

Abstract
Primary sclerosing cholangitis (PSC), a chronic inflammatory liver disease characterized by progressive bile duct destruction, develops as an extra-intestinal complication of inflammatory bowel disease (IBD) (Chapman, R.W. 1991. Gut. 32:1433–1435). However, the liver and bowel inflammation are rarely concomitant, and PSC can develop in patients whose colons have been removed previously. We hypothesized that PSC is mediated by long-lived memory T cells originally activated in the gut, but able to mediate extra-intestinal inflammation in the absence of active IBD (Grant, A.J., P.F. Lalor, M. Salmi, S. Jalkanen, and D.H. Adams. 2002. Lancet. 359:150–157). In support of this, we show that liver-infiltrating lymphocytes in PSC include mucosal T cells recruited to the liver by aberrant expression of the gut-specific chemokine CCL25 that activates α4β7 binding to mucosal addressin cell adhesion molecule 1 on the hepatic endothelium. This is the first demonstration in humans that T cells activated in the gut can be recruited to an extra-intestinal site of disease and provides a paradigm to explain the pathogenesis of extra-intestinal complications of IBD.
Keywords: chemokines, integrins, inflammation, hepatitis, colitis
Introduction
Primary sclerosing cholangitis (PSC) develops in 2.4–7.5% of patients with inflammatory bowel disease (IBD) and 70–85% of patients with PSC ultimately develop IBD (1). Any model to explain the development of hepatic complications in IBD needs to take into account the fact that colectomy does not alter the severity or course of PSC and that liver disease frequently runs an independent course from inflammation in the bowel (2). Thus, PSC can develop for the first time many years after IBD has become quiescent and even after previous proctocolectomy (3). We proposed that effector T cells generated in the organized lymphoid tissue of the gut during active IBD (4) persist as long-lived memory cells that may be recruited to the liver to trigger hepatic inflammation under specific circumstances.
Lymphocyte recruitment from the circulation to tissue is a highly regulated process dependent on sequential interactions with endothelial adhesion molecules and chemokines (5). Initial transient interactions between lymphocytes and endothelium tether the lymphocyte and induce it to roll on the vessel wall where it comes into contact with chemokines. In the presence of an appropriate chemokine, specific G protein–coupled receptors on the lymphocyte are activated, triggering high-affinity integrin binding to endothelial ligands leading to arrest and transendothelial migration into tissue in response to chemotactic signals (6).
Chemokines play a crucial role in orchestrating the recruitment and recirculation of leukocytes to lymphoid organs and peripheral tissues as well as to sites of inflammation (7). Tissue-specific combinations of chemokines and endothelial adhesion molecules provide a molecular “address” that can be recognized by particular subsets of circulating leukocytes (8, 9). Thus, the chemokines CCL19 and CCL21 and the peripheral node addressin are crucial for the structural and functional organization of secondary lymphoid tissues by recruiting and positioning specific subsets of lymphocytes (10). The best-defined tissue-specific recirculation of memory lymphocytes occurs in the skin and gut, which provide distinct barriers to the environment exposed to different types of antigen (11). The chemokine CCL17 is expressed by endothelium in the inflamed skin and can trigger the activation and adhesion of cutaneous leukocyte antigen positive skin-specific memory cells expressing high levels of CCR4 (12); CCR8 is selectively expressed on T cells in noninflamed skin where it interacts with CCL1 to promote immune surveillance (13). In contrast, CCL25 is constitutively expressed in epithelium and mucosal vessels in the small bowel (14), where it interacts with gut-homing B and T cells expressing its receptor, CCR9. CCR9 is coexpressed with α4β7 on gut-homing lymphocytes and promotes adhesion to mucosal addressin cell adhesion molecule 1 (MAdCAM-1) on mucosal vessels (6, 15). Several studies support a role for CCR9 and α4β7 in lymphocyte homing to the gut. Virtually all small intestinal intraepithelial and lamina propria lymphocytes are CCR9+ α4β7+ (16), and the few CCR9+ lymphocytes in peripheral blood expressing high levels of α4β7+ are mostly memory cells and increase in number during gut inflammation (17). IgA-secreting B cells in the small intestine are also CCR9+ and dependent on CCL25 for positioning in the gut (18). Perhaps the most compelling evidence comes from studies showing that only dendritic cells from mesenteric lymph nodes and Peyer's patches are able to imprint a CCR9+ α4β7+ phenotype on lymphocytes that directs homing to the gut after adoptive transfer in vivo (19).
In contrast, little is known about the signals that regulate lymphocyte homing to the noninflamed liver. CXCR3 ligands are important during inflammation and it has been proposed that the endothelial adhesion molecule vascular adhesion protein 1 (VAP-1), which is constitutively expressed on human liver endothelium, may be involved in trafficking (20). The first evidence of aberrant homing of gut-derived lymphocytes to the liver came from studies showing that α4β7+ lamina propria lymphocytes bind to liver endothelium in PSC via both constitutively expressed VAP-1 and aberrantly expressed MAdCAM-1 (21). However, efficient recruitment of T cells also requires the presence of an appropriate chemokine-mediated signal to activate integrin-mediated adhesion and transmigration. Because of the strong association between CCL25-CCR9 and gut homing, we investigated whether CCR9 was involved in the recruitment of mucosal lymphocytes to the liver in PSC.
Materials and Methods
Tissue.
Matched liver tissue and peripheral blood was obtained at the time of liver transplantation. Small bowel tissue was obtained from intestinal resections for Crohn's disease. All samples were collected with patient consent and local research ethics committee approval.
Immunohistochemistry and Dual-Color Coimmunofluorescence.
6-μm cryostat sections were fixed in acetone for 10 min. 15 cases of PSC and 5 cases each of normal liver, primary biliary cirrhosis (PBC), and alcoholic liver disease were studied. Sections were incubated with 20% goat serum before rabbit anti–human CCL25 polyclonal antibody (P134; 1 μg/ml; PeproTech) or mouse anti–human CXCL12 mAb (MAB350; 8 μg/ml; R&D Systems) overnight at 4°C. Control sections were incubated without primary antibody or rabbit immunoglobulin. Sections were incubated for 20 min with biotinylated secondary antibodies followed by streptavidin–horseradish peroxidase complex developed with diaminobenzidine and counterstained with hematoxylin.
Sections for dual immunofluorescence were incubated with 20% goat/rabbit serum for 30 min before primary antibodies raised against CD68, CD31, Cytokeratin 19 (DakoCytomation), LYVE-1 (Upstate Biotechnology), or CCL25. Control sections were incubated without primary antibody. Sections were stained with goat anti–rabbit FITC (Southern Biotechnology Associates, Inc.) and goat anti–mouse IgG1 or IgG2a Texas red (Southern Biotechnology Associates, Inc.) secondary antibodies and nuclei counterstained with DAPI. Immunofluorescence was assessed using AxioVision software (Carl Zeiss MicroImaging, Inc.).
Western Blotting.
Fresh tissue was homogenized, normalized for total protein using Coomassie blue and loaded on 8% SDS-PAGE gels. After electrophoresis and transfer onto Hybond membranes (Amersham Biosciences) blocked with 10% skimmed milk. CCL25 was detected using goat anti–rabbit horseradish peroxidase–conjugated antibody detected with the ECL system (Amersham Biosciences).
Real-Time PCR.
RNA was extracted from snap-frozen tissue using RNeasy Mini Kit (QIAGEN). mRNA was transcribed to cDNA, and real-time PCR was performed on a PE7700 ABI Prism machine. Each reaction was performed in triplicate using QuantiTect Probe RT-PCR kit (QIAGEN) according to the manufacturer's instructions. Reactions contained 400 nM of CCL25-specific 5′-CCACACCCAAGGTGTCTTTGA-3′ and 5′-GAGCACAGCCCACCCAAT-3′ primers (AltaBioscience) and 200 nM of CCL25-specific Taqman probe, 5′-FAM-ACTGCTGCCTGGCCTACCACTACCC-TAMRA-3′; Eurogentech). Data are given as fold increase in gene expression normalized to the 18S control and compared with normal skin tissue (normalized to 1).
Liver- and Gut-derived Lymphocyte Isolation.
To preserve chemokine receptor expression, liver- and gut-infiltrating lymphocytes were isolated by mechanical methods. Tissue was homogenized in a Stomacher 400 circulator and filtered through a fine mesh, and lymphocytes were separated using 33/77% (vol/vol) Percoll density gradient centrifugation (Amersham Biosciences).
PBL Isolation.
Lymphocytes were isolated from venous blood collected into EDTA tubes, diluted 1:1 with PBS, and centrifuged over Lymphoprep (Life Technologies).
Four-Color Flow Cytometry.
Lymphocytes were incubated with goat immunoglobulin before primary unconjugated mAb against α4β7 (ACT-1; Millennium) for 30 min at 4°C. Cells were washed in PBS, centrifuged, and labeled with goat anti–mouse RPE-Cy5. Cells were subsequently stained with fluorochrome-labeled primary mAb against CCR9-RPE (R&D Systems), CD8-ECD (Beckman Coulter), CD3-RPE-Cy5, CD11a-FITC (DAKO), CD45RO-FITC or CD45RA-Cy5 (Serotec). Control samples were labeled with isotype-matched immunoglobulin. Cells were fixed in 1% paraformaldehyde before analysis on a Coulter Epics XL flow cytometer (Beckman Coulter) using Summit software (DakoCytomation).
Lymphocyte Enrichment, Stimulation, and Intracellular Staining.
Normal PBLs and PSC liver-infiltrating lymphocytes (LILs) were immunomagnetically enriched to 95% purity for CD45RA, and CD45RA and CCR9, respectively, with EasySep (StemCell Technologies, Inc.) and either CCR9-RPE (R&D Systems) or CD45RA-FITC (Serotec) mAb. Purity was confirmed by flow cytometry. Enriched populations were stimulated with 50 ng/ml PHA and 500 ng/ml ionomycin, and cytokine export was blocked after 1 h with 5 mg/ml brefeldin A. Cells were labeled with anti-CD8ECD before incubation in Permeafix (BD Biosciences) and labeling with anti-IFNγ FITC or anti–IL-2 PE (BD Biosciences). Viability of cells was assessed using Viaprobe (BD Biosciences). Unstimulated and isotype-matched mAb were used as controls.
Transwell Chemotaxis of Lymphocytes.
The migration of LILs from seven PSC livers was assessed using 6.5-mm diameter, 3-μm pore transwell inserts (Corning; reference 22). Responses to CCL5 and CXCL12 were positive controls because large numbers of LILs express CCR5 and CXCR4 (23). 100 ng/ml recombinant human CCL25, 100 ng/ml rhCCL5, or 100 ng/ml rhCXCL12 was placed in the bottom of the well, and 5 × 106 lymphocytes were added to the upper chamber. Cells were collected from the top and bottom chambers after 2 h and counted by flow cytometry. Control wells contained medium alone.
Static Adhesion Assay.
Lymphocytes were phenotyped by flow cytometry for CCR9 expression and used in the static adhesion assay. 18-well Teflon-coated slides (Erie Scientific) were incubated with 10 μg/ml rhICAM-1, 10 μg/ml rhMAdCAM-1 (prepared as described previously; reference 24), or 1 μg/ml BSA. Some lymphocytes were preincubated with 100 ng/ml pertussis toxin or blocking mAb against α4β7. Adhesion was triggered by the addition of 10 ng/ml CCL25 or 100 nM/ml MnCl2. After nonadherent cells were removed, slides were fixed and mounted before counting adherent lymphocytes in three representative high power fields per well.
Results and Discussion
Approximately 20% of LILs in PSC expressed CCR9+, whereas <2% were detected in livers of organ donors or patients with other chronic inflammatory liver diseases (Fig. 1). The mean channel fluorescence of CCR9 on LILs in PSC was lower than on PBL, suggesting that CCR9 may have been down-regulated on LILs by ligand interactions. However, CCR9 expression on LILs remained functional and was still able to trigger static adhesion and induce chemotaxis. CCR9+ expression was strongly associated with coexpression of the gut integrin α4β7.
Figure 1.
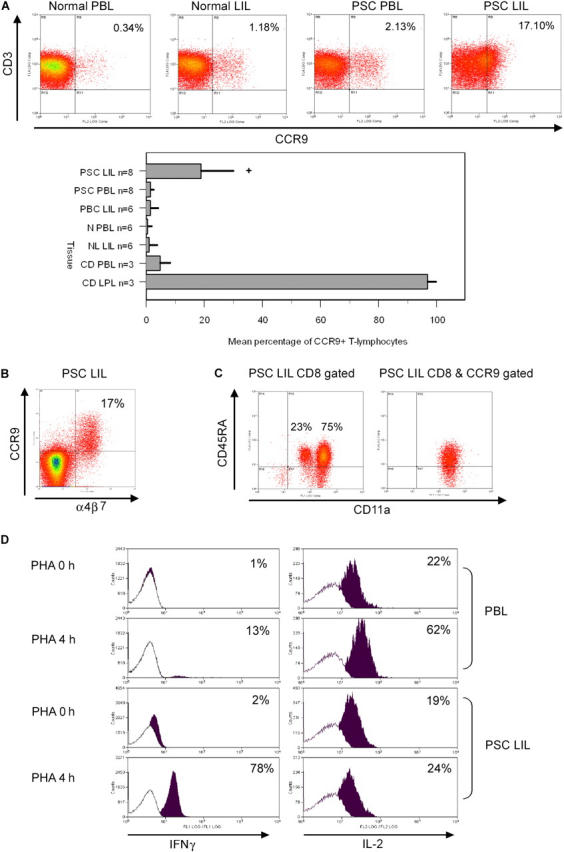
CCR9 expression and phenotyping of peripheral blood and tissue infiltrating lymphocytes. (A) 17% of liver-infiltrating T cells from PSC patients expressed CCR9+ compared with <1% normal PBLs and <2% PBLs from PSC patients (*, P < 0.01). 5% of PBLs and virtually all lamina propria CD3+ cells in Crohn's disease expressed CCR9, whereas <2% of LILs in PBC were CCR9+. The graph shows percentage of CCR9+/CD3+ cells as mean ± SEM for each group. (B) CCR9 is coexpressed with α4β7 on PSC LIL. (C) CCR9 was expressed on both CD8 and CD4+ LILs in PSC (CD8 63% vs. CD4 35%; not depicted). PSC CD8+ LILs are predominantly memory cells as demonstrated by their expression of high levels of CD11a (left). Gating of PSC LILs on CCR9 and CD8 reveals that all the CCR9+ cells are CD11ahigh and the majority also express CD45RA, confirming that they are long-lived memory cells. (D) PSC LILs were enriched for CD45RA+ and CCR9+ and their response to PHA stimulation was compared with that of CD45RA+ naive T cells from blood. After 4 h of PHA stimulation, intracellular cytokine staining of CD45RA+ PBLs showed a marked increase in IL-2, but minimal IFNγ production, consistent with a naive phenotype, whereas most CCR9+ CD45RA+ (CD11ahigh)-enriched PSC LILs stained for IFNγ consistent with memory cells. Isotype-matched controls are depicted as white histograms with positive cytokine staining as overlapping black histograms.
A functional role for CCR9 in PSC was supported by the strong CCL25 expression detected in the liver in PSC, but not in other chronic inflammatory diseases. CCL25 was detected in portal dendritic cells/macrophages and hepatic sinusoidal endothelium at areas of interface hepatitis in PSC, but was absent from other chronic inflammatory diseases (Fig. 2). The specificity of CCL25 staining for PSC was striking and we confirmed this using Western blotting with which we were unable to detect CCL25 in other liver diseases including primary biliary cirrhosis, another inflammatory biliary disease. Real-time PCR confirmed a 10-fold increase in CCL25 mRNA above control tissue. This is the first report of CCL25 expression outside the thymus or small intestine, and the lack of significant CCL25 up-regulation in other inflammatory liver diseases suggests that this aberrant expression is specific to PSC and likely to be involved in the pathogenesis of the disease.
Figure 2.
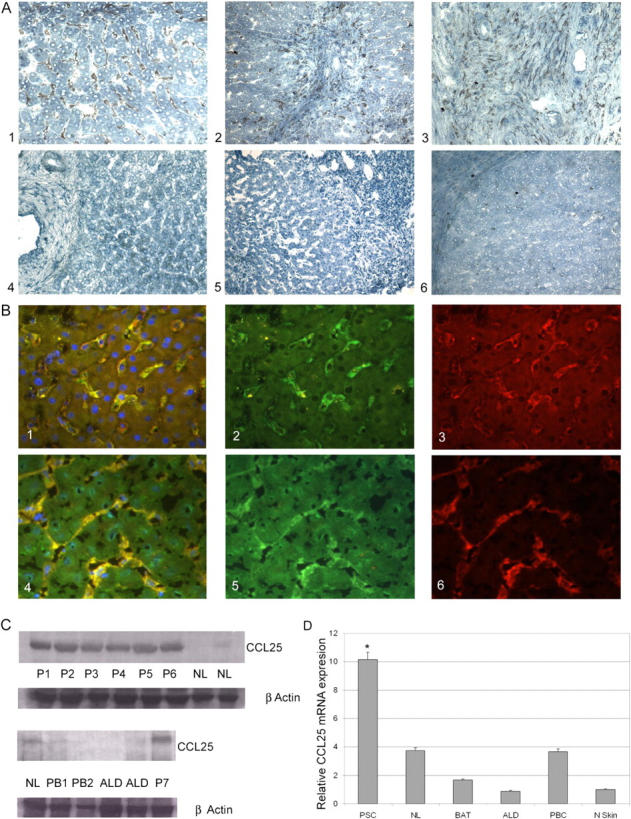
CCL25 expression in the liver. (A) The livers of patients with PSC demonstrated strong sinusoidal staining with CCL25 ab (A1, brown pigment), staining was particularly intense in periportal areas in association with areas of interface hepatitis (A2), and in portal tracts where macrophages/dendritic cells (confirmed by CD68 coexpression; not depicted) stained strongly (A3). There was no detectable CCL25 staining of hepatocytes, bile ducts, or vascular endothelium in normal liver (A4) or other chronic inflammatory diseases including primary biliary cirrhosis (A5). Staining with isotype-matched control antibodies was negative (A6). (B) Dual color immunofluorescence (B1/B4, yellow merged image) colocalized staining with CCL25 antibody (B2/B5, green) and CD31 antibody (B3, red) or LYVE-1 antibody (B6, red) to sinusoidal endothelial cells. Nulcei were counterstained with DAPI blue. (C) Western blotting confirmed the immunohistochemistry findings with detection of CCL25 in all PSC livers, but minimal detection in normal or other chronically inflamed livers. P1-7, PSC; NL, normal liver; PB1,2, PBC; ALD, alcoholic liver disease. Protein loading was normalized with β-actin staining. (D) Real-time PCR confirmed a 10-fold increase of CCL25 mRNA in PSC compared with control tissue (normal skin). *, P < 0.001. Modest amounts of CCL25 mRNA were detected in NL and other liver samples (BAT, biliary atresia; ALD and PBC) but the levels were not statistically significant compared with skin.
To determine that CCR9 is functionally active on LILs, we used adhesion and migration assays. CCR9+ LILs migrated preferentially to CCL25 rather than to CXCL12 or CCL5 and could also be triggered by CCL25 to bind immobilized MAdCAM-1 via α4β7 integrins (Fig. 3). CCL25-dependent migration and adhesion were both inhibited by pertussis toxin, suggesting that they were mediated by G proteins activated via CCR9. The CCR9+ T cells in the liver are primed/memory cells as demonstrated by their expression of high levels of CD11a (Fig. 1). The majority are also CD45RA+, suggesting that they belong to the recently described long-lived revertant memory population (25). We confirmed they are functional memory cells by demonstrating their ability to secrete IFNγ in response to PHA activation. The presence of functional CCR9 and α4β7 on liver-infiltrating memory T lymphocytes is strong evidence that they are of mucosal origin activated in the gut and subsequently recruited to the liver.
Figure 3.
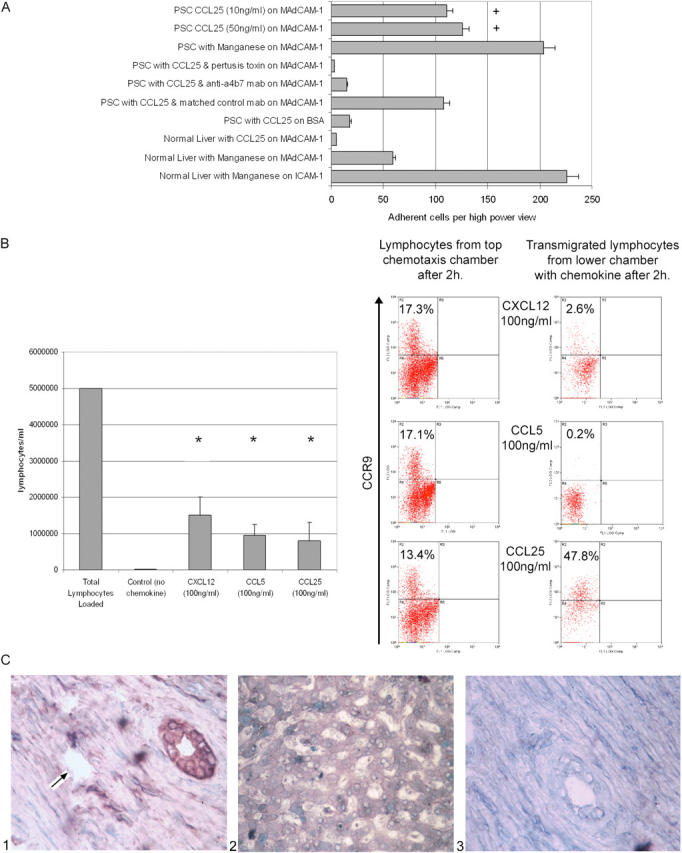
CCL25 triggers adhesion to MAdCAM-1 and chemotaxis of PSC LIL. (A) PSC LILs demonstrated a >20-fold dose-dependent increase in adhesion to MAdCAM-1 under the influence of 10 ng/ml CCL25 compared with normal LILs (+, P = 0.002), which was blocked by an antibody to α4β7 or preincubation with pertussis toxin. BSA served as a mock substrate, and MnCl2 was used as a nonsignaling trigger of integrin adhesion. Binding of normal lymphocytes to ICAM-1 in response to Mn confirms that these cells could be triggered to bind via integrins. There was no significant binding to MAdCAM-1 in the absence of CCL25 or Mn. (B) CCL25 (16 ± 7%), CXCL12 (29 ± 10%), and CCL5 (19 ± 6%) all stimulated chemotaxis of PSC liver-derived lymphocytes in invasion chambers in vitro (*, P < 0.02, n = 7). Phenotyping of transmigrated lymphocytes by flow cytometry confirmed that the CCR9+ LILs responded to CCL25, but not to CXCL12 or CCL5, suggesting that CCL25 is the dominant chemokine recruiting CCR9+ LIL. (C) Expression of CXCL12 is restricted to biliary epithelium (C1) and is not detected on portal (C1, arrow) or sinusoidal endothelium (C2) in PSC. No staining was seen in isotype matched control sections (C3). Thus, CXCL12 is unlikely to be important in recruiting CCR9+ cells to the liver, but might be involved in positioning/retaining recruited cells around bile ducts.
The overlapping expression of endothelial adhesion molecules and now tissue-specific chemokines between the liver and the gut suggests that, under particular circumstances, effector/memory lymphocytes are able to migrate to both sites as part of an entero-hepatic T cell recirculation. Such a mechanism may have evolved to provide surveillance against gut-derived antigens entering the liver via the portal circulation and is likely to be important in the pathogenesis of the immune-mediated hepatic complications of IBD. We propose that T cells activated in the gut during episodes of active IBD differentiate into effector cells that have the ability to bind to both hepatic and mucosal endothelium. Under noninflamed conditions, these cells may be able to enter the liver via interactions with VAP-1, an adhesion molecule that is constitutively expressed on liver endothelium and up-regulated on inflamed mucosal endothelium in IBD. Some of the effector T cells generated in the organized lymphoid tissue of the gut during active IBD (4) will revert to long-lived memory cells with the ability to recirculate to the liver and subsequently to trigger hepatic inflammation under the right conditions, even in the absence of active gut inflammation. The activation and expansion of these memory cells in the liver may result in the induction of MAdCAM-1 and CCL25 in the liver, promoting the recruitment of CCR9+ α4β7+ mucosal T cells and the development of established inflammation (1). This model, in which long-lived gut-derived memory T cells recirculate through the liver, could explain why IBD and PSC do not always occur at the same time and how PSC can be present for the first time in patients with IBD whose colons have been removed many years before (1). The alternative hypothesis, in which the extra-intestinal complications of IBD are driven by activated effector cells that are a consequence of the continuing release of factors by the inflamed colon, can explain the joint and skin complications that run concomitantly with active bowel disease (26) but not how PSC can develop de novo after colectomy or why the activity of liver disease does not parallel bowel inflammation.
PSC is associated with both ulcerative colitis and Crohn's disease, and usually develops in patients with colitis, suggesting that colonic inflammation is critical. Because CCL25 is largely confined to Peyer's patches and the small bowel mucosa, other gut-specific adhesion molecules might be involved (including CCR10) that may have a role in the recruitment of effector cells to the colon (14). However, recent papers show significant increases in CCR9+ lymphocytes in the blood during colitis, suggesting that colonic inflammation activates the CCR9 mucosal T cell pool (16, 17).
Our results demonstrate for the first time aberrant expression of CCL25 outside the gut or thymus associated with infiltration of CCR9+ T cells into the liver. The ability of CCL25 to activate migration and α4β7-mediated adhesion of LILs to MAdCAM-1 suggests that CCL25 and MAdCAM-1 cooperate in the recruitment of mucosal lymphocytes to the liver in PSC.
Acknowledgments
This work was supported by grants from the Medical Research Council, the Digestive Disorders Foundation, the European Commission, and a research grant from Pfizer Inc. to A. Miles.
M. Briskin was employed by Millennium Pharmaceuticals Inc. during this work. The authors have no other potential conflicting financial interests.
References
- 1.Chapman, R.W. 1991. Aetiology and natural history of primary sclerosing cholangitis–a decade of progress? Gut. 32:1433–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grant, A.J., P.F. Lalor, M. Salmi, S. Jalkanen, and D.H. Adams. 2002. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet. 359:150–157. [DOI] [PubMed] [Google Scholar]
- 3.Befeler, A.S., T.W. Lissoos, T.D. Schiano, H. Conjeevaram, K.A. Dasgupta, J.M. Millis, K.A. Newell, J.R. Thistlethwaite, and A.L. Baker. 1998. Clinical course and management of inflammatory bowel disease after liver transplantation. Transplantation. 65:393–396. [DOI] [PubMed] [Google Scholar]
- 4.Nagata, S., C. McKenzie, S.L. Pender, M. Bajaj-Elliott, P.D. Fairclough, J.A. Walker-Smith, G. Monteleone, and T.T. MacDonald. 2000. Human Peyer's patch T cells are sensitized to dietary antigen and display a Th cell type 1 cytokine profile. J. Immunol. 165:5315–5321. [DOI] [PubMed] [Google Scholar]
- 5.von Andrian, U.H., and C.R. Mackay. 2000. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 343:1020–1034. [DOI] [PubMed] [Google Scholar]
- 6.Pachynski, R.K., S.W. Wu, M.D. Gunn, and D.J. Erle. 1998. Secondary lymphoid-tissue chemokine (SLC) stimulates integrin α4β7-mediated adhesion of lymphocytes to mucosal addressin cell adhesion molecule-1 (MAdCAM-1) under flow. J. Immunol. 161:952–956. [PubMed] [Google Scholar]
- 7.Salmi, M., and S. Jalkanen. 1999. Molecules controlling lymphocyte migration to the gut. Gut. 45:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell, J.J., and E.C. Butcher. 2000. Chemokines in tissue-specific and microenvironment-specific lymphocyte homing. Curr. Opin. Immunol. 12:336–341. [DOI] [PubMed] [Google Scholar]
- 9.Warnock, R.A., S. Askari, E.C. Butcher, and U.H. von Andrian. 1998. Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J. Exp. Med. 187:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan, L., C.R. Reilly, Y. Luo, M.E. Dorf, and D. Lo. 2000. Cutting edge: ectopic expression of the chemokine TCA4/SLC is sufficient to trigger lymphoid neogenesis. J. Immunol. 164:3955–3959. [DOI] [PubMed] [Google Scholar]
- 11.Berlin, C., E.L. Berg, M.J. Briskin, D.P. Andrew, P.J. Kilshaw, B. Holzmann, I.L. Weissman, A. Hamman, and E.C. Butcher. 1993. α4β7 integrin mediates binding to the mucosal vascular addressin MAdCAM-1. Cell. 74:185–195. [DOI] [PubMed] [Google Scholar]
- 12.Campbell, J.J., G. Haraldsen, J. Pan, J. Rottman, S. Qin, P. Ponath, D.P. Andrew, R. Warnke, N. Ruffing, N. Kassam, et al. 1999. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature. 400:776–780. [DOI] [PubMed] [Google Scholar]
- 13.Schaerli, P., L. Ebert, K. Willimann, A. Blaser, R.S. Roos, P. Loetscher, and B. Moser. 2004. A skin-selective homing mechanism for human immune surveillance T cells. J. Exp. Med. 199:1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunkel, E.J., J.J. Campbell, G. Haraldsen, J. Pan, J. Boisvert, A.I. Roberts, E.C. Ebert, M.A. Vierra, S.B. Goodman, M.C. Genovese, et al. 2000. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J. Exp. Med. 192:761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zabel, B.A., W.W. Agace, J.J. Campbell, H.M. Heath, D. Parent, A.I. Roberts, E.C. Ebert, N. Kassam, S. Qin, M. Zovko, et al. 1999. Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J. Exp. Med. 190:1241–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosoe, N., S. Miura, C. Watanabe, Y. Tsuzuki, R. Hokari, T. Oyama, Y. Fujiyama, H. Nagata, and H. Ishii. 2003. Demonstration of functional role of TECK/CCL25 in T lymphocyte-endothelium interaction in inflamed and uninflamed intestinal mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 286:G458–G466. [DOI] [PubMed] [Google Scholar]
- 17.Papadakis, K.A., J. Prehn, S.T. Moreno, L. Cheng, E.A. Kouroumalis, R. Deem, T. Breaverman, P.D. Ponath, D.P. Andrew, P.H. Green, et al. 2001. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn's disease. Gastroenterology. 121:246–254. [DOI] [PubMed] [Google Scholar]
- 18.Kunkel, E.J., D.J. Campbell, and E.C. Butcher. 2003. Chemokines in lymphocyte trafficking and intestinal immunity. Microcirculation. 10:313–323. [DOI] [PubMed] [Google Scholar]
- 19.Mora, J.R., M.R. Bono, N. Manjunath, W. Weninger, L.L. Cavanagh, M. Rosemblatt, and U.H. von Andrian. 2003. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 424:88–93. [DOI] [PubMed] [Google Scholar]
- 20.Lalor, P.F., S. Edwards, G. McNab, M. Salmi, S. Jalkanen, and D.H. Adams. 2002. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J. Immunol. 169:983–992. [DOI] [PubMed] [Google Scholar]
- 21.Grant, A.J., P.F. Lalor, S.G. Hubscher, M. Briskin, and D.H. Adams. 2001. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium. Hepatology. 33:1065–1072. [DOI] [PubMed] [Google Scholar]
- 22.Campbell, J.J., E.P. Bowman, K. Murphy, K.R. Youngman, M.A. Siani, D.A. Thompson, L. Wu, A. Zlotnik, and E.C. Butcher. 1998. 6-C-kine (SLC), a lymphocyte adhesion-triggering chemokine expressed by high endothelium, is an agonist for the MIP-3beta receptor CCR7. J. Cell Biol. 141:1053–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shields, P.L., C.M. Morland, M. Salmon, S. Qin, S.G. Hubscher, and D.H. Adams. 1999. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J. Immunol. 163:6236–6243. [PubMed] [Google Scholar]
- 24.Tidswell, M., R. Pachynski, S.W. Wu, S.Q. Qiu, E. Dunham, N. Cochran, M.J. Briskin, P.J. Kilshaw, A.I. Lazarovits, D.P. Andrew, et al. 1997. Structure-function analysis of the integrin β7 subunit: identification of domains involved in adhesion to MAdCAM-1. J. Immunol. 159:1497–1505. [PubMed] [Google Scholar]
- 25.Faint, J.M., N.E. Annels, S.J. Curnow, P. Shields, D. Pilling, A.D. Hislop, L. Wu, A.N. Akbar, C.D. Buckley, P.A. Moss, et al. 2001. Memory T cells constitute a subset of the human CD8+CD45RA+ pool with distinct phenotypic and migratory characteristics. J. Immunol. 167:212–220. [DOI] [PubMed] [Google Scholar]
- 26.Salmi, M., K. Granfors, R. MacDermott, and S. Jalkanen. 1994. Aberrant binding of lamina propria lymphocytes to vascular endothelium in inflammatory bowel diseases. Gastroenterology. 106:596–605. [DOI] [PubMed] [Google Scholar]