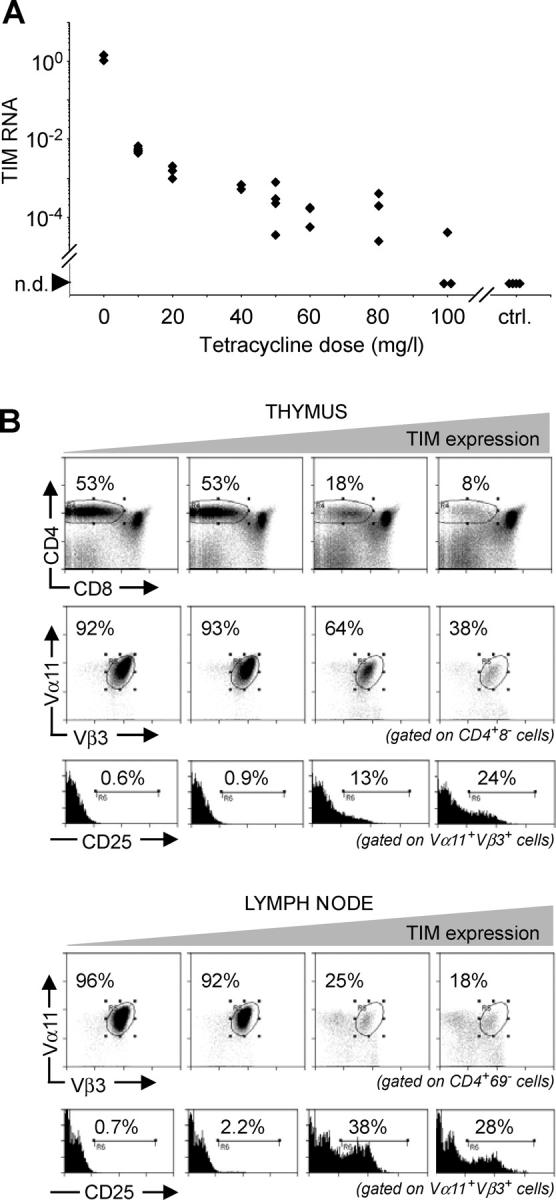
Increased proportion of CD4+CD25+ T reg cells upon increase in TIM expression. (A) The amount of TIM transcripts can be regulated by graded doses of tet. TA × TIM animals were treated for life with the indicated doses of tet. Thymic RNA was analyzed for TIM RNA transcripts via real-time PCR (TaqMan). Mice in the control group lacked either the transactivator or reporter transgene. n.d., not detected. (B) Thymus (top) and LNs (bottom) from TAND mice treated for life with tet were analyzed at 7 wk of age by flow cytometry for differentiation of AND-transgenic CD4+CD25+ T reg cells. Clonotype positive T cells were identified by staining with mAbs against the Vα11 and Vβ3 TCR chains. Animals shown in this panel were treated with, from left to right, 100, 60, 20, and 10 mg/l tet in the drinking water.
Increased Proportion of CD4+CD25+ AND T Reg Cells upon Encounter with Increasing Amounts of an Agonist Ligand.
The broad dynamic range of this tet system, and the expression of agonist ligand on the radio-resistant thymic stroma, provide an ideal system to ask how ligand expression levels might affect the reported differentiation of CD4+ TCR-transgenic thymocytes into CD4+CD25+ T reg cells upon encounter with agonist peptide/MHC ligands (26–29).
To this end, TA × TIM × AND mice, referred to as TAND mice, were treated for life with graded doses of tet and analyzed between 5 and 7 wk of age. RNA from one thymic lobe was used to quantitate TIM RNA transcripts, whereas the other lobe and LNs were analyzed by flow cytometry to assess the phenotype of AND-transgenic T cells. A reduction in the CD4+CD8− thymocyte population was observed with increasing amounts of the MCC epitope (Fig. 2, top). Furthermore, there was an agonist-dependent decrease in the percentage of CD4+CD8− thymocytes expressing the AND TCR, indicating that clonotypic thymocytes were eliminated via clonal deletion. Within the clonotype positive CD4+CD8− thymocyte population, an augmentation in the percentage of CD4+CD25+ thymocytes was found with increasing expression of the TIM transgene. A similar augmentation in CD4+CD25+ cells was observed within the CD4+ population in the peripheral lymphoid organs (Fig. 2, bottom). These findings suggested that the agonist MCC ligand directed differentiating AND thymocytes into the CD4+CD25+ T reg cell lineage in a dose-dependent fashion, extending previous observations (26–29) to a third TCR and a different H-2 haplotype.
Although the enrichment for CD4+CD25+ cells that occurred with increasing TIM expression was reminiscent of previously described T reg cells, we verified that the cells highlighted in our experiments were indeed of T reg cell phenotype. The transcription factor Foxp3 has been shown to be necessary for T reg cell differentiation and function in mice, and is considered to be a master gene defining this lineage (17–19). Levels of FoxP3 RNA were quantitated in sorted CD4+CD25+ and CD4+CD25− thymocytes isolated from a group of TAND mice treated with graded doses of tet (Fig. 3 A). In all cases, CD4+CD25+ thymocytes expressed higher levels of Foxp3 RNA than did their CD4+CD25− counterparts. TIM exerted little influence. As another criterion, we tested the expression of GITR, a member of the TNF receptor family enriched in CD4+CD25+ populations and instrumental in the inhibitory effect that CD4+CD25+ T cells can exert in vivo and in in vitro proliferation assays (41–43). The majority of CD4+CD25+ thymocytes in TAND mice expressed GITR at the cell surface (Fig. 3 B), with no apparent effect of the amount of TIM RNA expression on the number of GITR molecules. Finally, the ability of CD4+CD25+ thymocytes from TAND mice treated with low or intermediate doses of tet to inhibit proliferation of CD4+CD25− AND TCR-transgenic T cells was tested in the usual in vitro inhibition assay (44, 45). Using MCC peptide as the stimulating agent, we found that CD4+CD25+ thymocytes from TAND mice expressing the TIM transcript were able to inhibit the proliferation of CD4+CD25− responder cells in a dose-dependent fashion (Fig. 3 C), indicating that these thymocytes have regulatory capacity and can receive stimulation via the clonotypic receptor to exert their inhibitory effects.
Figure 3.
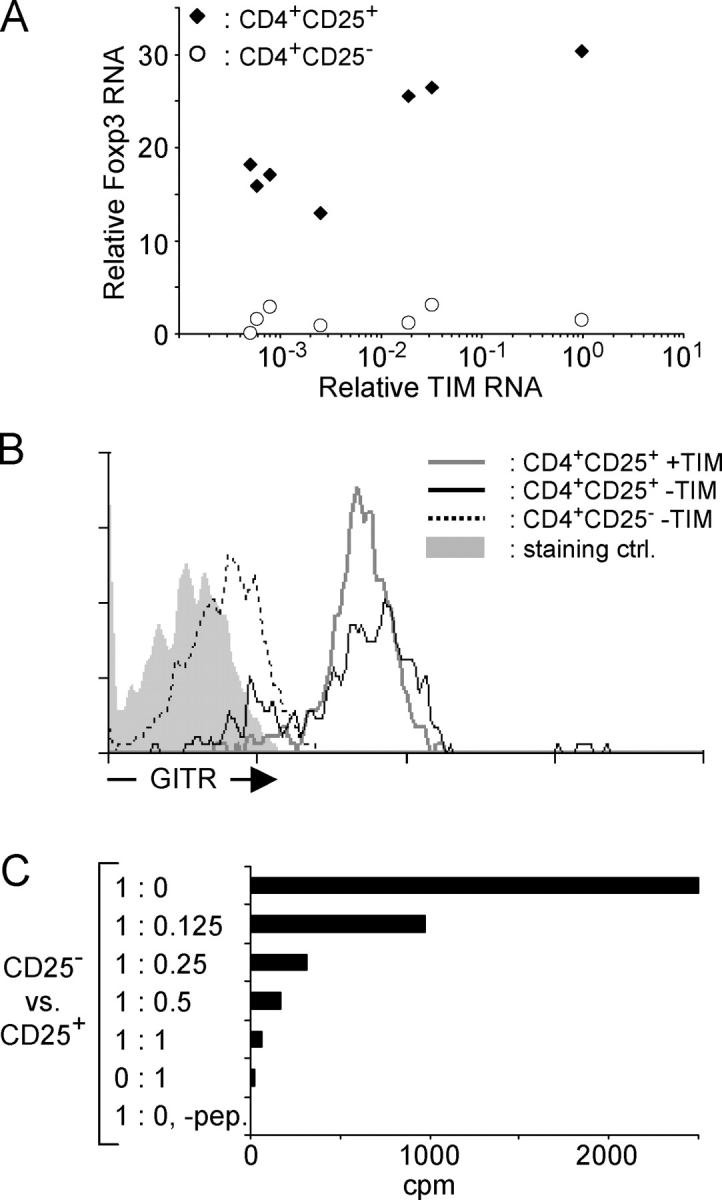
Characterization of CD4+CD25+ thymocytes in TAND mice. (A) RNA extracted from one thymic lobe of TAND animals treated with graded doses of tet was analyzed by real-time PCR for the relative amount of TIM RNA. CD4+CD25+ and CD4+CD25− thymocytes were sorted from the other lobe, and RNA purified from these cell populations was assayed by real-time PCR for the relative expression level of Foxp3 transcripts. Relative Foxp3 RNA levels were plotted as a function of TIM RNA transcripts levels. (B) CD4+CD25+ thymocytes in TAND mice express GITR. Vβ3+CD4+CD25+ thymocytes from animals treated for life with 250 mg/l (black line, no TIM message detected) or 40 mg/l (gray line) tet and Vβ3+CD4+CD25− thymocytes from animals treated with 250 mg/l tet (dotted black line) were analyzed for cell surface expression of GITR via flow cytometry. The solid gray histogram shows staining with affinity-purified control antibodies. (C) CD4+CD25+ thymocytes from a TAND mouse treated with 50 mg/l tet were purified via cell sorting using mAbs against CD4, CD8, and CD25. Staining with mAbs against both TCR chains was omitted to avoid potential stimulatory or inhibitory effects on the T reg cells. Increasing numbers of these cells were added to wells containing fixed numbers of CD4+CD25− AND T cells. Cultures were grown for 3 d in the presence of 10 μM MCC88–103 peptide, and [3H]thymidine was added during the last 16 h of culture.
The remaining CD4+CD25− thymocyte cell population in TAND mice expressing intermediate and high amounts of the TIM protein did not proliferate in response to the MCC peptide and did not suppress the proliferative response of AND T cells from a normal host (unpublished data). As shown in Fig. 3 A, these cells did not increase expression of the Foxp3 gene, compared with the CD4+ CD25− cells in TAND mice expressing no or very little TIM protein.
Thus, the CD4+CD25+ T cells that emerge in TAND mice are indeed cells with regulatory capacity, and the dose of agonist ligand these cells encountered during differentiation did not influence their phenotypic and functional properties in a major way. The TAND system provides a flexible system to study their origin.
Does Induction of CD4+CD25+ Clonotypic T Reg Cells Occur within a Window of Avidity?
To address whether the selection of the CD4+CD25+ cells takes place in a particular window of antigen dose relative to that which provokes clonal deletion, we analyzed a large group of triple transgenic TAND mice treated with a range of tet doses, to cover a wide spectrum of TIM expression on thymic stromal cells. As shown in Fig. 4 A, the proportion of CD4+CD25+ cells did increase considerably over the mid-range, consistent with the data of Fig. 2. At the highest TIM expression, CD25+CD4+ cells were eliminated, consistent with the deletion of T reg cells at very high antigen doses reported by Shih at el. (46). Yet, when we plotted the actual number of CD25+ and CD25−CD4+ cells as a function of the amount of TIM transgene expression (Fig. 4 B), it became clear that the number of CD4+CD25+ cells remained essentially flat, with no significant conversion from CD25− to CD25+ phenotypes. The relative increase in CD4+CD25+ cells occurred in the range of antigen dose where the deletion of conventional CD4+CD25− cells was evident. Thus, essentially all conventional CD4+CD25− cells underwent clonal deletion, rather than conversion to a CD25+ phenotype, and the increase seen in Fig. 2 was only a mirage.
Figure 4.

Enumeration of thymocyte populations in tet-treated TAND mice. (A) The proportion of clonotype positive CD4+CD25+ thymocytes amongst CD4+ thymocytes is dependent on the expression level of the TIM transgene. (B) Absolute number of clonotype positive CD4+ CD25− (open circles) and CD4+CD25+ (closed diamonds) thymocytes in tet-treated TAND mice, based on the analysis shown in Fig. 2, as a function of the relative expression level of TIM RNA in the thymus of these animals. Control animals lacked either the transactivator or reporter transgene. (C) Absolute numbers (as million cells/whole thymus) of clonotype positive (closed diamonds) and clonotype negative (open triangles) CD4+CD25+ thymocytes in thymi of tet-treated TAND mice. The relative increase in clonotype positive cells upon encounter with increasing amounts of the agonist ligand is very modest. Note that, at the highest doses attainable in this system, clonotype positive CD4+CD25+ thymocytes undergo clonal deletion. (D) Representative examples showing the percentage of clonotype positive CD4+CD25+ thymocytes are shown for a range of TIM doses (indicated by black arrows).
When the data were replotted on a different scale, a slight numeric increase in clonotype positive CD4+CD25+ cells was seen, from 1.5 to 4 × 105 on average (Fig. 4 C, closed symbols). However, clonotype negative CD4+ CD25+ thymocytes, most of which should be insensitive to TIM, showed a similar expansion (Fig. 4 C, open symbols). The proportion of clonotype positive cells in the CD4+CD25+ population, quite low in the absence of TIM, remained as such with TIM induction (Fig. 4 D). Hence, even the modest increase noticed in the clonotype positive T reg cell population was most likely attributable to expansion into thymic space vacated by conventional CD4+CD25− cells.
These data provide evidence that CD4+CD25+ T cells in the MCC system are more resistant to clonal deletion induced by an agonist peptide/MHC ligand than their CD4+CD25− counterparts. They also indicate that there is little or no induction of differentiation of CD4+CD25+ T reg cells in a thymus expressing MCC–peptide/MHC complexes, whatever the dose of the agonist ligand.
Discussion
The number and phenotype of AND thymocytes differentiating into CD4+CD25+ T reg cells was barely influenced by interaction with MCC–peptide/MHC complexes expressed on radio-resistant thymic stromal cells. This was not due to any requirement of a critical number of agonist complexes to be expressed by the stromal cells because diversion into the T reg cell pathway was absent over a wide range of expression levels of MCC–peptide/MHC complexes. Instead, CD4+CD25+ T reg cell thymocytes proved to be more resistant than their CD4+ CD25− counterparts to clonal deletion. This explains their relative enrichment in the presence of cognate ligand; as conventional CD4+ die off with increasing agonist, the apparent proportion of more resistant T reg cell increases, even though their numbers remain constant. That agonist ligand induces T reg cell differentiation is just a mirage.
The inefficient recruitment of AND-transgenic thymocytes into the CD4+CD25+ T reg cell lineage in TAND mice appears at odds with prior reports describing the differentiation of TCR-transgenic thymocytes into CD4+CD25+ T reg cells in response to encounter with agonist ligands displayed by stromal cells (26–29). Were these data misinterpreted by relying on apparent cell proportions rather than true cell counts? Indeed, reexamination of the published data shows that, in several instances, it was also the relative proportion of CD4+CD25+ cells, but not their absolute number, that increased in response to agonist ligand (26, 28). In the DO11.10 system, Kawahata et al. counted 2.0 ± 0.9 × 105 CD4+CD25+ cells in the absence of antigen, and only 2.7 ± 0.9 × 105 cells with antigen (28). In the HA system, there were only 1.5 × 105 CD4+CD25+ thymocytes in the presence of antigen, obviously not the number of CD4+ single positives expected from an efficient inductive process. Thus, deviation toward the CD4+CD25+ pathway also seems numerically very limited, bringing into question the reality of agonist induced differentiation in the HA and OVA systems as well.
However, there are several caveats to a summary dismissal of the notion of agonist-induced differentiation of T reg cells. First, one might argue that the TIM agonist ligand does induce differentiation along the CD4+CD25+ pathway, but that this is masked because of homeostatic control or negative feedback preventing the accumulation of >2–15 × 105 such cells per thymus. If that were the case, one would have expected that the proportion of cells expressing the AND clonotype would increase in the presence of TIM ligand. This prediction was not correct; although variable from mouse to mouse and quite lower than in conventional CD4+ cells, the proportion of clonotype positive cells did not correlate with agonist exposure (Fig. 4 D). A counter argument could be made that receptor editing, perhaps favored by FoxP3 expression, secondarily reduced clonotype expression, but it would have to be a coincidence that the original 50% ratio were preserved through a broad range of agonist concentration.
Second, it is possible that only specific epithelial niches can support agonist-induced differentiation of T reg cells. The issue may be complicated by the fact that a variety of promoters were used to drive expression of the transgenic neo-antigen in this and previous studies (SV40, RIP, Ld, and Ig-κ previously, MHC II here). The transgenic transactivator used here drives TIM expression in epithelial- but not in BM-derived macrophage/dendritic cells (Fig. 1 and reference 34), and should have been effective at eliciting T reg cells (26). Yet, it is possible that different compartments of epithelial cells may preferentially lead to clonal deletion rather than selection of T reg cells, and that this niche does not express TIM protein in TA × TIM mice.
Third, it is also possible that the particular affinity of the AND/MCC pair makes it particularly inefficient at eliciting T reg cell differentiation, whatever the amount presented, whereas other TCR/Ag systems would be more favorable.
Another argument made in support of the notion that self-agonists promote the CD4+CD25+ differentiation pathway is that CD4+CD25+ cells are often very rare or absent in TCR-transgenic RAG-deficient mice, but that they appear in the presence of an agonist ligand (27–29). This denotes a requirement for endogenous TCR chains for CD4+CD25+ selection. Yet there are two distinct interpretations for this finding: (a) as previously held, that nontransgenic TCR chains are required to yield the degree of self-reactivity that the transgenic clonotype is unable to provide; and (b) alternatively, that the transgenic clonotype is less efficient at interacting with the ligand that promotes CD4+CD25+ differentiation, but that it naturally leads to very efficient selection of conventional CD4+ cells. In this light, it is important to remember that all TCR-transgenic mouse strains used in this and other studies were derived from conventional CD4+ cells, with an additional “experimental bias” that favored lines with efficient selection into the CD4+ lineage. These lines also have grossly perturbed thymic architecture (47), perhaps eliminating or destroying a niche required for T reg cell differentiation. Thus, one can readily envision that the overwhelming positive selection of conventional CD4+ cells in TCR-transgenic RAG mice dwarfs and outcompetes T reg cell differentiation, and that agonist ligand only serves to relieve this competition. Here again, agonist-induced differentiation of T reg cells would also be a mirage.
If an agonist ligand per se does not promote the differentiation of CD4+CD25+ cells, then how do they arise? The model shown in Fig. 5 presents this different perspective on T reg cell differentiation, attempting to account for the observations made here and in work from other laboratories. The main tenets are that CD4+CD25+ cells are inefficiently selected in MHC II–restricted TCR-transgenic mice not because they miss a strong agonist signal, but because this pathway is poorly elicited by the MHC II ligands that elicit conventional CD4+ cells, and that there is competition between the lineages. In TCR-transgenic mice, the inefficient selection of CD4+CD25+ via the transgenic clonotype results in frequent reliance on endogenous TCR chains, and the cells are partly outcompeted by conventional CD4+ cells whose TCR matches well with MHC II ligands. This competition is relieved with the display of agonist on the thymic stroma, which preferentially eliminates the conventional lineage. In TCR-transgenic mice on a RAG-deficient background, as observed in other studies, the situation is more extreme; without an agonist, the CD4+CD25+ lineage is in an even more precarious situation, as it cannot resort to endogenous TCR chains for selection, and competition from the fully selectable conventional lineage is even stronger. But the presence of agonist has a more drastic impact because the conventional lineage cannot escape deletion by expressing endogenous chains.
Figure 5.

Factors determining the relative size of the CD4+CD25+ compartment. This model, which attempts to group information from previous studies and the present work, proposes that two main factors that determine the efficiency via which thymocyte precursors (gray circles) are directed toward the conventional CD4+CD25− pathway (blue circles) or the CD4+CD25+ T reg cell pathway (red circles). First, the poor ability of MHC II–restricted thymocytes to become T reg cells as noted quasi-universally in MHC II–restricted TCR-transgenic (Tg) systems. This is probably not unexpected, as there is an “experimental bias” in the Tg lines analyzed by the community, with a strong selection for lines that show robust MHC II selection to the conventional CD4+ compartment. Second, the relative resistance of CD4+CD25+ T reg cell thymocytes to clonal deletion induced by MHC II/p ligands is clearly established in this paper. The model proposes that the relative changes between populations can be accounted for by a competitive balance between these two forces: when T precursors express a TCR that promotes very efficient selection to the conventional CD4SP pathway, the CD4+CD25+ T reg cell lineage is underrepresented, and this is particularly true when the RAG deficiency prevents the rescue of the CD4+CD25+ T reg cell pathway through endogenous TCRs. In contrast, the presence of agonist ligand, which selectively eliminates conventional CD4+CD25− cells, brings forth the T reg cell pathway.
In this model, what promotes selection into the CD4+ CD25+ pathway? One can imagine that the ligand, or the selection niches, would be different. The selecting cells may be different from those that support conventional selection, providing an alternative costimulatory influence or MHC II molecules with a different peptide cargo or conformation than those selecting conventional cells. Bensinger et al. (48) have provided evidence that MHC II seems required for the selection of cells with T reg cell activity (yet MHC II–deficient mice contained paradoxically high numbers of CD4+CD25+ cells whose characteristics remain uncertain, and the functional assays used to test their T reg cell capabilities may have been complicated by reaction to alien MHC II molecules).
One of the key observations of this paper is that CD4+ CD25+ T reg cells are very resistant to clonal deletion induced by agonist peptide, considerably more so than conventional CD4+ cells. From a functional standpoint, this implies that a significant component of the repertoire of CD4+CD25+ T reg cells is self-reactive: a large proportion of TCRs generated by TCR rearrangement and pairing exhibits spontaneous reactivity to MHC molecules (15–60% depending on estimates; references 49–51). This self-reactivity is weeded out of the conventional repertoire by clonal deletion, but can persist in CD4+CD25+ T reg cells. Thus, the repertoire of CD4+CD25+ T reg cells molded by resistance to clonal deletion will be enriched in self-reactive specificities, as previously envisioned (26–29), but through a selective rather than an inductive process. From a mechanistic standpoint, the signal transduction pathways or pro-/antiapoptotic balance must be different in conventional T CD4+ cells or CD4+CD25+ T reg cells. In this respect, it is interesting that GITR has been implicated in T cell apoptosis; T cells in GITR-deficient mice are more sensitive to activation-induced cell death, whereas GITR-transgenic clones are more resistant (52, 53). Human Jurkat T cells cotransfected with hGITR and hGITR ligand were also resistant to activation-induced cell death (54). GITR might be one of the means through which CD4+CD25+ cells resist negative selection.
In conclusion, our data indicate that MCC agonist ligand does not directly promote differentiation of AND-transgenic precursors into CD4+CD25+ T reg cells, calling into question the paradigm of agonist-driven differentiation of CD4+CD25+ cells, and leading one to envision a conceptually different model. In fairness, it is unclear whether the observations made here can be generalized to other TCR/Ag pairs, and whether peculiarities of the AND/TIM system, such as affinity or expression niches, may condition the outcome. However, at the very least, the present data demonstrate that there is significant specificity to T reg cell differentiation and that induction by agonist ligands, if it occurs, is not available to cells expressing any TCR or encountering self-agonist in any epithelial niche. Ultimately, the solution will come from clearly elucidating the natural ligands that normally promote FoxP3 expression and differentiation toward the CD4+CD25+ pathway, and how they may differ from those that select conventional CD4+ T cells.
Acknowledgments
We thank V. Bruklich and T. Lipatova for help with maintaining the mouse colony, G. Losyev for flow cytometric analysis, and members of the T cell group for inspiring discussion.
This work was supported by a grant from the National Institutes of Health to D. Mathis and C. Benoist (no. 1 R01 AI51530-01), funds from the William T. Young Chair in Diabetes Research, and Joslin Diabetes and Endocrinology Research Center cores (grant no. 2 P30 DK36836-17). H.M. van Santen received a postdoctoral fellowship from the Cancer Research Institute and a Special Fellowship from the Leukemia and Lymphoma Society.
The authors have no conflicting financial interests.
Abbreviations used in this paper: CLIP, class II–associated invariant chain peptide; MCC, moth cytochrome c.; T reg cell, regulatory T cell; tet, tetracycline; TIM, tet-regulatable invariant chain with MCC.
References
- 1.Kappler, J.W., N. Roehm, and P. Marrack. 1987. T cell tolerance by clonal elimination in the thymus. Cell. 49:273–280. [DOI] [PubMed] [Google Scholar]
- 2.Kisielow, P., H. Bluthmann, U.D. Staerz, M. Steinmetz, and H. von Boehmer. 1988. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature. 333:742–746. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, M.S., E.S. Venanzi, L. Klein, Z. Chen, S. Berzins, S.J. Turley, H. von Boehmer, R. Bronson, A. Dierich, C. Benoist, and D. Mathis. 2002. Projection of an immunological self-shadow within the thymus by the aire protein. Science. 298:139–1401. [DOI] [PubMed] [Google Scholar]
- 4.Maloy, K.J., and F. Powrie. 2001. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2:816–822. [DOI] [PubMed] [Google Scholar]
- 5.Shevach, E.M. 2002. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389–400. [DOI] [PubMed] [Google Scholar]
- 6.Wood, K.J., and S. Sakaguchi. 2003. Regulatory T cells in transplantation tolerance. Nat. Rev. Immunol. 3:199–210. [DOI] [PubMed] [Google Scholar]
- 7.Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151–1164. [PubMed] [Google Scholar]
- 8.Asano, M., M. Toda, N. Sakaguchi, and S. Sakaguchi. 1996. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 184:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papiernik, M., M.L. de Moraes, C. Pontoux, F. Vasseur, and C. Penit. 1998. Regulatory CD4 T cells: expression of IL-2R alpha chain, resistance to clonal deletion and IL-2 dependency. Int. Immunol. 10:371–378. [DOI] [PubMed] [Google Scholar]
- 10.Itoh, M., T. Takahashi, N. Sakaguchi, Y. Kuniyasu, J. Shimizu, F. Otsuka, and S. Sakaguchi. 1999. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 162:5317–5326. [PubMed] [Google Scholar]
- 11.Baecher-Allan, C., J.A. Brown, G.J. Freeman, and D.A. Hafler. 2001. CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 167:1245–1253. [DOI] [PubMed] [Google Scholar]
- 12.Dieckmann, D., H. Plottner, S. Berchtold, T. Berger, and G. Schuler. 2001. Ex vivo isolation and characterization of CD4+CD25+ T cells with regulatory properties from human blood. J. Exp. Med. 193:1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jonuleit, H., E. Schmitt, M. Stassen, A. Tuettenberg, J. Knop, and A.H. Enk. 2001. Identification and functional characterization of human CD4+CD25+ T cells with regulatory properties isolated from peripheral blood. J. Exp. Med. 193:1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levings, M.K., R. Sangregorio, and M.G. Roncarolo. 2001. Human CD25+CD4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J. Exp. Med. 193:1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephens, L.A., C. Mottet, D. Mason, and F. Powrie. 2001. Human CD4(+)CD25(+) thymocytes and peripheral T cells have immune suppressive activity in vitro. Eur. J. Immunol. 31:1247–1254. [DOI] [PubMed] [Google Scholar]
- 16.Taams, L.S., J. Smith, M.H. Rustin, M. Salmon, L.W. Poulter, and A.N. Akbar. 2001. Human anergic/suppressive CD4(+)CD25(+) T cells: a highly differentiated and apoptosis-prone population. Eur. J. Immunol. 31:1122–1131. [DOI] [PubMed] [Google Scholar]
- 17.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 18.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 19.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 20.Godfrey, V.L., J.E. Wilkinson, and L.B. Russell. 1991. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am. J. Pathol. 138:1379–1387. [PMC free article] [PubMed] [Google Scholar]
- 21.Smyk-Pearson, S.K., A.C. Bakke, P.K. Held, and R.S. Wildin. 2003. Rescue of the autoimmune scurfy mouse by partial bone marrow transplantation or by injection with T-enriched splenocytes. Clin. Exp. Immunol. 133:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gambineri, E., T.R. Torgerson, and H.D. Ochs. 2003. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr. Opin. Rheumatol. 15:430–435. [DOI] [PubMed] [Google Scholar]
- 23.Bennett, C.L., J. Christie, F. Ramsdell, M.E. Brunkow, P.J. Ferguson, L. Whitesell, T.E. Kelly, F.T. Saulsbury, P.F. Chance, and H.D. Ochs. 2001. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27:20–21. [DOI] [PubMed] [Google Scholar]
- 24.Wildin, R.S., F. Ramsdell, J. Peake, F. Faravelli, J.L. Casanova, N. Buist, E. Levy-Lahad, M. Mazzella, O. Goulet, L. Perroni, et al. 2001. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 27:18–20. [DOI] [PubMed] [Google Scholar]
- 25.Shevach, E.M. 2001. Certified professionals: CD4+CD25+ suppressor T cells. J. Exp. Med. 193:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jordan, M.S., A. Boesteanu, A.J. Reed, A.L. Petrone, A.E. Holenbeck, M.A. Lerman, A. Naji, and A.J. Caton. 2001. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2:283–284. [DOI] [PubMed] [Google Scholar]
- 27.Apostolou, I., A. Sarukhan, L. Klein, and H. von Boehmer. 2002. Origin of regulatory T cells with known specificity for antigen. Nat. Immunol. 3:756–763. [DOI] [PubMed] [Google Scholar]
- 28.Kawahata, K., Y. Misaki, M. Yamauchi, S. Tsunekawa, K. Setoguchi, J. Miyazaki, and K. Yamamoto. 2002. Generation of CD4(+)CD25(+) regulatory T cells from autoreactive T cells simultaneously with their negative selection in the thymus and from nonautoreactive T cells by endogenous TCR expression. J. Immunol. 168:4399–4405. [DOI] [PubMed] [Google Scholar]
- 29.Walker, L.S., A. Chodos, M. Eggena, H. Dooms, and A.K. Abbas. 2003. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J. Exp. Med. 198:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furth, P.A., L. St. Onge, H. Böger, P. Gruss, M. Gossen, A. Kistner, H. Bujard, and L. Hennighausen. 1994. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc. Natl. Acad. Sci. USA. 91:9302–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kistner, A., M. Gossen, F. Zimmermann, J. Jerecic, C. Ullmer, H. Lübbert, and H. Bujard. 1996. Doxycycline-mediated, quantitative and tissue-specific control of gene expression in transgenic mice. Proc. Natl. Acad. Sci. USA. 93:10933–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaye, J., M.L. Hsu, M.E. Sauron, S.C. Jameson, N.R. Gascoigne, and S.M. Hedrick. 1989. Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature. 341:746–749. [DOI] [PubMed] [Google Scholar]
- 33.Landais, D., B.N. Beck, J.-M. Buerstedde, S. Degraw, D. Klein, N. Koch, D. Murphy, M. Pierres, T. Tada, K. Yamamoto, et al. 1986. The assignment of chain specificities for anti-Ia monoclonal antibodies using L cell transfectants. J. Immunol. 137:3002–3005. [PubMed] [Google Scholar]
- 34.Witherden, D., N. van Oers, C. Waltzinger, A. Weiss, C. Benoist, and D. Mathis. 2000. Tetracycline-controllable selection of CD4+ T cells: half-life and survival signals in the absence of major histocompatibility complex class II molecules. J. Exp. Med. 191:355–364. [DOI] [PubMed] [Google Scholar]
- 35.Viville, S., J. Neefjes, V. Lotteau, A. Dierich, M. Lemeur, H. Ploegh, C. Benoist, and D. Mathis. 1993. Mice lacking the MHC class II-associated invariant chain. Cell. 72:635–648. [DOI] [PubMed] [Google Scholar]
- 36.Lemeur, M., P. Gerlinger, C. Benoist, and D. Mathis. 1985. Correcting an immune-response deficiency by creating Eα gene transgenic mice. Nature. 316:38–42. [DOI] [PubMed] [Google Scholar]
- 37.Siebenkotten, I.M., C. Carstens, and N. Koch. 1998. Identification of a sequence that mediates promiscuous binding of invariant chain to MHC class II allotypes. J. Immunol. 160:3355–3362. [PubMed] [Google Scholar]
- 38.van Bergen, J., S.P. Schoenberger, F. Verreck, R. Amons, R. Offringa, and F. Koning. 1997. Efficient loading of HLA-DR with a T helper epitope by genetic exchange of CLIP. Proc. Natl. Acad. Sci. USA. 94:7499–7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwartz, R.H., B.S. Fox, E. Fraga, C. Chen, and B. Singh. 1985. The T lymphocyte response to cytochrome c. V. Determination of the minimal peptide size required for stimulation of T cell clones and assessment of the contribution of each residue beyond this size to antigenic potency. J. Immunol. 135:2598–2608. [PubMed] [Google Scholar]
- 40.Gossen, M., and H. Bujard. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA. 89:5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimizu, J., S. Yamazaki, T. Takahashi, Y. Ishida, and S. Sakaguchi. 2002. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3:135–142. [DOI] [PubMed] [Google Scholar]
- 42.McHugh, R.S., M.J. Whitters, C.A. Piccirillo, D.A. Young, E.M. Shevach, M. Collins, and M.C. Byrne. 2002. CD4(+) CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 16:311–323. [DOI] [PubMed] [Google Scholar]
- 43.Gavin, M.A., S.R. Clarke, E. Negrou, A. Gallegos, and A. Rudensky. 2002. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol. 3:33–41. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi, T., Y. Kuniyasu, M. Toda, N. Sakaguchi, M. Itoh, M. Iwata, J. Shimizu, and S. Sakaguchi. 1998. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10:1969–1980. [DOI] [PubMed] [Google Scholar]
- 45.Thornton, A.M., and E.M. Shevach. 1998. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shih, F.F., L. Mandik-Nayak, B.T. Wipke, and P.M. Allen. 2004. Massive thymic deletion results in systemic autoimmunity through elimination of CD4+CD25+ T regulatory cells. J. Exp. Med. 199:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goverman, J., T. Brabb, E.S. Huseby, and A.G. Farr. 1997. TCR signaling regulates thymic organization: lessons from TCR-transgenic mice. Immunol. Today. 18:204–208. [DOI] [PubMed] [Google Scholar]
- 48.Bensinger, S.J., A. Bandeira, M.S. Jordan, A.J. Caton, and T.M. Laufer. 2001. Major histocompatibility complex class II–positive cortical epithelium mediates the selection of CD4+25+ immunoregulatory T cells. J. Exp. Med. 194:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ignatowicz, L., J. Kappler, and P. Marrack. 1996. The repertoire of T cells shaped by a single MHC/peptide ligand. Cell. 84:521–529. [DOI] [PubMed] [Google Scholar]
- 50.Zerrahn, J., W. Held, and D.H. Raulet. 1997. The MHC reactivity of the T cell repertoire prior to positive and negative selection. Cell. 88:627–636. [DOI] [PubMed] [Google Scholar]
- 51.Merkenschlager, M., D. Graf, M. Lovatt, U. Bommhardt, R. Zamoyska, and A.G. Fisher. 1997. How many thymocytes audition for selection? J. Exp. Med. 186:1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ronchetti, S., G. Nocentini, C. Riccardi, and P.P. Pandolfi. 2002. Role of GITR in activation response of T lymphocytes. Blood. 100:350–352. [DOI] [PubMed] [Google Scholar]
- 53.Nocentini, G., L. Giunchi, S. Ronchetti, L.T. Krausz, A. Bartoli, R. Moraca, G. Migliorati, and C. Riccardi. 1997. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA. 94:6216–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gurney, A.L., S.A. Marsters, R.M. Huang, R.M. Pitti, D.T. Mark, D.T. Baldwin, A.M. Gray, A.D. Dowd, A.D. Brush, A.D. Heldens, et al. 1999. Identification of a new member of the tumor necrosis factor family and its receptor, a human ortholog of mouse GITR. Curr. Biol. 9:215–218. [DOI] [PubMed] [Google Scholar]