

Abstract
Rapid clearance of pathogens is essential for successful control of pyogenic bacterial infection. Previous experiments have shown that antibody to specific intracellular adhesion molecule-grabbing nonintegrin (SIGN)-R1 inhibits uptake of capsular polysaccharide by marginal zone macrophages, suggesting a role for SIGN-R1 in this process. We now demonstrate that mice lacking SIGN-R1 (a mouse homologue of human dendritic cell–SIGN receptor) are significantly more susceptible to Streptococcus pneumoniae infection and fail to clear S. pneumoniae from the circulation. Marginal zone and peritoneal macrophages show impaired bacterial recognition associated with an inability to bind T-independent type 2 antigens such as dextran. Our work represents the first evidence for a protective in vivo role for a SIGN family molecule.
Keywords: DC-SIGN, marginal zone macrophages, microbial infection, innate immunity, lectin
Introduction
Pathogen recognition by the immune system is crucial for the maintenance of protective immunity. Pattern recognition receptors (PRRs), including C-type lectins and Toll-like receptors, discriminate molecular patterns expressed by pathogens and facilitate differential recognition of pathogens and microbial products (for review see reference 1). These innate immune responses provide a critical, rapid defense mechanism that acts before the maturation of acquired immunity. However, there are occasions when PRRs become hijacked by pathogens and used to facilitate infection and circumvent immune detection. A recently identified example of this phenomenon is HIV-1 and Dengue virus usurping DC-specific intracellular adhesion molecule-grabbing nonintegrin (DC-SIGN; CD209) and DC-SIGNR molecules to facilitate infection (for review see reference 2). Therefore, these SIGN molecules appear to represent an immunological liability. This raises an unresolved question: what positive role, if any, do the SIGN molecules play in protective immunity?
Human DC-SIGN is expressed on the surface of human dendritic cells and is believed to play a key role in the initial interaction between DCs and naive T cells through interaction with ICAM-3 (3). However, it has also been demonstrated that HIV-1 exploits DC-SIGN, using it as a receptor to facilitate viral transport via DCs from mucosal surfaces to CD4+ T cells in secondary lymphoid tissue (4) through its lectin domain interacting with gp120 (5, 6). Recent studies have also highlighted a role for DC-SIGN in the infection of DCs by Ebola virus (7), human cytomegalovirus (8), Leishmania pifanoi (9), Mycobacterium tuberculosis (10), and Dengue virus (11). After the description of DC-SIGN, the closely related molecule DC-SIGNR (DC-SIGN2, L-SIGN) was reported, with its gene mapping within a few tens of kilobases of both DC-SIGN and CD23 (another C-type lectin; references 12, 13). DC-SIGNR has also been shown to bind HIV-1 (14) and Dengue virus (11), and it has been suggested that its expression on liver sinusoidal endothelial cells may facilitate clearance of antigenic proteins from the circulation (15).
The mouse genome encodes five DC-SIGN homologues that all map close to the mouse CD23 gene on mouse chromosome 8 (16–18). These mouse genes have been termed DC-SIGN and SIGN-R1–SIGNR4, with DC-SIGN mapping closest to CD23 as in the human (16). The mouse DC-SIGN family contains highly homologous carbohydrate recognition domains (CRDs), but the individual members differ in the numbers of neck repeats or the presence of a transmembrane domain (16). Expression studies also suggest that these molecules are differentially expressed in various tissues, suggesting that they may play tissue-specific roles (16–18). It is noteworthy that mouse DC-SIGN has been reported to be highly expressed by splenic DCs in a manner similar to that described for human DC-SIGN, whereas the other homologues are not as highly represented in the splenic DC compartment (16–18). Furthermore, recent studies have reported that, despite their having related mannose-binding motifs, the various mouse SIGN molecules display differential ligand specificity with the potential to recognize different pathogens (19, 20).
The highly organized microarchitecture of the spleen is intimately involved with the effective clearance of pathogens by the immune system. The spleen is divided into regions of white and red pulp separated by the marginal zones (MZs). The cellular composition of the marginal zone includes reticular cells, MZ B cells, DCs, metallophilic macrophages, and MZ macrophages (MZMs). It is in the MZ that the blood flow is slowed down, as the terminal arterioles open into venous sinuses, producing an environment for the efficient entrapment of blood-borne particles by resident phagocytes (21). The MZMs are highly phagocytic cells that are found in layers dispersed throughout the MZ and are defined by their expression of the cell surface molecules recognized by the antibodies ER-TR9 and MARCO (22, 23). Selective depletion of MZMs and metallophilic macrophages using clodronate liposomes identified that these cells are essential for trapping of microspheres and Listeria monocytogenes (24). Furthermore, MZMs have been identified as critical phagocytes for the uptake of neutral polysaccharides, such as Ficoll and dextran, which represent thymus independent type 2 (TI-2) antigens (25). Significantly, this uptake has been demonstrated to be inhibited by the ER-TR9 antibody (26). A recent paper from Kang et al. has indicated that antibody to SIGN-R1 can also inhibit capsular polysaccharide uptake by MZMs, but was unable to inhibit MZM uptake of pneumococci, leading the authors to suggest that SIGN-R1–independent recognition systems exist (27). SIGN-R1 expression has also been demonstrated on peritoneal macrophages, and in vitro assays suggest that they may play a role in mannose-mediated nonopsonic recognition of yeast cells (28). Significantly, peritoneal macrophages are strategically located to play an important role in protection against bacterial infection, acting as phagocytes and producing proinflammatory cytokines.
Little is known regarding the biological function of SIGN molecules. Given the subversion of human DC-SIGN and DC-SIGNR by several human pathogens, we wished to determine whether the SIGN-R1 molecule represents an immunological liability or a functionally protective immunoreceptor in vivo. To achieve this goal, we generated SIGN-R1−/− mice using homologous recombination. MZMs from the SIGN-R1−/− mice do not stain with ER-TR9 and fail to bind the TI-2 antigen dextran. Significantly, we demonstrate that SIGN-R1 is critical for survival after infection with the gram-positive bacterial pathogen Streptococcus pneumoniae. SIGN-R1 binds S. pneumoniae, facilitating efficient phagocytosis and bacterial clearance, leading to protection against septicemia.
Materials and Methods
Cloning of Mouse SIGN-R1.
EST accession no. AI386429 (IMAGE clone 597911) was identified as homologous to mCD23 and human DC-SIGN. Oligonucleotide primers (5′-ATCTCCCAAGGGAAGAATGAGTCC-3′ and 5′-CTGCAGGAAGGTCTGCTCTTCATC-3′) were generated using this EST sequence information and tested for the generation of DNA product from mouse genomic DNA. These primers produced a PCR product that was subcloned, sequenced, and used as a probe to screen mouse PAC genomic library RPCI-21 (HGMP UK; reference 29). RPCI-21 clone 424-C19 was identified and shotgun cloned after BamHI restriction digest into pUC or pBluescript. These colonies were rescreened for hybridization, and positives were sequenced using transposon integration (Genome Priming System; Biolabs). The genomic sequence was completed using sequence deposited in the public database from BAC clones RPCI-23-186C23 (accession no. AC073706) and RPCI-23-458N3 (accession no. AC073804). The mouse SIGN-R1 cDNA was generated by PCR using oligonucleotides 5′-CACCAGGGGACAGCGGCAACC-3′ and 5′-CTAGCCTTCAGTGCATGGGGTTGC-3′ with cDNA reverse transcribed from total splenocyte RNA as template.
Targeted Disruption of the Mouse SIGN-R1 Gene in ES Cells.
The targeting vector consisted of a 2.2-kb 5′ arm of homology and a 3.8-kb of 3′ homology. The replacement vector was constructed to insert the arms of homology on either side of a loxP-flanked neomycin resistance gene. The 5′ arm of homology was generated by PCR using primers 5′-CATTCAAGTGCGGCCGCATTCTAACCCCAGC-3′ and 5′-AATGTCGGATCCCAGGGGCAGAGTCGACACAGG-3′ to give a NotI site at the 5′ end and a BamHI site at the 3′ end. The 3′ arm of homology was generated by PCR using primers 5′-CATCATCTAGAGTGATGAAGAGCAGGTACATTTAGTG-3′ and 5′-CTACAATCTAGAATTGTACGTCAAATAATCC-3′ to give XbaI sites at either end of the fragment. The targeting vector was linearized with NotI and electroporated into E14.1 ES cells (30). Of 396 G418 resistant clones screened by Southern analysis, using a probe (probe A) produced with PCR primers (5′-GTGAGAAAGCAGCAGTCCAAGC-3′ and 5′-TCAAATCTTCAGGCCCTCCAG-3′), 3 were targeted correctly. Hybridization with a probe to the neomycin sequence confirmed the predicted size of the targeted fragment and that only a single integration had occurred. Targeted ES cells were microinjected into 3.5-d C57BL/6 blastocysts to generate chimeras. These mice were mated with C57BL/6 mice and transmitted the ES cell genotype through the germline. Mice homozygous for the disrupted SIGN-R1 gene were obtained by interbreeding the heterozygotes. Genotyping was undertaken routinely using a PCR screening protocol in which three oligonucleotides were combined in a single reaction tube to identify wild-type and targeted alleles. The oligonucleotides were 5′-CTATCAGGACATAGCGTTGGCTACC-3′, 5′-GCAGGAGAAGATCTACCAACAGCTG-3′, and 5′-GAAGAAGTAACAATTTCCTAGGAG-3′ and result in the generation of a 550-bp fragment from the wild-type allele and a 750-bp product from the targeted allele containing the neomycin gene. The SIGN-R1−/− and wild-type animals used in these experiments were maintained on a 129 × C57BL/6 (F2) background in a specific pathogen-free environment in accordance with UK Home Office guidelines. All animal experiments outlined in this paper were undertaken with the approval of the UK Home Office or the Department of Health and Children, Ireland.
Detection of SIGN-R1 RNA Transcripts in Spleen.
Total RNA was prepared from splenocytes using RNAzolB. RT-PCR primers for mouse SIGN-R1 were 5′-CTGAAAACTGAACTCTTGTCCAGG-3′ and 5′-CTGCTGCAGGAAGGTCTGCTC-3′. The internal oligonucleotide used for hybridization was 5′-CAGCTGGTACAGATGAAGACTG-3′. HPRT primers and conditions were as described previously (31). Additional SIGN-R1 primers were: 880, 5′-CTCCTAGGAAATTGTTACTTCTTC-3′; 969, 5′-ATGAGTGACTCCACAGAAGCC-3′; 1175, 5′-CTAGCCTTCAGTGCATGGGGTTGC-3′; 1266, 5′-CTGAAAACTGAACTCTTGTCCAGG-3′; 1267, 5′-CTGCTGCAGGAAGGTCTGCTC-3′; 2152, 5′-TTGGCTGGGCTCCTGCTGATC-3′; 2153, 5′-CTGCATGGACTCATTCTTCCC-3′; and 2154, 5′-TTCTCCTGCTTAGAATCATCC-3′.
Histochemistry and Fluorescence Microscopy.
For histochemistry, spleens were removed, embedded in OCT, frozen in liquid nitrogen, and kept at −80°C until sectioning. 10-μm cryosections were prepared and stored at −80°C until use. Sections were fixed for 10 min at room temperature with acetone and washed five times with PBS. After blocking (10% FCS in PBS, 30 min at room temperature), sections were incubated with ER-TR9 antibody for 1 h at room temperature, washed, and stained with Vectastain ABC Elite kit for horseradish peroxidase before being washed again and developed using a DAB kit (Vectastain). Sections were washed and mounted with VectaMount. For fluorescence analysis, sections were fixed and blocked as described before, incubated with the primary antibody (ER-TR9) for 1 h at room temperature, washed, and stained with affinity-purified, biotin-conjugated rabbit anti–rat immunoglobulin for 1 h at room temperature. After washing, the sections were incubated with Texas red–conjugated streptavidin (BioMeda) for 1 h at room temperature. Biotin-conjugated anti-CD11b (clone M1/70; BD Biosciences) or FITC-conjugated anti–MOMA-1 (Serotec) was also included at this point if appropriate for double staining. Sections were washed in PBS and mounted with Vectashield (Vectalabs). Visualization was performed using an MRC1024 confocal microscope (Bio-Rad Laboratories).
Inoculation with Dextran–Alexa Fluor 488.
Mice were inoculated into the tail vein with PBS containing 200 μg of dextran–Alexa Fluor 488 (molecular weight of 10,000; Molecular Probes). Coinoculation in the presence of 5% carbon particles (Indian ink) was also performed. After 2 h, the mice were killed; spleen was removed and embedded in OCT compound; and sections were prepared as described before. Visualization was performed using an MRC1024 confocal microscope (Bio-Rad Laboratories) or an Axioplan fluorescence microscope (Carl Zeiss MicroImaging, Inc.).
Phosphorylcholine (PC)-specific ELISA.
96-well plates were coated with 25 μg/ml PC conjugated to bovine sera albumin (Biosearch Technologies) overnight at 4°C. Plates were blocked with PBS/10% FCS and washed, and serial dilutions of serum were incubated overnight at 4°C. A positive control (pooled serum from seven wild-type mice 14 d after immunization with Pneumovax II; Aventis-Pasteur MSD) was included. Detection was performed using a biotinylated goat anti–mouse immunoglobulins (BD Biosciences). Horseradish peroxidase streptavidin (ICN Biomedicals) activity was revealed by 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) substrate (Sigma-Aldrich). Plates were read at 405 nm. Antibody titres were calculated relative to the positive control and compared using an unpaired Student's t test (Welch corrected).
Bacteria.
S. pneumoniae type 2 strain D39 and type 14 provided by J.S. Brown (Imperial College School of Medicine, London, England) and D. Goldblatt (University College Hospital, London, England) were cultured overnight on blood agar plates (5% CO2, 95% air, 37°C), inoculated into Todd-Hewitt broth (Oxoid Ltd.), supplemented with 0.5% yeast extract (Oxoid Ltd.), cultured for 4–5 h, washed, and resuspended at 109 CFU/ml (estimated by OD660 = 1); aliquots were stored at −70°C and made up in sterile PBS for use. Their concentration was verified by serial dilution and culture on blood agar plates.
S. pneumoniae Peritonitis.
Groups of 10–12 male or female, age-matched control and SIGN-R1−/− mice (8–12 wk of age) were inoculated intraperitoneally with 200 μl of PBS containing S. pneumoniae. Mice were observed at least every 6 h for the first 72 h and every 8 h until 96 h, and daily thereafter. During observation, mice were scored by a blinded observer for the presence or absence of physical signs of progressive sepsis in mice. Sickness was scored according to the presence of piloerection (1 point), plus hunched posture (2 points) and lack of spontaneous movement (3 points). Mice that became moribund were considered to have reached the end point of the experiment and were killed. Tail bleeds were performed at 26 or 30 h after infection, and blood was cultured for bacterial growth for 24 h. Survival data was analyzed using the Kaplan-Meier graphs and Log rank tests. All animal experiments were performed in accordance with UK Home Office regulations.
Phagocytosis Assay.
S. pneumoniae type 2 was cultured to log phase in Todd-Hewitt broth with 0.5% yeast extract (Oxoid Ltd.), heat inactivated at 60°C for 1 h, and labeled with FITC (Sigma-Aldrich). FITC-labeled S. pneumoniae were incubated in PBS or a 1-in-10 dilution of heat-inactivated nonimmune serum at 37°C for 1 h before washing. Nonimmune serum used for opsonization was taken from 2–10 control or age-matched SIGN-R1−/− mice. Peritoneal macrophages or RAW-297 cells were adhered to plastic, and aliquots of serum-opsonized or nonopsonized FITC-labeled pneumococci were added at 4°C (to assess binding) and 37°C (to assess phagoctyosis) for 30 min. Adhered macrophages were washed, harvested, and analyzed by flow cytometry (FACScalibur; Becton Dickinson) as described previously (32). Peritoneal macrophages were identified by scatter characteristics and Mac-1 staining. The percentage of FITC positive macrophages was used as a measure of phagocytosis. Duplicate or triplicate wells were processed for each serum sample, and results were compared using the Student's t test.
Binding Assay.
SIGN-R1 or control vector transduced NIH3T3 fibroblasts (28) were incubated with FITC-labeled S. pneumoniae type 2 at 4°C for 1 h. Samples were washed and analyzed by flow cytometry (FACScalibur; Becton Dickinson). The percentage of FITC positive cells and the geometric mean fluorescence were used as a measure of pneumococcal binding. Triplicate wells were processed, and results were compared using the Student's t test.
Inoculation with FITC-labeled S. pneumoniae.
Mice were inoculated into the tail vein with PBS containing 107 FITC-labeled S. pneumoniae. After 30 min, the mice were killed; spleens were removed and embedded in OCT compound; and sections were prepared as described before. Counterstaining with biotin-conjugated MARCO antibody (Serotec) and Texas red–conjugated streptavidin (BioMeda) was performed as described before, to delineate MZMs. Visualization was performed using an MRC1024 confocal microscope (Bio-Rad Laboratories).
Results
Cloning of Mouse SIGN-R1 and Inactivation of the Mouse SIGN-R1 Gene.
A full-length mouse SIGN-R1 cDNA was generated using primers designed from the mouse SIGN-R1 genomic sequence and cross-referenced to the human DC-SIGN sequence. The clone encoded a type II transmembrane protein with an extracellular domain consisting of a neck region made up of four repeats of 28 amino acids (aa) and a CRD (Fig. 1, a and b). The overall aa sequence encoded by mouse SIGN-R1 showed ∼40% sequence identity to human DC-SIGN and DC-SIGNR. However, more significantly, the CRD of mouse SIGN-R1 showed ∼75% identity to the CRD of human DC-SIGN and DC-SIGNR, and 42% identity to the CRD of mouse CD23. Within the CRD, the residues defining mannose-binding specificity are conserved between mouse SIGN-R1 and human DC-SIGN as are the residues required for calcium-dependent binding of carbohydrates, features that are also shared with mouse CD23 (12). During the course of our work, a family of five SIGN-related genes were characterized as mapping to mouse chromosome 8 close to the CD23 gene (16). This juxtaposition to CD23 is analogous to that observed in human where CD23, DC-SIGN, and DC-SIGNR are all closely linked within a 85-kb region on chromosome 19p13 (12, 13). Little is known about the biological significance of any of these SIGN-related molecules.
Figure 1.

Inactivation of the mouse SIGN-R1 gene by homologous recombination. (a) Structure of the human DC-SIGN locus. (b) Structure of the mouse SIGN-R1 locus, the targeting vector, and the predicted homologous recombination event are shown. NeoR, neomycin resistance cassette; TK, thymidine kinase cassette; B, BamHI. (c) RT-PCR analysis of SIGN-R1 expression. RNA was prepared from splenocytes and PCR was performed. −/−, SIGN-R1−/−. (d) RT-PCR analysis of SIGN-R1 expression using PCR primers throughout the cDNA to verify gene deletion. (1) Wild-type. (2) SIGN-R1−/−. (3) Wild-type. (4) SIGN-R1−/−. (e) Staining with ER-TR9 (anti–SIGN-R1) monoclonal antibody using horseradish peroxidase detection. (f) Co-staining with ER-TR9 monoclonal antibody (Texas red) and MOMA monoclonal antibody (FITC) (identifying marginal metallophilic macrophages). Original magnification, 10.
To study the in vivo roles of mouse SIGN-R1 in the development of immune responses, we have generated SIGN-R1−/− mice using gene targeting (Fig. 1 b). The correct insertion of this construct would result in the deletion of nucleotides encoding aa 215–232 in exon 8. Mouse SIGN-R1−/− mice were healthy and displayed no overt phenotypic abnormalities and, as detected by flow cytometry, the immune cell composition appeared normal (unpublished data). Analysis of the SIGN-R1−/− mice failed to detect SIGN-R1 RNA transcripts from spleen cells using reverse transcriptase–polymerase chain reaction assays, though SIGN-R1 RNA was readily detected in the spleens of wild-type mice (Fig. 1 c). To verify that gene disruption had ablated SIGN-R1 transcription, we designed PCR primer pairs throughout the coding region (Fig. 1 d) and RT-PCR analysis failed to detect a SIGN-R1–specific product in RNA prepared from SIGN-R1−/− splenocytes using any of the PCR primer pairs. In contrast, all PCR products were detected in RNA prepared from wild-type splenocytes (Fig. 1 d). Analysis of transcripts for SIGN-R3, which maps to within ∼100 kb of SIGN-R1 (16) and is also expressed in the spleen, showed normal expression levels in SIGN-R1−/− mice when compared with wild-type controls (unpublished data). These data demonstrate that interruption of the SIGN-R1 sequence has not resulted in the production of aberrant SIGN-R1 transcription products that might lead to truncated SIGN-R1 protein or the disruption of a neighboring SIGN-R gene. To corroborate these results, we used antibody staining to assess the cell surface expression of SIGN-R1. Recently, it has been demonstrated that the monoclonal antibody ER-TR9, which has been used previously as a specific MZM marker (22), recognizes mouse SIGN-R1 (33). Therefore, to verify the absence of SIGNR1 expression from the cell surface, spleen sections were stained with ER-TR9. As shown in Fig. 1 e, wild-type spleen sections display clear ER-TR9 staining of cells in the MZ, whereas no staining is observed in sections prepared from SIGN-R1−/− spleen. Co-staining with the fluorescently labeled antibodies ER-TR9 and MOMA (which recognizes marginal metallophilic macrophages of the MZ; reference 34) confirmed that SIGN-R1 expression has been deleted in our mice (Fig. 1 f).
MZMs from SIGN-R1−/− Mice Fail to Take Up the Polysaccharide Dextran.
MZMs play a central role in the removal of TI-2 antigens, and previous studies using ER-TR9 and a polyclonal rabbit anti–SIGN-R1 antibody (33) have reported that these antibodies block the uptake of polysaccharides such as Ficoll (26) and dextran (33) by MZMs. To test whether SIGN-R1−/− mice had a deficit in TI-2 polysaccharide uptake, we injected wild-type and SIGN-R1−/− mice intravenously with ALEXA-conjugated dextran. After 2 h, spleen sections were prepared and the presence of ALEXA-dextran was determined by microscopy. As expected, ALEXA-dextran was visualized in the MZs of wild-type mice (Fig. 2, a, c, e, and g). In contrast, we were unable to detect ALEXA-dextran uptake in the SIGN-R1−/− mice (Fig. 2, b, d, f, and h).
Figure 2.
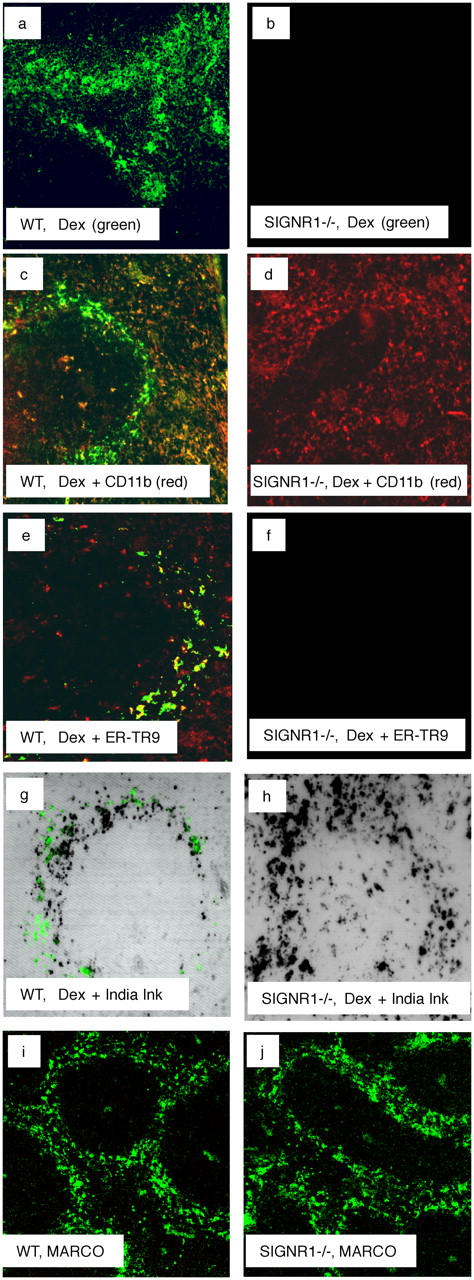
Analysis of MZMs and ALEXA-dextran uptake in the spleens of wild-type (WT) and SIGN-R1−/− mice. (a and b) ALEXA-dextran (Dex, green) localized to the MZs in spleen sections 2 h after i.v. inoculation. (c and d) Spleen sections demonstrating ALEXA-dextran uptake counterstained with anti-CD11b (Texas red). (e and f) Spleen sections demonstrating ALEXA-dextran uptake counterstained with ER-TR9 antibody (anti–SIGN-R1; Texas red). (g and h) Spleen sections showing results of coinoculation with ALEXA-dextran (green) and Indian ink (black spots). (i and j) Spleen sections demonstrating anti-MARCO antibody (FITC) staining of MZM. All original magnifications,10.
To confirm that MZMs were still present in the SIGN-R1−/− mice, and capable of phagocytosis, we coinjected wild-type and SIGN-R1−/− mice with Indian ink, which has been shown to be taken up specifically by MZMs (35), and ALEXA-dextran. As shown in Fig. 2 (g and h), we detected Indian ink uptake in both the wild-type and SIGN-R1−/− mice, indicating that MZMs remain in the SIGN-R1−/− mice, but are specifically unable to mediate the uptake of ALEXA-dextran. Double-staining with ER-TR9 and MARCO (a marker of MZM; reference 23) results in convergent staining of the MZMs (reference 27 and unpublished data). As we detected normal staining with MARCO in the spleens of both wild-type and SIGN-R1−/−, this implies that SIGN-R1 is not necessary for normal MZM development (Fig. 2, i and j). These data demonstrate that SIGN-R1 is not required for the development or localization of MZMs, but is specifically required for the binding of dextran, a prototypical TI-2 antigen. It is noteworthy that, in the absence of SIGN-R1, we did not detect any compensatory uptake of ALEXA-dextran by MZ B cells, DCs, or reticular cells.
SIGN-R1−/− Mice Succumb to Sublethal S. pneumoniae Infection.
TI-2 antigens are highly represented on gram-positive bacteria such as S. pneumoniae. These bacteria are important human and mouse pathogens causing pneumonia, meningitis, peritonitis, and septicemia. To test whether SIGN-R1 is functionally important during infection with S. pneumoniae, we infected wild-type and SIGN-R1−/− mice with a sublethal intraperitoneal dose of the virulent S. pneumoniae serotype 2. Less than 18% of the SIGN-R1−/− mice survived infection as compared with >60% of wild-type mice (P < 0.005; Fig. 3 a). The survival curves also demonstrate that the absence of SIGN-R1 results in more rapid mortality. Although 90% of the wild-type mice were alive at 48 h, only 20% of the SIGN-R1−/− had survived (Fig. 3 a). These data were corroborated by the sickness scores collected during the course of infection, which clearly demonstrated that the onset of clinically apparent disease occurred more rapidly in SIGN-R1−/− mice than in controls (Fig. 3, b and c). Strikingly, SIGN-R1−/− mice showed persistent septicemia after infection, compared with controls. At 30 h after infection, 95% of the SIGN-R1−/− mice had circulating bacterial loads of >25 × 103 CFU/ml of blood, whereas only 10% of wild type had similar levels of circulating bacteria, and 50% had no detectable bacteria (Fig. 3 d). To exclude any possibility that these results arose due to strain background effects, we have verified these data in preliminary experiments using SIGN-R1−/− mice and wild-type mice generated by backcrossing for six generations to the C57BL/6 background. We found 100% survival of wild type (n = 10) and 40% survival of SIGN-R1−/− (n = 7) mice 48 h after intraperitoneal infection with 103 S. pneumoniae serotype 2, and 90% survival of wild type (n = 10) and 0% survival of SIGN-R1−/− (n = 7) mice 48 h after intraperitoneal infection with 104 S. pneumoniae serotype 2, fully validating our earlier findings.
Figure 3.

Absence of SIGN-R1 increases susceptibility to pneumococcal peritonitis. (a) Survival after S. pneumoniae type 2 infection. SIGN-R1−/− mice (n = 12) or control mice (n = 12) were inoculated intraperitoneally with 103 S. pneumoniae type 2. SIGN-R1−/− have reduced survival (P = 0.005). The experiment shown is representative of two, and p-values were obtained using a Log rank test. (b and c) Physical signs of illness in mice with pneumococcal peritonitis. After inoculation with S. pneumoniae (type 2), mice were monitored by a blinded observer for the signs of systemic illness. The presence or absence of piloerection (1 point), with additional hunched posture (2 points), and lack of spontaneous movement (3 points) was scored in control and SIGN-R1−/− mice inoculated with 103 S. pneumoniae at 24 h (b) and 30 h (c). The experiment is representative of two. (d) 30 h after inoculation with S. pneumoniae, tail bleeds were performed on control (n = 12) and SIGN-R1−/− mice (n = 10), and blood was cultured for bacterial growth. More SIGN-R1−/− mice were bacteremic (P = 0.014); the p-value was obtained using a Fisher's exact test. (e) Survival after S. pneumoniae type 14 infection. SIGN-R1−/− mice (n = 10) or control mice (n = 10) were inoculated intraperioneally with 105 S. pneumoniae type 14. SIGN-R1−/− have reduced survival (P = 0.018). Numbers denote individual mice. p-values were obtained using a Log rank test. (f and g) Physical signs of illness in mice with pneumococcal peritonitis. After inoculation with S. pneumoniae (type 14), mice were monitored by a blinded observer for the signs of systemic illness (as described before). (h) 26 h after inoculation with S. pneumoniae, tail bleeds were performed on control (n = 10) and SIGN-R1−/− mice (n = 10), and blood was cultured for bacterial growth. More SIGN-R1−/− mice were bacteremic (P = 0.023).
To extend our findings to other S. pneumoniae strains, we have also performed infections with the less virulent S. pneumoniae serotype 14, which has been shown previously to associate with SIGN-R1 through its capsular polysaccharides (27). As for S. pneumoniae serotype 2, increased mortality was also observed in SIGN-R1−/− mice infected with S. pneumoniae serotype 14 when compared with wild-type controls (Fig. 3 e). This experiment also demonstrated that sickness scores and bacteremia correlate with survival; those animals showing the highest bacteremia are the first to display higher sickness scores and subsequently die (Fig. 3, e–h).
These data clearly demonstrate that SIGN-R1 is important for the efficient clearance of S. pneumoniae from infected mice and that, in the absence of SIGN-R1, mice develop severe bacteremia leading to rapid and increased mortality.
Impaired Bacterial Clearance Is Not Due to Reduced Natural Antibody.
Although the adaptive antibody response would not be elicited within the time scale of this S. pneumoniae infection, the levels of natural antibody have been shown to be critical to survival (36). Natural antibodies are preexisting antibodies found in the sera of naive mice and are involved in responses to a range of pathogens. Natural antibodies exist in germ-free mice and these are thought to be selected by self-antigens (37), though some natural antibodies require the presence of exogenous antigen or gut flora for their production (38). Natural antibodies have a restricted range of specificities and are generally lower affinity antibodies against bacterial antigens such as PC and phosphatidylcholine (39). They contribute to innate immunity through the binding of bacteria, facilitating complement activation and Fc receptor-mediated clearance, which can lead to phagocytosis and destruction of the bacterium (40). Both B-1 and MZ B cells have been shown to generate anti-PC antibodies, and these natural antibodies can confer protection against S. pneumoniae infections in mice (36). To assess whether the inability to clear the infection in SIGN-R1−/− mice was due to a defect in their production of natural antibodies, we analyzed the levels of anti-PC antibodies in naive wild-type and SIGN-R1−/− mice; no significant difference was found (Fig. 4 a). Furthermore, we would predict that if SIGN-R1 was altering levels of natural antibody, serum from SIGN-R1−/− mice might be less efficient at opsonizing encapsulated bacteria and promoting Fc receptor-dependent phagocytosis by cells such as macrophages. To test this, we used serum from naive SIGN-R1−/− mice and wild-type mice to opsonize FITC-conjugated S. pneumoniae. These bacteria were incubated with the RAW mouse macrophage cell line for 30 min. Phagocytosis of bacteria was measured by flow cytometry, and opsonization with serum from SIGN-R1−/− mice resulted in a similar proportion of macrophages engulfing bacteria as serum from the wild-type mice (Fig. 4 b). Similar results were obtained when peritoneal macrophages from wild-type mice were used in place of RAW cells (Fig. 4 c).
Figure 4.
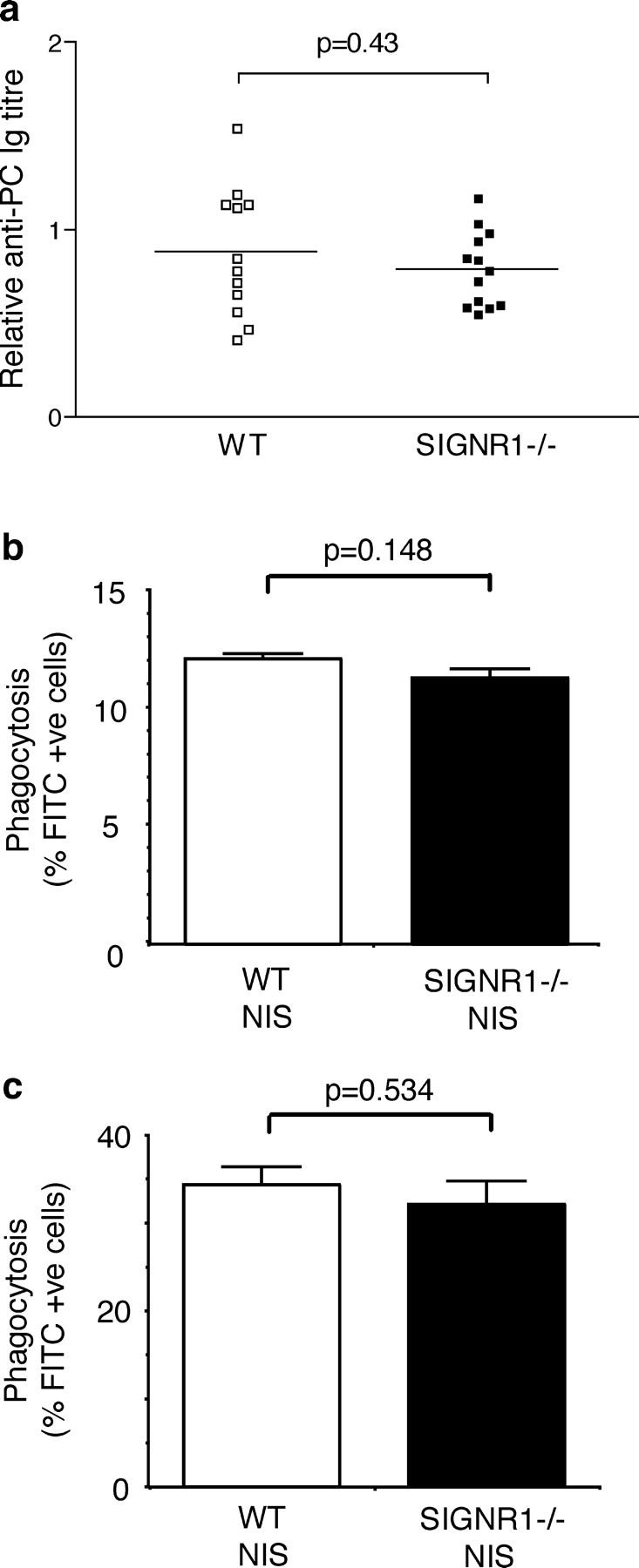
Absence of SIGN-R1 does not impair natural antibody levels or opsonization of S. pneumoniae. (a) Natural anti-PC antibody levels in control and SIGN-R1−/− mice. Sera from naive WT and SIGN-R1−/− mice were assayed for natural antibodies against PC by ELISA. Each point represents the value from an individual mouse. p-values were determined using an unpaired Student's t test (Welch corrected). (b and c) Nonimmune serum from control and SIGN-R1−/− mice provide equivalent opsonization. The RAW-297 macrophage cell line (b) or BALB/c peritoneal macrophages (c) were incubated at 37°C with FITC-labeled S. pneumoniae opsonized with heat-inactivated nonimmune serum (NIS) from wild-type or SIGN-R1−/− mice, followed by flow cytometric analysis. Phagocytosis is expressed as percentage of FITC positive cells. Values represent mean of triplicates, the experiments shown are representative of two, and p-values were obtained using an unpaired Student's t test.
Impaired S. pneumoniae Recognition by Peritoneal Macrophages from SIGN-R1−/− Mice.
Peritoneal macrophages are uniquely located to play a crucial role in the defensive response to bacterial infection; a recent paper has suggested that SIGN-R1 expression by peritoneal macrophages may be important for the recognition of yeast particles (28). Given the extremely rapid susceptibility of the SIGN-R1−/− mice to S. pneumoniae infection, we wished to determine whether S. pneumoniae recognition by peritoneal macrophages was impaired by the absence of SIGN-R1. Initially, we wished to confirm that SIGN-R1 was binding S. pneumoniae. To test this, FITC-labeled S. pneumoniae were incubated with retrovirally transduced NIH3T3 cells expressing SIGN-R1 (28) or parental NIH3T3 cells. The cells expressing SIGN-R1 bound S. pneumoniae significantly more efficiently than the parental line, demonstrating that SIGN-R1 binds directly to S. pneumoniae (Fig. 5 a). We assessed whether binding or phagocytosis of S. pneumoniae was abnormal in SIGN-R1−/− peritoneal macrophages. Peritoneal macrophages were incubated for 30 min with FITC-labeled S. pneumoniae at 4°C to assess surface binding, or 37°C to assess phagocytosis, before analysis by flow cytometry. We found that peritoneal macrophages from SIGN-R1−/− mice were significantly impaired (threefold reduction) in their ability to bind S. pneumoniae at 4°C (Fig. 5 b). Furthermore, fewer than half as many macrophages from SIGN-R1−/− mice phagocytosed S. pneumoniae at 37°C, compared with controls (Fig. 5 c); internalization was confirmed by confocal microscopy (Fig. 5 d). This reduction in phagocytosis in SIGN-R1−/− macrophages was proportional to the reduction in surface binding seen at 4°C (Fig. 5 b), suggesting that the observed reduction in phagocytosis was secondary to reduced recognition of S. pneumoniae in the absence of SIGN-R1.
Figure 5.
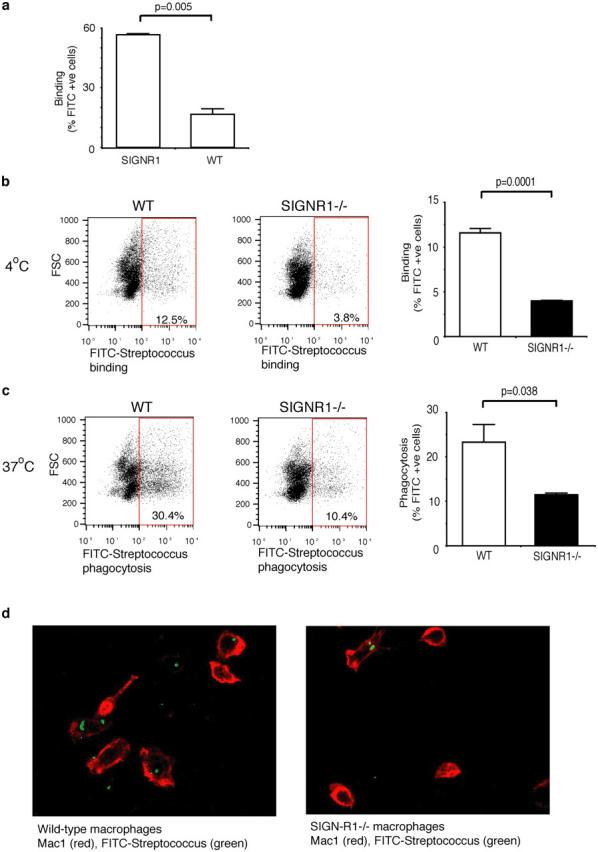
Impaired recognition of S. pneumoniae by SIGN-R1−/− peritoneal macrophages. (a) S. pneumoniae bind preferentially to SIGN-R1–transduced NIH3T3 (SIGNR1) cells compared with wild-type (WT) NIH3T3. Cells were incubated for 1 h at 4°C with FITC-labeled pneumococci, washed, and analyzed by flow cytometry. Values represent the mean of triplicates, and p-values were obtained using an unpaired Student's t test. (b–d) Pooled peritoneal macrophages from wild-type (n = 2) or SIGN-R1−/− mice (n = 2) were incubated for 30 min with FITC-labeled S. pneumoniae at 4°C to assess binding (b), or 37°C to assess phagocytosis (c), followed by flow cytometric analysis. Binding or phagocytosis are expressed as percentage of FITC positive cells. SIGN-R1−/− peritoneal macrophages show reduced binding and phagocytosis of S. pneumoniae. Representative FACS profiles are shown. Values represent mean of triplicates, the experiments shown are representative of two, and p-values were obtained using an unpaired Student's t test. (d) Confocal images of wild-type and SIGN-R1−/− peritoneal macrophages labeled with Mac-1 allophycocyanin after incubation with FITC-labeled S. pneumoniae at 37°C for 45 min.
Reduced Binding of S. pneumoniae to MZMs in SIGN-R1−/− Mice.
Given that SIGN-R1 is necessary for normal binding of S. pneumoniae by peritoneal macrophages in vitro, we sought to determine if the same was true of MZMs in vivo because they have been identified as critical for the uptake of certain polysaccharide antigens. Live FITC-conjugated S. pneumoniae were injected intravenously into SIGN-R1−/− and wild-type mice and spleen sections were assessed for the distribution of bacteria. 30 min after administration, S. pneumoniae were found localized almost exclusively in the MZs of the spleens of wild-type mice, with few bacteria detected in the red pulp (Fig. 6, left). In contrast, in the spleens from SIGN-R1−/− mice, S. pneumoniae were no longer restricted to the MZs, but were disseminated throughout the red pulp (Fig. 6, right). These results demonstrate that SIGN-R1 is important in trapping bacteria in the MZ.
Figure 6.
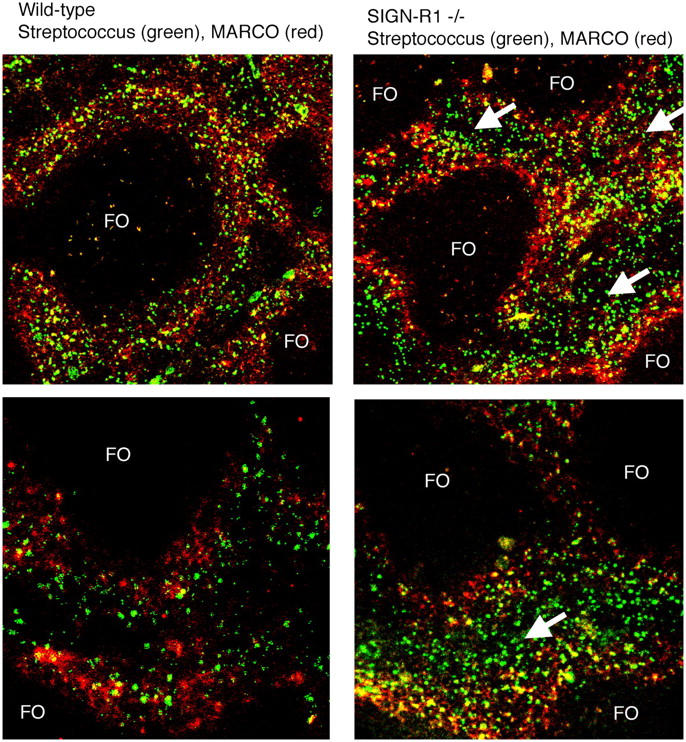
Impaired S. pneumoniae trapping by SIGN-R1−/− MZM. Wild-type (left) and SIGN-R1−/− (right) mice were injected intravenously with 107 FITC-labeled S. pneumoniae. 30 min later, spleens were snap frozen, and cryosections were prepared and counterstained with anti-MARCO antibody (Texas red) to delineate MZMs. Arrows indicate areas where S. pneumoniae are located in the red pulp and are not restricted to the MZ. FO, follicles. Original magnification, 10 (top), 20 (bottom). Representative sections from two mice per group are shown.
Thus, SIGN-R1 is required for the binding and efficient clearance of S. pneumoniae by macrophages. Absence of SIGN-R1 leads to impaired trapping of bacteria in the MZs, increased septicemia, and reduced survival in response to streptococcal infection.
Discussion
Here, we have shown that SIGN-R1 is a key receptor required for innate immunity to the gram-positive pathogen S. pneumoniae. In the absence of SIGN-R1, SIGN-R1−/− mice show markedly increased mortality after infection with S. pneumoniae. This impaired survival is accompanied by a failure of the SIGN-R1−/− mice to eradicate S. pneumoniae from the blood. In contrast, wild-type animals show rapid clearance of the infection and show significantly improved survival. We demonstrate that this failure to clear the bacteria is not due to changes in natural antibody levels, but is consistent with the observed failure of the SIGN-R1−/− MZMs and peritoneal macrophages to bind and consequently phagocytose S. pneumoniae efficiently and, thus, remove these bacteria from the circulation. These data provide the first evidence that SIGN-R1 expressed on macrophages plays an essential role in host defense against S. pneumoniae by mediating recognition and clearance of these bacteria.
It has long been recognized that the macrophages of the MZ are highly phagocytic and are organized strategically within the spleen to optimize their capacity to filter blood-borne particles. Lying between the white pulp and the red pulp, and arranged at the border of the sinus, MZMs were first characterized by their capacity to recognize and internalize TI-2 antigens. We demonstrate that SIGN-R1, expressed on MZMs and macrophages of the peritoneum, is a key receptor for the recognition and efficient clearance of pathogenic bacteria such as S. pneumoniae by the innate immune system. It is also noteworthy that, although SIGN-R1 is a member of a family of SIGN molecules (16–18), its function in S. pneumoniae clearance is not compensated for by the other family members.
The germline-encoded PRR recognize antigens on pathogens that are critical to survival and that do not undergo significant alteration due to evolutionary pressure. Recent papers have demonstrated, using in vitro assay systems, that human DC-SIGN and DC-SIGNR have been commandeered by several human pathogens and used to facilitate rather than protect against infection (2). Although the human SIGN molecules appear to represent an immunological liability in some circumstances, our experiments with SIGN-R1 suggest that SIGN molecules play functionally important roles in the rapid clearance of specific pathogens by the innate immune system. Our paper clearly demonstrates for the first time a protective role for a SIGN family member.
Acknowledgments
We thank T. Langford and the MRC CBS staff for technical assistance, Dr. T. Geijtenbeek (Amsterdam) for supplying the ER-TR9 antibody, and Dr. P. Taylor (Oxford) for supplying SIGN-R1–transduced NIH3T3 cells.
P.G. Fallon is supported by the Wellcome Trust and Science Foundation, Ireland. P. Smith is supported by a grant from the Leukaemia Research Fund. M.R. Clatworthy is funded by a Wellcome Trust Clinical Training Fellowship (065770) and The Sackler Fund. K.G.C. Smith is supported by a Wellcome Research Leave Award for Clinical Academics (grant no. 067543AIA) and the Medical Research Council (grant no. 9805187).
The authors have no conflicting financial interests.
A. Lanoue and M.R. Clatworthy contributed equally to this work.
M.J. Townsend's present address is Dept. of Immunology and Infectious Diseases, Harvard School of Public Health, Rm. 205, 651 Huntingdon Ave., Boston, MA 02115.
Abbreviations used in this paper: aa, amino acids; CRD, carbohydrate recognition domain; MZ, marginal zone; MZM, MZ macrophage; PC, phosphorylcholine; PRR, pattern recognition receptor; SIGN, specific intracellular adhesion molecule-grabbing nonintegrin; TI-2, thymus independent type 2.
References
- 1.Gordon, S. 2002. Pattern recognition receptors: doubling up for the innate immune response. Cell. 111:927–930. [DOI] [PubMed] [Google Scholar]
- 2.Van Kooyk, Y., and T.B. Geijtenbeek. 2003. DC-SIGN: escape mechanism for pathogens. Nat. Rev. Immunol. 3:697–709. [DOI] [PubMed] [Google Scholar]
- 3.Geijtenbeek, T.B., R. Torensma, S.J. van Vliet, G.C. van Duijnhoven, G.J. Adema, Y. van Kooyk, and C.G. Figdor. 2000. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 100:575–585. [DOI] [PubMed] [Google Scholar]
- 4.Geijtenbeek, T.B., D.S. Kwon, R. Torensma, S.J. van Vliet, G.C. van Duijnhoven, J. Middel, I.L. Cornelissen, H.S. Nottet, V.N. KewalRamani, D.R. Littman, C.G. Figdor, and Y. van Kooyk. 2000. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 100:587–597. [DOI] [PubMed] [Google Scholar]
- 5.Pohlmann, S., G.J. Leslie, T.G. Edwards, T. Macfarlan, J.D. Reeves, K. Hiebenthal-Millow, F. Kirchhoff, F. Baribaud, and R.W. Doms. 2001. DC-SIGN interactions with human immunodeficiency virus: virus binding and transfer are dissociable functions. J. Virol. 75:10523–10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtis, B.M., S. Scharnowske, and A.J. Watson. 1992. Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc. Natl. Acad. Sci. USA. 89:8356–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarez, C.P., F. Lasala, J. Carrillo, O. Muniz, A.L. Corbi, and R. Delgado. 2002. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 76:6841–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halary, F., A. Amara, H. Lortat-Jacob, M. Messerle, T. Delaunay, C. Houles, F. Fieschi, F. Arenzana-Seisdedos, J.F. Moreau, and J. Dechanet-Merville. 2002. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity. 17:653–664. [DOI] [PubMed] [Google Scholar]
- 9.Colmenares, M., A. Puig-Kroger, O. Muniz Pello, A.L. Corbi, and L. Rivas. 2002. Dendritic-cell specific ICAM-3 grabbing nonintegrin (DC-SIGN, CD209), a C-type surface lectin in human dendritic cells, is a receptor for Leishmania amastigotes. J. Biol. Chem. 277:36766–36796. [DOI] [PubMed] [Google Scholar]
- 10.Geijtenbeek, T.B., S.J. Van Vliet, E.A. Koppel, M. Sanchez-Hernandez, C.M. Vandenbroucke-Grauls, B. Appelmelk, and Y. Van Kooyk. 2003. Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tassaneetrithep, B., T.H. Burgess, A. Granelli-Piperno, C. Trumpfheller, J. Finke, W. Sun, M.A. Eller, K. Pattanapanyasat, S. Sarasombath, D.L. Birx, et al. 2003. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197:823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soilleux, E.J., R. Barten, and J. Trowsdale. 2000. DC-SIGN; a related gene, DC-SIGNR; and CD23 form a cluster on 19p13. J. Immunol. 165:2937–2942. [DOI] [PubMed] [Google Scholar]
- 13.Mummidi, S., G. Catano, L. Lam, A. Hoefle, V. Telles, K. Begum, F. Jimenez, S.S. Ahuja, and S.K. Ahuja. 2001. Extensive repertoire of membrane-bound and soluble dendritic cell-specific ICAM-3-grabbing nonintegrin 1 (DC-SIGN1) and DC-SIGN2 isoforms. Inter-individual variation in expression of DC-SIGN transcripts. J. Biol. Chem. 276:33196–33212. [DOI] [PubMed] [Google Scholar]
- 14.Pohlmann, S., E.J. Soilleux, F. Baribaud, G.J. Leslie, L.S. Morris, J. Trowsdale, B. Lee, N. Coleman, and R.W. Doms. 2001. DC-SIGNR, a DC-SIGN homologue expressed in endothelial cells, binds to human and simian immunodeficiency viruses and activates infection in trans. Proc. Natl. Acad. Sci. USA. 98:2670–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cambi, A., and C.G. Figdor. 2003. Dual function of C-type lectin-like receptors in the immune system. Curr. Opin. Cell Biol. 15:539–546. [DOI] [PubMed] [Google Scholar]
- 16.Park, C.G., K. Takahara, E. Umemoto, Y. Yashima, K. Matsubara, Y. Matsuda, B.E. Clausen, K. Inaba, and R.M. Steinman. 2001. Five mouse homologues of the human dendritic cell C-type lectin, DC-SIGN. Int. Immunol. 13:1283–1290. [DOI] [PubMed] [Google Scholar]
- 17.Caminschi, I., K.M. Lucas, M.A. O'Keeffe, H. Hochrein, Y. Laabi, T.C. Brodnicki, A.M. Lew, K. Shortman, and M.D. Wright. 2001. Molecular cloning of a C-type lectin superfamily protein differentially expressed by CD8alpha(−) splenic dendritic cells. Mol. Immunol. 38:365–373. [DOI] [PubMed] [Google Scholar]
- 18.Parent, S.A., T. Zhang, G. Chrebet, J.A. Clemas, D.J. Figueroa, B. Ky, R.A. Blevins, C.P. Austin, and H. Rosen. 2002. Molecular characterization of the murine SIGNR1 gene encoding a C-type lectin homologous to human DC-SIGN and DC-SIGNR. Gene. 293:33–46. [DOI] [PubMed] [Google Scholar]
- 19.Takahara, K., Y. Yashima, Y. Omatsu, H. Yoshida, Y. Kimura, Y.S. Kang, R.M. Steinman, C.G. Park, and K. Inaba. 2004. Functional comparison of the mouse DC-SIGN, SIGNR1, SIGNR3 and Langerin, C-type lectins. Int. Immunol. 16:819–829. [DOI] [PubMed] [Google Scholar]
- 20.Galustian, C., C.G. Park, W. Chai, M. Kiso, S.A. Bruening, Y.S. Kang, R.M. Steinman, and T. Feizi. 2004. High and low affinity carbohydrate ligands revealed for murine SIGN-R1 by carbohydrate array and cell binding approaches, and differing specificities for SIGN-R3 and langerin. Int. Immunol. 16:853–866. [DOI] [PubMed] [Google Scholar]
- 21.Kraal, G. 1992. Cells in the marginal zone of the spleen. Int. Rev. Cytol. 132:31–74. [DOI] [PubMed] [Google Scholar]
- 22.Dijkstra, C.D., E. Van Vliet, E.A. Dopp, A.A. van der Lelij, and G. Kraal. 1985. Marginal zone macrophages identified by a monoclonal antibody: characterization of immuno- and enzyme-histochemical properties and functional capacities. Immunology. 55:23–30. [PMC free article] [PubMed] [Google Scholar]
- 23.Elomaa, O., M. Kangas, C. Sahlberg, J. Tuukkanen, R. Sormunen, A. Liakka, I. Thesleff, G. Kraal, and K. Tryggvason. 1995. Cloning of a novel bacteria-binding receptor structurally related to scavenger receptors and expressed in a subset of macrophages. Cell. 80:603–609. [DOI] [PubMed] [Google Scholar]
- 24.Aichele, P., J. Zinke, L. Grode, R.A. Schwendener, S.H. Kaufmann, and P. Seiler. 2003. Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J. Immunol. 171:1148–1155. [DOI] [PubMed] [Google Scholar]
- 25.Mosier, D.E., J.J. Mond, and E.A. Goldings. 1977. The ontogeny of thymic independent antibody responses in vitro in normal mice and mice with an X-linked B cell defect. J. Immunol. 119:1874–1878. [PubMed] [Google Scholar]
- 26.Kraal, G., H. Ter Hart, C. Meelhuizen, G. Venneker, and E. Claassen. 1989. Marginal zone macrophages and their role in the immune response against T-independent type 2 antigens: modulation of the cells with specific antibody. Eur. J. Immunol. 19:675–680. [DOI] [PubMed] [Google Scholar]
- 27.Kang, Y.S., J.Y. Kim, S.A. Bruening, M. Pack, A. Charalambous, A. Pritsker, T.M. Moran, J.M. Loeffler, R.M. Steinman, and C.G. Park. 2004. The C-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of Streptococcus pneumoniae in the marginal zone of mouse spleen. Proc. Natl. Acad. Sci. USA. 101:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor, P.R., G.D. Brown, J. Herre, D.L. Williams, J.A. Willment, and S. Gordon. 2004. The role of SIGNR1 and the beta-glucan receptor (Dectin-1) in the nonopsonic recognition of yeast by specific macrophages. J. Immunol. 172:1157–1162. [DOI] [PubMed] [Google Scholar]
- 29.Osoegawa, K., M. Tateno, P.Y. Woon, E. Frengen, A.G. Mammoser, J.J. Catanese, Y. Hayashizaki, and P.J. de Jong. 2000. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 10:116–128. [PMC free article] [PubMed] [Google Scholar]
- 30.McKenzie, G.J., C.L. Emson, S.E. Bell, S. Anderson, P. Fallon, G. Zurawski, R. Murray, and A.N.J. McKenzie. 1998. Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 9:423–432. [DOI] [PubMed] [Google Scholar]
- 31.Reiner, S., S. Zheng, D. Corry, and R. Locksley. 1993. Constructing polycompetitor cDNAs for quantitative PCR. J. Immunol. Methods. 165:37–46. [DOI] [PubMed] [Google Scholar]
- 32.Clatworthy, M.R., and K.G. Smith. 2004. FcγRIIb balances efficient pathogen clearance and the cytokine-mediated consequences of sepsis. J. Exp. Med. 199:717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang, Y.S., S. Yamazaki, T. Iyoda, M. Pack, S.A. Bruening, J.Y. Kim, K. Takahara, K. Inaba, R.M. Steinman, and C.G. Park. 2003. SIGN-R1, a novel C-type lectin expressed by marginal zone macrophages in spleen, mediates uptake of the polysaccharide dextran. Int. Immunol. 15:177–186. [DOI] [PubMed] [Google Scholar]
- 34.Kraal, G., and M. Janse. 1986. Marginal metallophilic cells of the mouse spleen identified by a monoclonal antibody. Immunology. 58:665–669. [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuno, K., H. Fujii, and M. Kotani. 1986. Splenic marginal-zone macrophages and marginal metallophils in rats and mice. Cell Tissue Res. 246:263–269. [DOI] [PubMed] [Google Scholar]
- 36.Briles, D.E., C. Forman, S. Hudak, and J.L. Claflin. 1982. Antiphosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae. J. Exp. Med. 156:1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochsenbein, A.F., T. Fehr, C. Lutz, M. Suter, F. Brombacher, H. Hengartner, and R.M. Zinkernagel. 1999. Control of early viral and bacterial distribution and disease by natural antibodies. Science. 286:2156–2159. [DOI] [PubMed] [Google Scholar]
- 38.Bos, N.A., H. Kimura, C.G. Meeuwsen, H. De Visser, M.P. Hazenberg, B.S. Wostmann, J.R. Pleasants, R. Benner, and D.M. Marcus. 1989. Serum immunoglobulin levels and naturally occurring antibodies against carbohydrate antigens in germ-free BALB/c mice fed chemically defined ultrafiltered diet. Eur. J. Immunol. 19:2335–2339. [DOI] [PubMed] [Google Scholar]
- 39.Rothstein, T.L. 2002. Cutting edge commentary: two B-1 or not to be one. J. Immunol. 168:4257–4261. [DOI] [PubMed] [Google Scholar]
- 40.Mold, C., B. Rodic-Polic, and T.W. Du Clos. 2002. Protection from Streptococcus pneumoniae infection by C-reactive protein and natural antibody requires complement but not Fc gamma receptors. J. Immunol. 168:6375–6381. [DOI] [PubMed] [Google Scholar]