

Abstract
Heat shock protein 70 (Hsp70) is a potent survival protein whose depletion triggers massive caspase-independent tumor cell death. Here, we show that Hsp70 exerts its prosurvival function by inhibiting lysosomal membrane permeabilization. The cell death induced by Hsp70 depletion was preceded by the release of lysosomal enzymes into the cytosol and inhibited by pharmacological inhibitors of lysosomal cysteine proteases. Accordingly, the Hsp70-mediated protection against various death stimuli in Hsp70-expressing human tumor cells as well as in immortalized Hsp70 transgenic murine fibroblasts occurred at the level of the lysosomal permeabilization. On the contrary, Hsp70 failed to inhibit the cytochrome c–induced, apoptosome-dependent caspase activation in vitro and Fas ligand–induced, caspase-dependent apoptosis in immortalized fibroblasts. Immunoelectron microscopy revealed that endosomal and lysosomal membranes of tumor cells contained Hsp70. Permeabilization of purified endo/lysosomes by digitonin failed to release Hsp70, suggesting that it is physically associated with the membranes. Finally, Hsp70 positive lysosomes displayed increased size and resistance against chemical and physical membrane destabilization. These data identify Hsp70 as the first survival protein that functions by inhibiting the death-associated permeabilization of lysosomes.
Keywords: cathepsins, cell death, neoplasms, tumor necrosis factor, immunoelectron microscopy
Introduction
Defects in signaling pathways leading to the classic mitochondrion- and caspase-mediated apoptosis are frequent in primary tumors (1, 2). As a result, alternative cell death programs are gaining increasing interest among cancer researchers. Until recently, lysosomes have been considered “suicide bags” that, through the release of unspecific enzymes, cause autolysis and damage neighboring cells during uncontrolled tissue damage. However, accumulating data now show that lysosomes also function as death signal integrators in many controlled death paradigms (3, 4). Lysosomal proteases and cathepsins translocate from the lysosomal lumen to the cytosol in response to a wide variety of death stimuli such as TNF (5, 6), Fas (7), p53 activation (8), microtubule stabilizing agents (9), oxidative stress (7, 10), staurosporine (11), growth factor deprivation (7), and lysosomotropic agents (10, 12). Once released to the cytosol, cathepsins, especially cysteine cathepsins B and L and aspartyl cathepsin D, may trigger the mitochondrial outer membrane permeabilization followed by caspase- or apoptosis-inducing factor–mediated apoptosis (5, 8, 11–13) or mediate caspase- and/or apoptosis-inducing factor–independent programmed cell death (PCD) with apoptosis- or necrosis-like morphology (6, 14). Because the latter pathway can circumvent most of the known resistance mechanisms occurring in tumor cells, lysosomal membrane permeabilization (LMP) appears to be a promising target for novel cancer drugs.
The heat shock protein 70 (Hsp70) family of proteins consists of both constitutively expressed and stress-inducible molecular chaperones that are localized to different intracellular compartments (15). The major stress-inducible Hsp70 (also called Hsp72) is highly expressed in the cytosol and plasma membrane of primary tumors of various origins, whereas its expression in unstressed normal cells is very low and restricted to the cytosol (15–17). The role of Hsp70 in tumorigenesis is supported by experimental data showing that its high expression is required for the growth of human tumor xenografts in immunodeficient mice (18), and that it enhances the tumorigenic potential of rodent cells in syngenic animals (19). Furthermore, its high expression correlates with poor therapeutic outcome in human breast cancer (20). The molecular mechanism underlying the tumorigenic potential of Hsp70 is as yet unclear, but it may be explained by its ability to confer resistance to both apoptosis- and necrosis-like PCD induced by diverse stimuli (21, 22). In vitro studies have suggested that Hsp70 may directly interfere with the apoptosis signaling machinery by binding to the apoptotic protease-activating factor-1 (22) or apoptosis-inducing factor (23) and thereby inhibit the apoptosome-mediated activation of caspases or apoptosis-inducing factor–induced nuclear changes, respectively. However, most studies using cellular death models suggest that Hsp70-mediated inhibition of caspase-mediated PCD occurs upstream of mitochondrial outer membrane permeabilization and apoptosome formation (24–26). Furthermore, Hsp70 can effectively rescue cells from caspase-independent PCD induced by TNF, heat shock, serum starvation, or oxidative stress (6, 23, 24, 27, 28), and the depletion of Hsp70 induces caspase-independent apoptosis-like PCD in various human tumor cell lines (18, 29). Here, we aim at determining the mechanism through which Hsp70 regulates tumor cell survival with special focus on LMP and caspase-independent cell death.
Materials and Methods
Cell Culture.
WEHI-V2 and WEHI-V3 are vector-transfected and WEHI-hsp-2 and WEHI-hsp-18 Hsp70-transfected single cell clones of WEHI-S murine fibrosarcoma cells (28). ME-180v and ME-180as are single cell clones of ME-180 human cervix carcinoma cells transfected with vector and Hsp70 antisense (as) cDNA, respectively (28). HeLa human cervix carcinoma cell line was provided by J. Lukas (Danish Cancer Society, Copenhagen, Denmark). CX+ (>90% of cell positive) and CX− (<20% of cells positive) cells are stable sublines of CX2 human colon carcinoma cells sorted according to the Hsp70 surface expression (17). HBL-100 is an immortalized breast epithelial cell line provided by P. Briand (Danish Cancer Society, Copenhagen, Denmark). Murine embryonic fibroblasts (MEFs) explanted from days 14–16 wild type or homozygote Hsp70 transgenic CBA × C57Bl/6 mice (provided by G. Kollias, Hellenic Pasteur Institute, Athens, Greece, and G. Pagoulatos, University of Ioannina, Ioannina, Greece; reference 30) embryos were passaged once a week (6–8,000 cells/cm2) with a change of medium on day three until they reached senescence. Senescent cells were maintained by medium change twice a week, and passaging was restarted upon immortalization. Immortalized MEFs (iMEFs) were used at passages 15–20. DMEM (Invitrogen) supplemented with 10% heat-inactivated calf serum (Biological Industries), 0.1 mM of nonessential amino-acids (Invitrogen), 110 mg/ml Na-pyruvate (Merck) and antibiotics were used as growth medium for MEFs. Other cells were propagated in RPMI 1640 (Invitrogen) supplemented with 6% heat-inactivated calf serum and antibiotics (complete medium) at 37°C in a humidified air atmosphere with 5% CO2. All cells were repeatedly tested and found negative for mycoplasma by Hoechst (H-33342; Molecular Probes) staining and immunofluorescence microscopy.
Cell Death Induction and Determination.
Ad.asHsp70 and Ad.β-Gal are adenoviral (Ad.) shuttle vectors carrying bases 475–796 of the published human Hsp70 sequence in as orientation and β-galactosidase (β-Gal) cDNA, respectively (29). The infections were performed as described previously using the lowest multiplicity of infection resulting in a 100% infection (29). Recombinant human TNF was provided by A. Cerami (Kenneth S. Warren Laboratories, Tarrytown, NY), recombinant murine TNF was obtained from R&D Systems, and etoposide and hydrogen peroxide were obtained from Sigma-Aldrich. To obtain Fas ligand–containing supernatant, confluent Neuro2 cells were provided with fresh serum-free medium, and after 24 h at 37°C, the supernatant was collected, centrifuged at 600 g for 10 min, and stored in aliquots at −80°C. The blue light was delivered to CX cells by a custom-made lamp house consisting of a 100-W Hg gas discharge lamp, a bp filter (450–500 nm), and a mirror system (Leica) and to iMEFs by the 488-nm laser of the Axiovert 100M confocal microscope (Carl Zeiss MicroImaging, Inc.). γ-irradiation was delivered to exponentially growing cells by a 137Cs source at a dose rate of 1 Gy/10 s.
z-Val-Ala-DL-Asp-CH2F (zVAD-fmk) obtained from Bachem, PD150606 obtained from Calbiochem-Novabiochem, CA-074-Me obtained from Peptides International, z-Phe-Ala-CH2F (zFA-fmk) obtained from Enzyme System Products, Mu-Leu-HphVSPh (LHVS; provided by J. Palmer, Celera, San Francisco, CA; reference 31), and Ac-Asp-Glu-Val-Asp-aldehyde (DEVD-CHO) obtained from BIOMOL Research Laboratories, Inc. were dissolved in DMSO. When using the protease inhibitors, DMSO concentration of all samples was adjusted to 0.2%. The experiments with ME-180 transfectants were performed in RPMI 1640 supplemented with 0.5% FCS after a preincubation of cells in this medium for 18–24 h. Experiments with other cells were performed in complete medium.
3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction and lactate dehydrogenase (LDH) release assays (cytotoxicity detection kit; Roche) were used to analyze the survival of the cells as described previously (6). Staining with annexin V conjugated with fluorescein isothiocyanate (Bender Medsystems) and flow cytometry (FACSCalibur™; Becton Dickinson) analysis were used to detect apoptotic cells exposing phosphatidylserine on the outer leaflet of the plasma membrane.
Measurement of Enzyme Activities.
To measure cytosolic enzyme activities, subconfluent cells were treated with an extraction buffer (250 mM sucrose, 20 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 1 mM pefabloc, pH 7.5) containing 15 μg/ml digitonin for 12–15 min on ice. The digitonin concentration and treatment times were optimized to result in the total release of the cytosolic LDH activity without disruption of lysosomes. To measure the total cellular cysteine cathepsin activity, cells were treated with the aforementioned extraction buffer containing 200 μg/ml digitonin for 12–15 min on ice and, when indicated, incubated with the indicated protease inhibitors or recombinant human Hsp70 (rhHsp70; StressGen Biotechnologies) for 30 min on ice before the measurement. The effector caspase and cysteine cathepsin activities were estimated by adding one volume of 20 μM Ac-DEVD–7-amino-trifluoromethylcoumarin (AFC) (BIOMOL Research Laboratories, Inc.) in caspase reaction buffer (100 mM Hepes, 20% glycerol, 0.5 mM EDTA, 0.1% CHAPS, 5 mM dithiothreitol (DTT), and 1 mM pefabloc, pH 7.5) or 20 μM zFR-AFC (Enzyme System Products) in cathepsin reaction buffer (50 mM sodium acetate, 4 mM EDTA, 8 mM DTT, and 1 mM pefabloc, pH 6.0), respectively. The Vmax of the liberation of AFC (excitation, 400 nm; emission, 489 nm) was measured for 20 min at 30°C with a Spectramax Gemini fluorometer (Molecular Devices). β-N-acetyl-glucosaminidase (NAG) activity was estimated by adding three volumes of 0.2 M sodium citrate buffer, pH 4.5, containing 300 μg/ml 4-methylumbelliferyl-2-acetamido-2-deoxy-β-d-glucopyranoside (Sigma-Aldrich). The Vmax of the liberation of methylumbelliferyl (excitation, 356 nm; emission, 444 nm) was measured for 20 min at 30°C with a Spectramax Gemini fluorometer. LDH activity of the cytosol determined by a cytotoxicity detection kit (Roche) was used as an internal standard with which protease activities were normalized.
In Vitro Apoptosome Assay.
Subconfluent cultures of HeLa cells were harvested by scraping on ice, washed in ice-cold PBS, and resuspended in equal volume of ice-cold isotonic lysis buffer (20 mM Hepes-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, 1 mM DTT, 10 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 100 μg/ml pefabloc). After a 30-min incubation on ice, the cells were lysed by 30 strokes of a Dounce homogenizer and centrifuged at 750 g for 10 min. The obtained supernatant was further centrifuged at 10,000 g for 10 min and at 20,000 g for 30 min. The clarified supernatant was removed carefully, stored in aliquots at −80°C, and used at protein concentrations ranging from 5 to 10 mg/ml. The apoptosome was activated by the addition of 1 mM dATP (dissolved in ddH20 and adjusted to pH 7.0; ICN Biomedicals) and 1 μM horse heart cytochrome c (Sigma-Aldrich) to the cytosolic HeLa cell extract (protein concentration; 5–10 mg/ml) containing 100 μM DEVD-AFC (BIOMOL Research Laboratories, Inc.). When indicated, 2 μM recombinant human Hsp70 (StressGen Biotechnologies) was added to the cytosolic extract before the addition of cytochrome c and dATP. After a 30-min incubation at 37°C, the Vmax of the liberation of AFC was measured as mentioned before. After 2 h at 37°C, the samples from the in vitro apoptosome assay were mixed with 0.25 volumes of 4 × Laemmli sample buffer and the immunodetection of proteins separated by SDS-PAGE was performed as described in the next paragraph.
Immunoblot Analysis, Immunocytochemistry, and Electron Microscopy.
The primary antibodies used included murine monoclonal antibodies against human Hsp70 (Multimmune GmbH), cathepsin B (Oncogene Research Products), cathepsin L and DFF45/ICAD (Transduction Laboratories), caspase-3 and -9 (BD Biosciences), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Biogenesis), as well as polyclonal goat antibodies against rat cathepsin B (32), rabbit antibodies against human cathepsin D (no. 06-467; Upstate Biotechnology), and rabbit antiserum against Hsp70 raised (Neosystem) by immunizing a rabbit with an ovalbumin-coupled peptide (YTKDNNLLGRFELSG) corresponding to amino acids 450–463 in the human Hsp70 sequence (P08107). Immunodetection of proteins (20 μg/lane) separated by 8–10% SDS-PAGE and transferred to nitrocellulose was performed using ECL Western blotting reagents (Amersham Biosciences), and the indicated primary antibodies and appropriate secondary antibodies were obtained from DakoCytomation.
To visualize cathepsins and cytochrome c, cells were fixed in −20° methanol for 10 min at 25°C and 4% formaldehyde for 30 min followed by 0.1% Triton for 10 min at 25°C, respectively. After blocking for 30 min in 10% FCS, slides were stained with the indicated primary antibodies and biotinylated (cathepsin B; DakoCytomation) or Alexa Fluor488–conjugated anti–mouse IgG (Molecular Probes). The indicated nuclei were stained in the end with 2 μl/ml ethidium bromide for 1–2 min at 25°C preceded by the 10-min treatment with 0.5 mg/ml RNase A at 37°C. Biotinylated IgG was visualized by peroxidase-conjugated streptavidin–biotin complex and diaminobenzidine/H2O2 according to the manufacturer's instructions (SteptABC; Vector Laboratories) and images were taken with a digital camera mounted on a BX60 microscope (Olympus). Confocal images were obtained with an Axiovert 100M microscope equipped with LSM 510 system (Carl Zeiss MicroImaging, Inc.).
For immunoelectron microscopy, CX+ and CX− tumor cells were fixed in 8% paraformaldehyde in 250 mM Hepes buffer for 1 h. After two washes, free aldehyde groups were quenched with 50 mM NH4Cl for 10 min. For cryoprotection, the cell pellets were incubated in 2.1 M sucrose in 17% polyvinylpyrrolodone at 20°C for 30 min before freezing in liquid nitrogen. Ultrathin sections (70 nm) were cut at −100°C on an Ultracut E microtome (FC4E; Reichart-Jung) using a glass knife and mounted on 150-mesh Parlodion (Mallinckrodt Specialty Chemicals)–coated nickel grids. Immunogold labeling of Hsp70 was performed with 10-nm gold particles and of cathepsin D with 5-nM gold particles. In brief, the grids were rinsed and blocked in 0.1% acetylated BSA buffer before incubation with murine anti-Hsp70 antibody (1:100) and/or rabbit anti-cathepsin D antibody (1:200) for 18 h at 4°C. After washing, the grids were incubated individually (single staining) or subsequently (double staining) for 3 h at 25°C in 1% BSA buffer containing 1:75 dilutions of Aurion goat anti–mouse IgG/IgM (10 nm GP) and goat anti–rabbit IgG (5 nm GP; both from Amersham Biosciences). Nonspecific binding was blocked by extensive washing in 0.1% acetylated BSA buffer. An additional fixation in 2% glutaraldehyde in PBS was performed after immunostaining. The sections were stained in uranyl acetate/methyl cellulose and viewed in an EM 10CR electron microscope (Carl Zeiss MicroImaging, Inc.). This method is optimized to the localization of antigens that are localized in both intracellular and extracellular compartments.
Fractionation of iMEFs.
Subconfluent cultures of iMEF-Hsp-4 cells were harvested by scraping on ice, washed in ice-cold PBS, and resuspended (20 × 106 cells/ml) in ice-cold SCA buffer (20 mM Hepes-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, 1 mM DTT, 10 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 1 mM pefabloc). After a 30-min incubation on ice, cells were lysed by 30–40 strokes in a Dounce homogenizer and centrifuged at 750 g for 5 min. The pellet was collected as the heavy membrane fraction (intact cells and nuclei). The supernatant obtained was centrifuged at 750 g for 5 min, the pellet was discarded, and the new supernatant was divided into two tubes and further centrifuged at 20,000 g for 20 min. The resulting supernatants were collected as the cytosolic fraction. The pellets were resuspended in 450 μl SCA buffer with or without 800 μg/ml digitonin, incubated for 30 min on ice, and centrifuged at 20,000 g for 20 min. The pellets were collected as the light membrane fractions (cellular organelles including endosomes and lysosomes) and the supernatants as the supernatants of the light membrane fraction (without digitonin; washing buffer and with digitonin; the contents of permeabilized cellular organelles). All fractions were mixed with 1:3 volumes of 3× Laemmli sample buffer and subjected to immunoblot analysis.
Lysosomal Volume and Permeability Assessment.
To label lysosomes, cells were incubated with 5 μM acridine orange (Molecular Probes) for 15 min at 37°C and washed twice with PBS before the indicated treatments or measurements. Acridine orange is a metachromatic fluorochrome and a weak base that exhibits red fluorescence when highly concentrated in acidic lysosomes and green fluorescence when outside the lysosomes. Total lysosomal volume and lysosomal integrity were evaluated by assessing the red fluorescence (FL2) and green fluorescence (FL1), respectively, by flow cytometry using a flow cytometer (Becton Dickinson) or confocal microscopy (Axiovert 100M microscope equipped with LSM 510 system; Carl Zeiss MicroImaging, Inc.).
Results
Hsp70 Depletion Triggers LMP Followed by Cathepsin-mediated PCD.
We have shown recently that the depletion of Hsp70 by adenoviral transfer of antisense Hsp70 cDNA (Ad.asHsp70) induces massive caspase-independent PCD in diverse tumor cell lines, but not in nontumorigenic epithelial cells or fibroblasts (18, 29). To find clues to the tumor-specific death pathway suppressed by Hsp70, we analyzed the ability of various protease inhibitors to rescue tumor cells from the death induced by Hsp70 depletion. Remarkably, the inhibition of cysteine cathepsin activity by broad spectrum cysteine cathepsin inhibitors zFA-fmk (6) and LHVS (31) or high concentrations of zVAD-fmk (6) conferred significant protection against Ad.asHsp70-induced death in MCF-7 and MDA-MB-468 breast cancer cells (Fig. 1, A and B). A more specific inhibition of cathepsin B by CA-074-Me conferred smaller, but significant, protection in MCF-7 cells. Due to an unspecific toxicity CA-074-Me and LHVS in the long-term assay, they could not be applied to MDA-MB-486 and MCF-7 cells, respectively. As we have shown earlier, selective inhibition of caspases by DEVD-CHO at concentrations up to 200 μM (not depicted) or low concentrations of zVAD-fmk (<50 μM) did not increase the survival of Ad.asHsp70-treated MCF-7 cells (Fig. 1 A), whereas such treatments conferred complete protection against TNF-induced apoptosis in MCF-7 cells (33). Also, 50 μM PD150606 capable of inhibiting calpain activation in MCF-7 cells was unable to rescue cells from Ad.asHsp70 (unpublished data).
Figure 1.

Ad.asHsp70 induces LMP and cathepsin-mediated PCD. (A and B) MCF-7 (A) and MDA-MB-468 (B) breast cancer cells were left untreated or infected with Ad.β-Gal or Ad.asHsp70. 6 h later, cells were trypsinized and plated on a 96-well plate at a density of 6,000 cells/well. After an additional 6 h (MCF-7) or 18 h (MDA-MB-468), the indicated protease inhibitors at indicated concentrations were added, and 72 h after the infection, the survival of the cells was determined by the MTT assay. The survival of Ad.β-Gal–treated cells is expressed as a percentage of noninfected cells and that of Ad.asHsp70-infected cells as a percentage of Ad.β-Gal–infected cells treated with the same inhibitors. (C) The cysteine cathepsin activity in WEHI-S cell lysates (4 × 105 cells/ml) containing indicated concentrations of recombinant Hsp70 were measured using zFR-AFC as a fluorogenic probe. The specificity of the assay for cysteine cathepsins was demonstrated by the ability of 1 μM zFA-fmk, but not 1 μM zVAD-fmk, to attenuate the protease activity. (D–F) MCF-7 cells were stained with antibodies against human cathepsin B (D and E) or L (F) 65 h after the infection with Ad.β-Gal (D) or Ad.asHsp70 (E and F) and visualized using biotinylated (D and E) or FITC-coupled (F) secondary antibodies. Representative fields are shown, and cells with cathepsins released into the cytosol are marked with arrows. (G–I) MCF-7 (G and H) and HBL-100 (I) cells were infected with Ad.β-Gal or Ad.asHsp70, and the cysteine cathepsin (G and I) and NAG (H) activities of digitonin-extracted cytosols and total cellular lysates were analyzed at the indicated time points. The results are expressed as percentages of the cytosolic activity of the total activity and corrected for the LDH activity. The values represent means of triplicate determinations. *, P < 0.02l; **, P < 0.01. All experiments were repeated two to four times with essentially similar results.
Hsp70 could protect tumor cells from cysteine cathepsin-mediated PCD either via the direct inhibition of the cysteine cathepsin activity or the inhibition of the LMP. To test the first possibility, we measured the cysteine cathepsin activity in tumor cell lysates incubated with or without recombinant Hsp70. Although 1 μM zFA-fmk and high concentrations (100 μM) of zVAD-fmk effectively inhibited the cysteine cathepsin activity, Hsp70 at concentrations up to 5 μM was without an effect (Fig. 1 C). Moreover, Hsp70 did not directly bind the most abundant cysteine cathepsins (B or L) as judged by the inability of Hsp70 to coimmunoprecipitate cysteine cathepsins or vice versa (unpublished data). To investigate the second possibility, we analyzed the subcellular distribution of cathepsins B and L using specific antibodies and confocal microscopy. In contrast with untreated and β-Gal–infected cells that displayed mainly perinuclear punctuate distribution of cathepsins consistent with their lysosomal localization, cathepsins B and L translocated from cytosolic granules to the cytosol and nucleus in Ad.asHsp70-treated cancer cells (Fig. 1, D–F). To evaluate the kinetics and the extent of the lysosomal permeabilization, we also measured the activities of lysosomal enzymes in cytosolic fractions obtained by digitonin-based plasma membrane permeabilization (6). Although the total cellular cysteine cathepsin activity remained essentially unchanged, the cytosolic fractions of Ad.asHsp70-infected cells contained ∼8, 20, and 25% of the total cellular cysteine cathepsin activity 24, 48, and 60 h after the Ad.asHsp70 infection, respectively (Fig. 1 G). The cytosolic enzyme activity in vehicle-treated and Ad.β-Gal–infected cells remained <2% of the total activity throughout the experiment (Fig. 1 G). Notably, the gross morphological changes indicative of PCD (detachment, shrinkage, and chromatin condensation) and the decrease in cell survival as measured by MTT reduction or LDH release assays were first detectable several hours after the appearance of cathepsin activity in the cytosol, ∼60 h after the Ad.asHsp70 infection (Fig. 1 A and reference 29). NAG, a 250-kD enzyme present in the lysosomes of untreated cells, was also released into the cytosol of MCF-7 cells upon Ad.asHsp70 infection (Fig. 1 H). However, the kinetics of the NAG release were clearly slower than that of cysteine cathepsins. Nontumorigenic immortalized breast epithelial cells (HBL-100) that are resistant to the Hsp70 depletion (29) showed no increase in the cytosolic cysteine cathepsin (Fig. 1 I) or NAG (not depicted) activities 24–96 h after the Ad.asHsp70 infection.
Hsp70 Inhibits TNF- and Etoposide-induced LMP.
Hsp70 confers effective protection against TNF-induced cell death in ME-180 cervix carcinoma and WEHI-S fibrosarcoma cells without inhibiting the activation of caspases (28). Although TNF-induced death of ME-180 cervix carcinoma cells is strictly caspase dependent, that of WEHI-S fibrosarcoma cells is completely independent of caspase activity and can even be enhanced by pan-caspase and caspase-8 inhibitors (6). However, both death pathways depend on the activity of cathepsin B, which is released from the lysosomal compartment into the cytosol upon TNF treatment either in a caspase-dependent (ME-180 cells) or caspase-inhibited (WEHI-S) manner (6). Next, we studied whether Hsp70 interfered with LMP and cathepsin release in TNF-induced WEHI-S and ME-180 cells. WEHI-S cells expressing Hsp70 resisted not only the death induced by TNF (Fig. 2 A) but also the enhanced cytotoxicity induced by the combination of TNF plus 1 μM zVAD-fmk (Fig. 2 C). The Hsp70-mediated protection was associated with a complete inhibition of the appearance of the cysteine cathepsin activity in the cytosol (Fig. 2, B and D). Also, in ME-180v cells expressing high levels of endogenous Hsp70, TNF failed to induce the translocation of cathepsins and death, whereas ME-180as cells that express low levels of Hsp70 due to the transfection of an as Hsp70 cDNA that responded to TNF by releasing cysteine cathepsins into the cytosol before the lysis of the plasma membrane (Fig. 2, E and F).
Figure 2.

Hsp70 inhibits TNF-induced lysosomal release of cysteine cathepsins in tumor cells. Vector and Hsp70-transfected WEHI-S clones (A–D) or vector- and asHsp70-transfected ME-180 cells (E and F) were treated as indicated and analyzed for the cytosolic cysteine cathepsin activity (arbitrary units) as a measure of LMP (B, D, and F) and for the released LDH activity (percentage of total activity) as a measure of cytotoxicity (A, C, and E). The values represent means of a triplicate determination ± SD. The experiments were repeated twice with essentially similar results.
To extend the analysis to other cell types and death stimuli, we established iMEFs originating from wild-type (iMEF wt-1 and -5) and Hsp70 transgenic mice (iMEF hsp-3 and -4). The iMEFs from the transgenic animals expressed considerably higher levels of Hsp70 than the cells from the wild-type animals (Fig. 3 A). However, the expression level was below that observed in most human cancer cell lines. The moderate expression level of Hsp70 was sufficient to confer significant protection against cell death induced by TNF plus cycloheximide (CHX) or etoposide (a topoisomerase inhibitor), but not against that induced by Fas ligand plus CHX (Fig. 3 B). Next, we studied the involvement of LMP, mitochondria, cysteine cathepsins, and caspases in the death programs inhibited by Hsp70. Inhibition of cysteine cathepsin activity in iMEFs by 50 μM zFA-fmk conferred significant protection against death induced by TNF plus CHX or etoposide, whereas inhibition of caspases by 10 μM zVAD-fmk protected iMEFs only against TNF plus CHX (Fig. 3 C). Both stimuli induced an early LMP in wild-type iMEFs as demonstrated by the release of cysteine cathepsin activity into the cytosol (Fig. 3, D and E). In both cases, the LMP was almost completely attenuated in iMEFs originating from Hsp70 transgenic animals. Both stimuli also induced mitochondrial outer membrane permeabilization as demonstrated by the release of cytochrome c from the mitochondrial intermembrane space into the cytosol (Fig. 3 F) and effector caspase activation (not depicted) in wild-type iMEFs. Both cytochrome c release and effector caspase activation were significantly attenuated in Hsp70-expressing iMEFs. Furthermore, zFA-fmk, a pharmacological cysteine cathepsin inhibitor, effectively inhibited TNF- and etoposide-induced cytochrome c release in iMEFs, indicating that cytochrome c release requires cathepsin activity (Fig. 3 F). This is consistent with our earlier data showing that TNF plus CHX fails to induce cytochrome c release and effector caspase activation in cathepsin B–deficient iMEFs (34). Thus, Hsp70 interfered with these cell death programs by inhibiting LMP, which occurred upstream of mitochondrial outer membrane permeabilization and effector caspase activation.
Figure 3.

Hsp70 inhibits TNF- and etoposide-induced death upstream of LMP and mitochondrial outer membrane permeabilization in iMEFs. (A) An immunoblot demonstrating the moderate expression level of the Hsp70 transgene in iMEFs originating from Hsp70 transgenic animals (hsp-3 and -4). iMEFs from wild-type animals (wt-1 and -5) express barely detectable levels of Hsp70 (visible in longer exposure). Proteins from MCF-7 cells serve as a positive control, and immunoblot for GAPDH demonstrates the fairly equal loading of the protein. (B and C) The sensitivity of the iMEFs to TNF (1 ng/ml murine TNF plus 4 μM CHX for 18 h), etoposide (Eto; 50 μM for 48 h), and Fas ligand (2.5% Fas ligand–containing supernatant plus 4 μM CHX for 18 h) in the absence or presence of 10 μM zVAD-fmk (zVAD) or 50 μM zFA-fmk (zFA) was evaluated by MTT reduction assay, and the survival is expressed as the percentage of untreated cells. (D and E) iMEFs were treated with 1 ng/ml TNF plus 4 μM CHX (D) or 50 μM etoposide (E) for indicated times and analyzed for the cytosolic cysteine cathepsin activity as a measure of LMP. The values represent percentages of total activity. (F) iMEFs were grown on coverslips, treated with 1 ng/ml TNF plus 4 μM CHX for 8 h or 50 μM etoposide for 48 h, fixed, and analyzed for cytochrome c release by immunostaining. When indicated cells were pretreated for 1 h with 50 μM zFA-fmk (zFA). A minimum of 500 cells were counted, and the values represent the percentages of cells with diffuse (extra-mitochondrial) cytochrome c staining. (B–E) The values represent the means of a triplicate determination ± SD. All assays were repeated at least once with essentially similar results, and additional experiments with five (C) or two (D–F) independent iMEF clones gave similar results.
The aforementioned data showing that Ad.asHsp70 triggers LMP and that Hsp70 inhibits LMP and cathepsin release in TNF-treated WEHI-S and ME-180 cells as well as in TNF- or etoposide-treated iMEFs support the previously published data showing that Hsp70 prevents PCD either upstream of mitochondrial outer membrane permeabilization (iMEFs), downstream of effector caspase activation (ME-180), or independent of both (WEHI-S and Ad.asHsp70-treated cancer cells). Accordingly, recombinant Hsp70 had no effect on the activation of apoptosome as demonstrated by its inability to inhibit cytochrome c plus dATP-induced effector caspase activation as well as processing of caspases or their substrates in an in vitro assay system optimized and successfully used to screen small molecule libraries for apoptosome inhibitors (Fig. 4 and reference 33). Furthermore, cytochrome c plus dATP induced similar caspase activation in cytosols with high versus low Hsp70 content (i.e., cytosols from WEHI-hsp-2 vs. WEHI-v2 and iMEF-wt-5 vs. iMEF-hsp-4; unpublished data).
Figure 4.

Hsp70 does not inhibit the apoptosome-mediated activation of caspases. Cytosolic extracts prepared from HeLa cells were incubated for 90 min with 1 μM cytochrome c and 1 mM dATP (Cc/dATP) in the presence or absence of 2 μM of recombinant human Hsp70, 1 μM zVAD-fmk, or 0.1 μM DEVD-fmk. (A) Caspase-3–like activity was assessed by spectrofluorometric quantification of DEVD-AFC cleavage and presented as pmol per min. (B) Thereafter, samples were analyzed by immunoblotting using antibodies specific against the indicated proteins. *, a background band. Vertical lines indicate that intervening lanes have been spliced out. The experiment was repeated twice with similar results.
Hsp70 Localizes to the Lysosomal Membranes.
In addition to its localization in the cytosol of unstressed normal cells, most cultured tumor cells as well as freshly isolated tumors present Hsp70 on the cell surface (16, 17). Using an Hsp70-specific antibody that recognizes the extracellular epitope of the membrane-associated Hsp70, we studied whether cells expressing Hsp70 on the plasma membrane also have it localized in intracellular membranes. For this purpose, we used the CX2 colon carcinoma cells separated by fluorescence-associated cell sorter into stable CX+ and CX− subclones with >80% and <20% of cells positive for surface Hsp70, respectively (17). Notably, the total level of Hsp70 in CX+ and CX− cells was indistinguishable (17). Immunoelectron microscopy revealed high levels of Hsp70 on the plasma membrane and in the membranes of the intracellular vacuolar structures of CX+ cells (Fig. 5 A). Costaining with cathepsin D showed a colocalization of cathepsin D and Hsp70 in the intracellular vesicles as well as on the cell surface in CX+ (Fig. 5, B and C), but not in CX− cells (Fig. 5 D). Quantification of the costaining in three CX− and four CX+ cells revealed that, on average, 10% ± 1% and 62% ± 25% of cathepsin D positive vesicles contained Hsp70, respectively (Fig. 5 E). Thus, tumor-associated surface expression of Hsp70 was associated with the localization of Hsp70 to the endosomal/lysosomal compartment.
Figure 5.

Hsp70 is localized on lysosomal membranes. Representative cryo-ultramicrotomy images of Hsp70 membrane positive CX+ (A–C) and membrane negative CX− (D) tumor cells labeled either with a murine anti-Hsp70 antibody alone (A) or together with rabbit anti-cathepsin D antibodies (B–D) are shown. (A) Localization of Hsp70 (10 nm gold particles) can be detected in the cytosol, plasma membrane and intracellular vesicles of CX+ tumor cells. (B and C) Colocalization of Hsp70 (visualized by 10-nm gold particles) and cathepsin D (visualized by 5-nm gold particles) in intracellular vesicles and on the plasma membrane can be detected in CX+ cells. (D) In CX− tumor cells, only minimal amounts of Hsp70 were detectable in cathepsin D positive vesicles. (E) The percentage of Hsp70 positive vesicles of all cathepsin D positive vesicles was calculated in pictures of 3 CX− and 4 CX+ cells. The number of cathepsin D positive vesicles/picture is indicated (top of bars). (F) iMEF-hsp-4 cells were fractionated as described in Materials and Methods, and the proteins from the indicated fractions originating from the indicated amount of cells were analyzed by immunoblotting with rabbit and goat antibodies against Hsp70 and cathepsin B, respectively. Where indicated, the samples were incubated with a buffer containing 800 μg/ml digitonin to permeabilize the membranes. Cyt, cytoplasm; HM, heavy membrane; LM, light membrane; S-LM, supernatant of the LM.
To address more directly whether Hsp70 is attached to the membranes of the endosomal/lysosomal compartment, next we fractionated the iMEFs originating from the Hsp70 transgenic animals and performed immunoblot analysis of proteins from various cellular fractions. As expected, most of the Hsp70 was found in the cytosolic and heavy membrane fractions (Fig. 5 F). However, considerable amount of Hsp70 was detected in the light membrane fraction (cellular organelles including endosomes and lysosomes) together with most of the cellular cathepsin B. Permeabilization of the organelles in the light membrane fraction with digitonin resulted in the total release of cathepsin B into the supernatant, whereas a large part of Hsp70 remained in the pellet, suggesting that it was bound to the membranes of the intracellular organelles.
To examine whether the lysosomal localization of Hsp70 had any effect on the lysosomal stability permeability and size, we first measured the longest diameters of 19 and 13 cathepsin D positive vesicles in CX+ and CX− cells, respectively, and calculated the theoretical volumes of the vesicles. As demonstrated in the Fig. 6 A, the average volume of cathepsin D positive vesicles was almost four times larger in CX+ than in CX− cells. Immunoelectron microscopy does not allow the estimation of the number of the vesicles/cell and, therefore, next we measured the total volume of the acidic compartment in CX+ and CX− cells by flow cytometry after staining with acridine orange, a lysosomotrophic base that accumulates in the acidic lysosomal compartment and emits red fluorescence when in high concentration. Based on three independent measurements, the mean intensity of the red fluorescence was 2.13 ± 0.47 times higher (P = 0.003) in CX+ cells than in CX− cells (Fig. 6 B). Similar results were obtained when comparing Hsp70-transfected and vector-transfected WEHI-S cells (1.89 ± 0.20 times higher; P = 0.016; Fig. 6 C) as well as Hsp70-transgenic and wild-type iMEFs (1.77 ± 0.29 times higher; P = 0.01; Fig. 6 D). The increased volume of the acidic compartment as well as the larger size of individual lysosomes in Hsp70-expressing cells was also evident when cells stained with acridine orange were visualized by confocal microscopy (Fig. 6 E). Furthermore, the increase in the volume of the acidic compartment correlated with the increased total cysteine cathepsin activity (Fig. 6, F and G) and protein level as analyzed by immunoblotting (not depicted), suggesting that, indeed, Hsp70 stabilized lysosomal membranes.
Figure 6.

Hsp70 increases the volume of the acidic compartment and total cellular cysteine cathepsin activity. (A) The diameters of cathepsin D positive vesicles in representative cryo-ultramicrotomy images (magnification, 23,000) of CX+ (n = 19) and CX− (n = 13) tumor cells labeled with cathepsin D antibody were measured. The values present theoretical volumes of the vesicles calculated with the formula 4/3 × πr3 ± SD. P < 0.05. (B–E) CX+ and CX− (B), WEHI-hsp-2 and WEHI-V-2 (C), and iMEF-hsp-4 and iMEF-wt-5 (D and E) cells were labeled with acridine orange, and the total volume of the acidic compartment (VAC) was measured by flow cytometry (B–D) or visualized by confocal microscopy (E). (F and G) The total cysteine cathepsin activities in cell lysates of CX+ and CX− cells (F) and vector-transfected (V-2 and V-3) and Hsp70-transfected (Hsp-2 and Hsp-18) WEHI-S clones (G) were measured as described in Materials and Methods. The values represent means of triplicate determinations. All experiments were repeated two to four times with essentially similar results.
Next, we compared the stability of CX+ and CX− lysosomes by analyzing the LMP after exposure to oxidative stress (H2O2) or photolysis (blue light, 450–500 nm). For this purpose, lysosomes were stained and activated with acridine orange before the challenge, and the permeabilization of lysosomes was analyzed by measuring the emission of green fluorescence indicative of the leakage of acridine orange from the acidic compartment to the cytosol (10). The Hsp70 positive lysosomes of CX+ cells were significantly more resistant to H2O2 than Hsp70 negative lysosomes of CX− cells (Fig. 7 A). Although 30-min treatment with 5 μM H2O2 triggered a 1.9-fold increase in the green fluorescence in CX− cells, >10 times higher concentrations were required for a similar effect in CX+ cells. The decreased permeability of the lysosomes correlated with the increased survival of the CX+ cells as analyzed by the MTT reduction assay 24 h after the H2O2 treatment (Fig. 7 B). Furthermore, CX+ cells were clearly more resistant to γ-irradiation–induced apoptosis than CX− cells (Fig. 7 C). Furthermore, lysosomes of CX+ cells tolerated longer exposures to blue light than those of CX− cells (Fig. 7 D). Also, the lysosomes of Hsp70-transgenic iMEFs tolerated blue light better than those of wild-type iMEFs (Fig. 7 E). Counting of acridine orange positive (red) lysosomes before and immediately after 25-s exposure to blue light showed that, whereas 36.7 ± 7.5% of lysosomes in wild-type cells (nine cells) had permeabilized, only 16.2 ± 10.4% of lysosomes in Hsp70-transgenic iMEFs had lost the red staining (P < 0.001).
Figure 7.
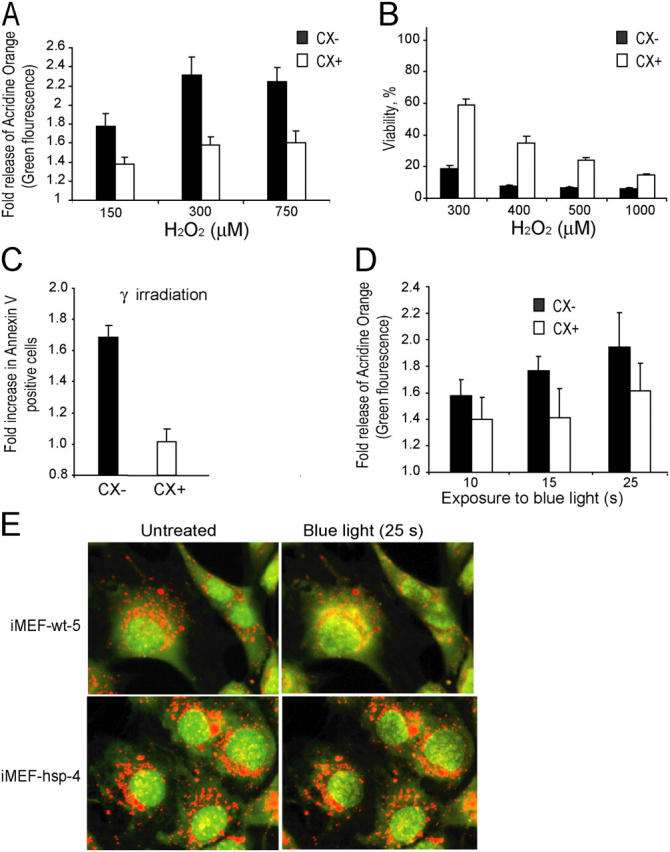
Hsp70 protects lysosomal membranes against permeabilization induced by oxidative stress or photolysis. (A–D) CX− (black bars) and CX+ (white bars) cells were treated for 30 min (A) or 24 h (B) with indicated concentrations of H2O2, exposed to 2 × 10 Gy γ-irradiation (C) or indicated times of blue light (D). Cells prestained with acridine orange were analyzed immediately afterwards (A) or 45 min after the treatment (D) for lysosomal leakage by flow cytometry (green fluorescence). The values represent the fold increases in the mean intensity of the green fluorescence. (B) The viability of the cells was analyzed 24 h after the treatment by the MTT reduction assay and is expressed as the percentage of untreated cells. (C) The number of apoptotic cells was analyzed by annexin V staining and is expressed as fold increase as compared with the untreated cells. (E) Wild-type and Hsp70-expressing iMEFs prestained with acridine orange were exposed to the 488-nm laser of the confocal microscope (10-s preexposure and 15-s imaging). Representative cells are shown. Notice the better preservation of the lysosomes in Hsp70-expressing cells. The experiments were repeated twice with similar results. The values represent averages ± SD of three independent experiments (A–D) or averages of 12 measurements.
Discussion
The data presented here demonstrate that Hsp70 is localized to the membranes of the endosomal/lysosomal compartment of tumor cells and that it inhibits lysosomal permeabilization induced by such diverse stimuli as cytokines, anticancer drugs, γ-irradiation, oxidative stress, and photolysis. Furthermore, the mere depletion of Hsp70 from tumor cells triggered LMP and cathepsin-mediated PCD. Thus, the main mechanism by which Hsp70 confers a survival advantage to tumor cells appears to be the inhibition of the permeabilization of lysosomal membranes and/or membranes of other vesicles containing cathepsins. This may explain the widely demonstrated ability of Hsp70 to protect tumor cells against the diverse death stimuli that all trigger LMP, but may kill cells in a caspase-dependent or -independent manner (15, 35, 36). Solid tumors commonly show increased expression levels, plasma membrane association, and secretion of cathepsins (37). Such changes have been considered tumor promoting because they often correlate positively with invasive and metastatic capacity of tumors. However, accumulating data suggest that the increased expression and altered trafficking of lysosomal proteases may also form an “Achilles heel” for the tumor because stress-induced leakage of cathepsins from the lysosomal compartment to the cytosol can trigger PCD (35, 36, 38). Data presented here clearly demonstrate that tumor cells can escape the death induced by lysosomal proteases by increasing the expression of Hsp70, which effectively inhibits the release of lysosomal proteases into the cytosol. The membrane localization of Hsp70 appears to be crucial for its ability to inhibit LMP. This is demonstrated here using CX+ and CX− sublines of CX2 colon cancer cells that differ from each other only in their membrane expression, but not in the total level, of Hsp70 (17). Hsp70 plasma membrane expression has been found in >80% of freshly isolated human biopsy samples from solid tumors of various origins and leukemic blasts from patients with acute myelogenous leukemia (16, 39). Because normal tissues and bone marrows of healthy human individuals do not express Hsp70 on the cell surface, Hsp70 can be considered a tumor-selective membrane structure. Supporting our data, showing that Hsp70 is directly associated with the membranes, it has been shown previously that it binds lipids in vitro (40). How Hsp70 localizes to the lysosomes and plasma membrane of tumor cells is still an open question. The inability of inhibitors of endoplasmic reticulum (Golgi route; i.e., brefeldin A and colchicine) to inhibit the plasma membrane localization of Hsp70 suggests that the transport occurs via an alternative cytoplasmic protein export route (41). Interestingly, only the cells with surface expression of Hsp70 had Hsp70 also in the membranes of endosomal/lysosomal compartment and showed plasma membrane expression of cathepsin D. Thus, Hsp70 may be transported to the plasma membrane together with cathepsins via the lysosomal exocytosis and back to the cells via endocytosis. It should be noted here that Hsc70, the cognate member of the Hsp70 family, also associates with lysosomes (42). Together with numerous cochaperones, it assists the delivery of proteins to be degraded in the lysosomal compartment. Thus, Hsp70 may also be involved in the delivery of proteins to the lysosomes and the protective effect could be due to the removal of toxic proteins or protein aggregates.
Interestingly, both increased expression and secretion of cathepsins as well as sensitivity to the lysosomal death pathway are associated with an aggressive tumor phenotype (34, 37). We are presently studying whether the lysosomal localization of Hsp70 and cathepsin secretion are coregulated during tumorigenesis. Because the mere depletion of Hsp70 from tumor cells, but not from normal cells, triggered LMP, one might speculate that Hsp70 is required for the stability of the tumor-specific subpopulation of lysosomes, possibly the same subpopulation that is responsible for the transport of cathepsins to the plasma membrane and extracellular space where they contribute to the invasiveness of the tumor. Thus, the better understanding of the mechanisms underlying the lysosomal localization and function of Hsp70 may provide promising targets for the future cancer therapy.
Acknowledgments
We thank D. Wissing, B. Poulsen, and I. Fossar Larsen for excellent technical assistance and G. Pagoulatos, G. Kollias, J. Palmer, A. Cerami, J. Lukas, and P. Briand for invaluable research tools.
This work was supported by grants from the Danish Cancer Society, the Danish Medical Research Council, the European Commission 5th framework program, the Novo Foundation, and the Association for International Cancer Research.
The authors have no conflicting financial interests.
J. Nylandsted and M. Gyrd-Hansen contributed equally to this work.
Abbreviations used in this paper: β-Gal, β-galactosidase; Ad., adenoviral; AFC, 7-amino-trifluoromethylcoumarin; as, antisense; CHX, cycloheximide; Hsp70, heat shock protein 70; iMEF, immortalized MEF; LDH, lactate hydrogenase; LHVS, Mu-Leu-HphVSPh; LMP, lysosomal membrane permeabilization; MEF, murine embryonic fibroblast; MTT, 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide; NAG, β-N-acetyl-glucosaminidase; PCD, programmed cell death; zFA-fmk, z-Phe-Ala-CH2F; zVAD-fmk, z-Val-Ala-DL-Asp-CH2F.
References
- 1.Hanahan, D., and R.A. Weinberg. 2000. The hallmarks of cancer. Cell. 100:57–70. [DOI] [PubMed] [Google Scholar]
- 2.Leist, M., and M. Jäättelä. 2001. Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2:589–598. [DOI] [PubMed] [Google Scholar]
- 3.Jäättelä, M., and J. Tschopp. 2003. Caspase-independent cell death in T lymphocytes. Nat. Immunol. 4:416–423. [DOI] [PubMed] [Google Scholar]
- 4.Ferri, K.R., and G. Kroemer. 2001. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3:E255–E263. [DOI] [PubMed] [Google Scholar]
- 5.Guicciardi, M.E., J. Deussing, H. Miyoshi, S.F. Bronk, P.A. Svingen, C. Peters, S.H. Kaufmann, and G.J. Gores. 2000. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 106:1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foghsgaard, L., D. Wissing, D. Mauch, U. Lademann, L. Bastholm, M. Boes, F. Elling, M. Leist, and M. Jäättelä. 2001. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 153:999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunk, U.T., and I. Svensson. 1999. Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep. 4:3–11. [DOI] [PubMed] [Google Scholar]
- 8.Yuan, X.M., W. Li, H. Dalen, J. Lotem, R. Kama, L. Sachs, and U.T. Brunk. 2002. Lysosomal destabilization in p53-induced apoptosis. Proc. Natl. Acad. Sci. USA. 99:6286–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broker, L.E., C. Huisman, S.W. Span, J.A. Rodriguez, F.A. Kruyt, and G. Giaccone. 2004. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 64:27–30. [DOI] [PubMed] [Google Scholar]
- 10.Brunk, U.T., H. Dalen, K. Roberg, and H.B. Hellquist. 1997. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic. Biol. Med. 23:616–626. [DOI] [PubMed] [Google Scholar]
- 11.Bidere, N., H.K. Lorenzo, S. Carmona, M. Laforge, F. Harper, C. Dumont, and A. Senik. 2003. Cathepsin D triggers Bax activation, resulting in selective AIF relocation in T lymphocytes entering the early commitment phase to apoptosis. J. Biol. Chem. 278:31401–31411. [DOI] [PubMed] [Google Scholar]
- 12.Boya, P., K. Andreau, D. Poncet, N. Zamzami, J.L. Perfettini, D. Metivier, D.M. Ojcius, M. Jäättelä, and G. Kroemer. 2003. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J. Exp. Med. 197:1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberg, K., K. Kagedal, and K. Ollinger. 2002. Microinjection of cathepsin d induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 161:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vancompernolle, K., F. Van Herreweghe, G. Pynaert, M. Van de Craen, K. De Vos, N. Totty, A. Sterling, W. Fiers, P. Vandenabeele, and J. Grooten. 1998. Atractyloside-induced release of cathepsin B, a protease with caspase-processing activity. FEBS Lett. 438:150–158. [DOI] [PubMed] [Google Scholar]
- 15.Jäättelä, M. 1999. Heat shock proteins as cellular lifeguards. Ann. Med. 31:261–271. [DOI] [PubMed] [Google Scholar]
- 16.Hantschel, M., K. Pfister, A. Jordan, R. Scholz, R. Andreesen, G. Schmitz, H. Schmetzer, W. Hiddemann, and G. Multhoff. 2000. Hsp70 plasma membrane expression on primary tumor biopsy material and bone marrow of leukemic patients. Cell Stress Chaperones. 5:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Multhoff, G., C. Botzler, L. Jennen, J. Schmidt, J. Ellwart, and R. Issels. 1997. Heat shock protein 72 on tumor cells: a recognition structure for natural killer cells. J. Immunol. 158:4341–4350. [PubMed] [Google Scholar]
- 18.Nylandsted, J., W. Wick, U.A. Hirt, K. Brand, M. Rohde, M. Leist, M. Weller, and M. Jäättelä. 2002. Eradication of glioblastoma, and breast and colon carcinoma xenografts by hsp70 depletion. Cancer Res. 62:7139–7142. [PubMed] [Google Scholar]
- 19.Jäättelä, M. 1995. Overexpression of hsp70 confers tumorigenicity to mouse fibrosarcoma cells. Int. J. Cancer. 60:689–693. [DOI] [PubMed] [Google Scholar]
- 20.Vargas-Roig, L.M., F.E. Gago, O. Tello, J.C. Aznar, and D.R. Ciocca. 1998. Heat shock protein expression and drug resistance in breast cancer patients treated with induction chemotherapy. Int. J. Cancer. 73:468–475. [DOI] [PubMed] [Google Scholar]
- 21.Jäättelä, M. 2004. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 23:2746–2756. [DOI] [PubMed] [Google Scholar]
- 22.Xanthoudakis, S., and D.W. Nicholson. 2000. Heat-shock proteins as death determinants. Nat. Cell Biol. 2:E163–E165. [DOI] [PubMed] [Google Scholar]
- 23.Ravagnan, L., S. Gurbuxani, S.A. Susin, C. Maisse, E. Daugas, N. Zamzami, T. Mak, M. Jäättelä, J.M. Penninger, C. Garrido, and G. Kroemer. 2001. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 3:839–843. [DOI] [PubMed] [Google Scholar]
- 24.Creagh, E.M., R.J. Carmody, and T.G. Cotter. 2000. Heat shock protein 70 inhibits caspase-dependent and -independent apoptosis in Jurkat T cells. Exp. Cell Res. 257:58–66. [DOI] [PubMed] [Google Scholar]
- 25.Gabai, V.L., K. Mabuchi, D.D. Mosser, and M.Y. Sherman. 2002. Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol. Cell. Biol. 22:3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosser, D.D., A.W. Caron, L. Bourget, A.B. Meriin, M.Y. Sherman, R.I. Morimoto, and B. Massie. 2000. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol. Cell. Biol. 20:7146–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabai, V.L., J.A. Yaglom, V. Volloch, A.B. Meriin, T. Force, M. Koutroumanis, B. Massie, D.D. Mosser, and M.Y. Sherman. 2000. Hsp72-mediated suppression of c-Jun N-terminal kinase is implicated in development of tolerance to caspase-independent cell death. Mol. Cell. Biol. 20:6826–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jäättelä, M., D. Wissing, K. Kokholm, T. Kallunki, and M. Egeblad. 1998. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 17:6124–6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nylandsted, J., M. Rohde, K. Brand, L. Bastholm, F. Elling, and M. Jäättelä. 2000. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA. 97:7871–7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angelidis, C.E., C. Nova, I. Lazaridis, D. Kontoyiannis, G. Kollias, and G.N. Pagoulatos. 1996. Overexpression of HSP70 in transgenic mice results in increased cell thermotolerance. Transgenics. 2:111–117. [Google Scholar]
- 31.Palmer, J.T., D. Rasnick, J.L. Klaus, and D. Bromme. 1995. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 38:3193–3196. [DOI] [PubMed] [Google Scholar]
- 32.Ekdahl, C.T., P. Mohapel, E. Weber, B. Bahr, K. Blomgren, and O. Lindvall. 2002. Caspase-mediated death of newly formed neurons in the adult rat dentate gyrus following status epilepticus. Eur. J. Neurosci. 16:1463–1471. [DOI] [PubMed] [Google Scholar]
- 33.Lademann, U., K. Cain, M. Gyrd-Hansen, D. Brown, D. Peters, and M. Jäättelä. 2003. Diarylurea compounds inhibit caspase activation by preventing the formation of the active 700-kilodalton apoptosome complex. Mol. Cell. Biol. 23:7829–7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fehrenbacher, N., M. Gyrd-Hansen, B. Poulsen, U. Felbor, T. Kallunki, M. Boes, E. Weber, M. Leist, and M. Jäättelä. 2004. Sensitization to the lysosomal death pathway upon immortalization and transformation. Cancer Res. In press. [DOI] [PubMed] [Google Scholar]
- 35.Brunk, U.T., J. Neuzil, and J.W. Eaton. 2001. Lysosomal involvement in apoptosis. Redox Rep. 6:91–97. [DOI] [PubMed] [Google Scholar]
- 36.Leist, M., and M. Jäättelä. 2001. Triggering of apoptosis by cathepsins. Cell Death Differ. 8:324–326. [DOI] [PubMed] [Google Scholar]
- 37.Roshy, S., B.F. Sloane, and K. Moin. 2003. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 22:271–286. [DOI] [PubMed] [Google Scholar]
- 38.Mathiasen, I.S., and M. Jäättelä. 2002. Triggering caspase-independent cell death to combat cancer. Trends Mol. Med. 8:212–220. [DOI] [PubMed] [Google Scholar]
- 39.Gehrmann, M., H. Schmetzer, G. Eissner, T. Haferlach, W. Hiddemann, and G. Multhoff. 2003. Membrane-bound heat shock protein 70 (Hsp70) in acute myeloid leukemia: a tumor specific recognition structure for the cytolytic activity of autologous NK cells. Haematologica. 88:474–476. [PubMed] [Google Scholar]
- 40.Arispe, N., M. Doh, and A. De Maio. 2002. Lipid interaction differentiates the constitutive and stress-induced heat shock proteins Hsc70 and Hsp70. Cell Stress Chaperones. 7:330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Multhoff, G., and L.E. Hightower. 1996. Cell surface expression of heat shock proteins and the immune response. Cell Stress Chaperones. 1:167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiang, H.-L., S. Terlecky, C.P. Plant, and J.F. Dice. 1989. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 246:382–385. [DOI] [PubMed] [Google Scholar]