

Abstract
Oxidation of low density lipoprotein (LDL) generates a variety of oxidatively modified lipids and lipid-protein adducts that are immunogenic and proinflammatory, which in turn contribute to atherogenesis. Cells undergoing apoptosis also display oxidized moieties on their surface membranes, as determined by binding of oxidation-specific monoclonal antibodies. In the present paper, we demonstrated by mass spectrometry that in comparison with viable cells, membranes of cells undergoing apoptosis contain increased levels of biologically active oxidized phospholipids (OxPLs). Indeed, immunization of mice with syngeneic apoptotic cells induced high autoantibody titers to various oxidation-specific epitopes of oxidized LDL, including OxPLs containing phosphorylcholine, whereas immunization with viable thymocytes, primary necrotic thymocytes, or phosphate-buffered saline did not. Reciprocally, these antisera specifically bound to apoptotic cells through the recognition of oxidation-specific epitopes. Moreover, splenocyte cultures from mice immunized with apoptotic cells spontaneously released significant levels of T helper cell (Th) 1 and Th2 cytokines, whereas splenocytes from controls yielded only low levels. Finally, we demonstrated that the OxPLs of apoptotic cells activated endothelial cells to induce monocyte adhesion, a proinflammatory response that was abrogated by an antibody specific to oxidized phosphatidylcholine. These results suggest that apoptotic cell death generates oxidatively modified moieties, which can induce autoimmune responses and a local inflammatory response by recruiting monocytes via monocyte–endothelial cell interaction.
Keywords: apoptosis, lipid peroxidation, autoimmunity, inflammation, oxidized LDL
Introduction
Apoptotic cells have been increasingly recognized as targets of autoantibodies that arise in a broad spectrum of autoimmune diseases. The evidence that apoptotic cells are targeted by autoantibodies was provided by studies showing that autoantibodies from humans and mice with systemic lupus erythematosus (SLE) recognize autoantigens that are prominently clustered in the surface blebs of apoptotic cells (1, 2). Several studies have also demonstrated that antiphospholipid antibodies (aPLs), which arise in a variety of autoimmune disorders such as the primary aPL syndrome and SLE, recognize the plasma membranes of apoptotic cells, but not viable cells (3, 4). In addition, mice immunized with syngeneic apoptotic cells have been shown to generate autoantibodies to apoptotic cells and cellular components, such as ssDNA and cardiolipin (5), implying that apoptotic cells not only serve as targets for antibodies but also provide autoantigens that trigger autoimmune responses. Moreover, evidence suggests that defective clearance and, hence, abnormal accumulation of apoptotic cells may contribute to such autoimmune response (6, 7). However, the exact molecular nature of the antigenic determinants or immunostimulatory molecules that induce such autoimmune response remains elusive.
Atherosclerosis is now considered a chronic inflammatory disease of the vascular wall; in addition, immune mechanisms modulate its progression (8, 9). Oxidized low density lipoprotein (Ox[LDL]) has been demonstrated to play a key role in atherogenesis because of its proinflammatory and immunogenic properties (10, 11). Oxidation of LDL generates a variety of oxidatively modified lipids and lipid-protein adducts. For example, when polyunsaturated fatty acids in phospholipids of LDL undergo peroxidation, reactive decomposition products, such as the oxidized phospholipid (OxPL) backbone and malondialdehyde (MDA) are formed. These products can covalently modify protein and lipid moieties in LDL, leading to formation of neoself-epitopes, which we have termed “oxidation-specific” neoepitopes and demonstrated to be highly immunogenic (11). Antibodies to these oxidation-specific epitopes can be found in humans as well as in mice. We documented previously that hypercholesterolemic apolipoprotein E–deficient (apoE −/−) mice with atherosclerosis have robust antibody titers to OxLDL (12). Using copper OxLDL or MDA-modified LDL (MDA-LDL) as selecting antigens, we cloned monoclonal IgM autoantibodies to oxidation-specific epitopes of OxLDL from the spleens of naive apoE −/− mice, termed EO antibodies (13). Previous studies have shown that these monoclonal anti-OxLDL antibodies, such as the prototypic EO6, also recognize the plasma membranes of apoptotic cells but not viable cells (14), implying that some oxidation-specific epitopes on OxLDL are also present on membranes of cells undergoing apoptosis, which are known to be under increased oxidative stress. EO6 can block the uptake of OxLDL by macrophage scavenger receptors (15), and especially CD36 (16). Similarly, EO6 partially blocks the uptake of apoptotic cells by macrophages, implying that such OxPLs are ligands mediating apoptotic cell clearance as well (14). Indeed, Hazen et al. have demonstrated that a variety of phosphorylcholine (PC)-containing OxPLs are CD36 ligands (17).
Of considerable interest, EO6 has been demonstrated recently to be identical to highly conserved T15 clonospecific natural antibodies specific for PC (18). These T15 antibodies recognize PC as a component of the capsular polysaccharide of microorganisms, such as Streptococcus pneumoniae, and participate in the innate immune response to microorganisms in mice (19). We have shown previously that EO6/T15 bind to OxPLs containing the PC headgroup, such as 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-PC (POVPC), but did not bind to native, nonoxidized phosphatidylcholines even though they contained the same PC moiety (15, 18, 20). Thus, the PC headgroup is a cryptic epitope of phosphatidylcholine in native LDL or viable cell membranes, and the PC epitope becomes accessible for binding by these anti-PC antibodies after oxidation-induced conformational changes occur in the phosphatidylcholine molecule in OxLDL or apoptotic cells. In point of fact, it is the PC head group of aldol condensates of POVPC dimers, trimers, or larger aggregates that appears to be the actual epitope of free POVPC (20).
In the present paper, we demonstrate that immunization of mice with syngeneic apoptotic cells induced humoral immune responses to oxidation-specific epitopes of OxLDL and apoptotic cells. In addition, we demonstrate that cells undergoing apoptosis contain increased levels of biologically active OxPLs and that these apoptotic cells activate endothelial cells to induce monocyte adhesion, which can be blocked by EO6. These data indicate that cells undergoing apoptosis generate oxidation-specific neoepitopes, including biologically active OxPLs, which in turn serve as dominant autoantigens as well as provide “proinflammatory” signals, mediating autoimmune and inflammatory responses.
Materials and Methods
Mice and Cell/Immunogen Preparation.
All experimental protocols were approved by the Animal Subjects Committee at the University of California, San Diego. Thymocytes were isolated from 4–6-wk-old National Institutes of Health (NIH)/Swiss-Webster and C57BL/6 mice (Harlan Sprague Dawley, Inc.). To obtain apoptotic cells, thymocytes were incubated with 1 μM dexamethasone (Sigma-Aldrich) or 50 ng/ml PMA (Sigma-Aldrich) in RPMI 1640/10% FCS for 15 h. Under these conditions, ∼80% of thymocytes have undergone apoptosis as judged by annexin V binding and among this population, ∼60% of thymocytes express oxidation-specific epitopes on their surface membranes as determined by binding of EO6 or EO14, which are autoantibodies specific for OxLDL and MDA-LDL, respectively (13). The apoptotic thymocytes were harvested and washed twice with PBS before the use. For monocyte adhesion and chemokine assays, we used serum deprivation for 18 h or UV irradiation (20 mJ/cm2) to generate apoptotic thymocytes, which also express OxPLs, as determined by EO6 binding (unpublished data).
Lipid Extraction and Analysis by Mass Spectrometry.
Total lipids were extracted from thymocytes by chloroform/methanol as described previously (15, 21). The OxPLs in these lipids extracts were analyzed by liquid chromatography multiple reaction monitoring (LC/MRM) as described previously (21). A detailed description of these procedures can be found in the online supplemental Materials and Methods (available at http://www.jem.org/cgi/content/full/jem.20031763/DC1).
Immunization Protocol.
Three groups of 10-wk-old NIH/Swiss-Webster mice were immunized in the presence of adjuvant with the following: (a) PBS, (b) viable thymocytes, or (c) dexamethasone-treated thymocytes. 100 μl of 20 × 106 thymocytes in PBS or PBS alone was mixed with an equal volume of Freund's adjuvant, and used to immunize each mouse. For primary immunization, immunogens with CFA were injected subcutaneously into the inguinal area, whereas booster immunizations were given with IFA by i.p. injection 2, 4, and 6 wk after the primary. Plasma samples were obtained immediately before primary immunizations and 1 wk after final booster immunizations.
Four groups of 10-wk-old C57BL/6 mice were also immunized in the absence of adjuvant with the following: (a) PBS, (b) viable thymocytes, (c) dexamethasone-treated thymocytes, or (d) necrotic thymocytes. The primary necrotic thymocytes were generated by three cycles of freezing and thawing of initially viable cells. After the final thaw, the entire cell mixture was suspended in PBS. Each immunogen was injected i.p. every 2 wk for 8 wk. Plasma samples were obtained as described before.
Chemiluminescent Immunoassay.
Titers of autoantibodies in murine sera to oxidation-specific epitopes were determined using a chemiluminescent enzyme immunoassay as described in online supplemental Materials and Methods.
Competition Immunoassay.
Pooled or representative immune sera from mice immunized with apoptotic cells were diluted in PBS containing 1% BSA and incubated with increasing concentrations of indicated competitors for 18 h at 4°C, and the binding to OxLDL was determined by chemiluminescent immunoassay. The apparent binding affinities of the IgG and the avidity of IgM (22) of the antiserum to OxLDL were determined by competitive inhibition assay to calculate the dissociation constants (Kds) of antibodies according to the Klotz method (MW of apoB = 500,000; reference 23). Calculations are based on the concentration of soluble OxLDL at which binding of antiserum to immobilized OxLDL was inhibited by ∼50%. To demonstrate specificity of immune sera binding to apoptotic cells through recognition of oxidation-specific epitopes, immune sera diluted in 1% BSA/PBS were preabsorbed as described before. Immune complexes were pelleted, and supernatants were tested for remaining binding activity to apoptotic cells by flow cytometry analysis.
Immunofluorescence Microscopy and Flow Cytometry Analysis.
Normal, viable thymocytes or dexamethasone-induced apoptotic thymocytes were harvested and incubated with immune sera diluted in 1% BSA/PBS (buffer A) for 30 min at 4°C. After the incubation, cells were washed and incubated with 15 μg/ml of fluorescein-conjugated F(ab)2 fragments against mouse IgM or IgG (Jackson ImmunoResearch Laboratories) in buffer A for 30 min and then washed. For immunofluorescence microscopy study, cells were incubated with 1 μg/ml of Hoechst dye (Sigma-Aldrich) for 10 min, fixed with 2% paraformaldehyde, and spun down on glass slides using cytospin (ThermoShandon). Images were captured by deconvolution microscopy using a DeltaVision deconvolution microscopic system operated by SoftWorx software (Applied Precision). For flow cytometry analysis, cells were incubated with 1 μg/ml of propidium iodide (PI) for 10 min and immediately analyzed by a FACScan instrument (Becton Dickinson). Data from each experiment were analyzed using CELLQuest software.
Splenocyte Culture and Cytokine Assay.
3 d before sacrifice, mice were boosted i.p. with the respective immunogens without Freund's adjuvant. Splenocytes from the mice were taken up in DME with 10% FCS, 10 mM Hepes, and 2 mM gentamycin, and seeded at 5 × 105 nucleated cells per well into 96-well tissue culture plates (Falcon). After 72 h of incubation, the levels of cytokines in cell culture supernatants were determined by chemiluminescent immunoassays using matched monoclonal capturing antibodies and biotinylated detecting antibodies against murine cytokines according to the manufacturer's protocol (R&D Systems).
Monocyte Adhesion Assay.
Monocyte adhesion assay was performed as described previously (24). In brief, porcine aortic endothelial cells (PAECs) or human coronary artery endothelial cells (HCAECs) were cultured in 48-well tissue culture plates in medium 199 supplemented with 15% FCS and antibiotics until confluent. Stimulants (normal, viable thymocytes, apoptotic thymocytes, or LPS) were added to each well with confluent endothelial cells and incubated for 4 h at 37°C. After the incubation of stimulants with endothelial cells, stimulants were removed and endothelial cells were washed six times with PBS. THP-1 cells (a monocyte-like cell line) in RPMI 1640 medium with 10% FCS were seeded at 105 cells per well and incubated for 30 min at 37°C. Wells were washed gently to remove all but the firmly adherent THP-1 cells. The number of adherent THP-1 cells was determined in a 10× high-power field per well.
Flow Cytometry and Chemokine Assays.
HCAECs were cultured in 6-well or 96-well tissue culture plates for flow cytometry and chemokines assays, respectively, and confluent HCAECs were treated with complete culture media alone or apoptotic thymocytes as stimulant for 4 h at 37°C in the complete culture media. For flow cytometry analyses, HCAECs were harvested with EDTA and stained with antibodies against ICAM, VCAM, E-selectin, or CS-1 fibronectin (BD Biosciences) and analyzed using flow cytometry as described before. For chemokine analysis, the culture supernatants were collected and assayed for IL-8 protein levels (R&D Systems) as described before.
Online Supplemental Material.
Procedures for lipid extraction from cells and for analysis of these lipids extracts by LC/MRM, as well as detailed methods for chemiluminescent immunoassays are available in online supplemental material available at http://www.jem.org/cgi/content/full/jem.20031763/DC1.
Results
Cells Undergoing Apoptosis Generate Proinflammatory OxPLs.
We showed previously that apoptotic cells are recognized by monoclonal autoantibody EO6, which specifically recognizes the PC moiety of OxPLs, such as POVPC and 1-palmitoyl-2-epoxyisoprostane-sn-glycero-3-PC (PEIPC), as well as by antibody EO14, which recognizes MDA-modified epitopes (14). These recent observations suggested that membranes of apoptotic cells contain apoptotic cell–associated molecular patterns (7) that are derived from phospholipid oxidation. To definitively identify PC-containing OxPLs in membranes of cells undergoing apoptosis, we analyzed lipid fractions extracted from normal and apoptotic thymocytes by LC/MRM. Consistent with the immunologic characterization, lipid fractions of dexamethasone or PMA-treated apoptotic thymocytes had a significantly increased content of biologically active OxPLs including POVPC, PEIPC, 1-palmitoyl-2-glutaroyl-sn-glycero-3-PC, and 1-palmitoyl-2-lyso-sn-glycero-3-PC (lyso-PC) compared with lipid fractions from viable thymocytes (Fig. 1). As we have shown recently that EO6 and EO14 recognize surface membrane determinants that newly arise on apoptotic cells (14), the current data indicate that apoptotic cells contain increased levels of these oxidized moieties, which may also have redistributed to facilitate their immune recognition.
Figure 1.

LC/MRM analysis of OxPLs from cellular lipid extracts. Quantification of OxPLs obtained from lipid extracts of viable or apoptotic thymocytes induced either by dexamethasone (DEXA) or by PMA treatment was performed using LC/MRM. POVPC, 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-PC; PGPC, 1-palmitoyl-2-glutaroyl-sn-glycero-3-PC; PEIPC, 1-palmitoyl-2-epoxyisoprostane-sn-glycero-3-PC; lyso-PC, 1-palmitoyl-2-lyso-sn-glycero-3-PC. Amounts of the four OxPLs were determined using 1,2-dimyristoyl-sn-glycero-3-PC as a standard. Data are expressed as micrograms per milligram of the parent lipid 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (PAPC) ± SD from three separate extractions. *, P < 0.001 compared with viable thymocytes.
Normal Mice Immunized with Syngeneic Apoptotic Thymocytes Develop Autoantibodies against Oxidation-specific Epitopes.
To determine whether these stress-induced oxidation-specific epitopes on apoptotic cells could induce specific immune responses, we immunized NIH/Swiss-Webster mice with syngeneic apoptotic thymocytes emulsified in Freund's adjuvant. After the final boost, plasma was obtained and tested for antibody binding to several models of oxidation-specific epitopes, including copper OxLDL, POVPC-modified LDL (POVPC-LDL), and MDA-modified LDL (MDA-LDL). As seen in Fig. 2, all mice immunized with apoptotic cells developed high IgM and high IgG titers to oxidation-specific epitopes of OxLDL. Moreover, mice immunized with apoptotic cells also developed IgM (Fig. 2 A) and to a lesser extent IgG (Fig. 2 B) titers to PC-KLH, suggesting the induction of anti-PC antibodies, which represent a family of autoantibodies including the natural anti-PC antibody T15/EO6 (18, 25, 26). In contrast, mice immunized only with PBS in adjuvant or with viable thymocytes in adjuvant developed lower or undetectable antibody titers to these antigens, respectively (Fig. 2, A and B).
Figure 2.
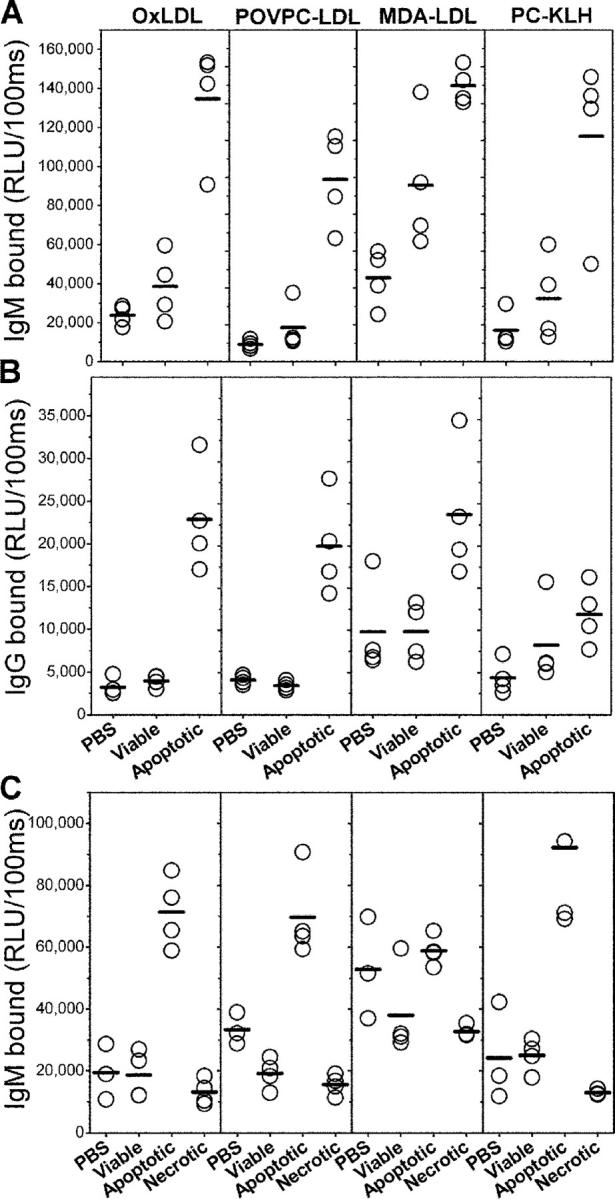
Binding of immune sera to oxidatively modified epitopes. Chemiluminescent immunoassay for the binding of IgM (A) and IgG (B) from pooled immune sera of NIH/Swiss-Webster mice to indicated antigens. NIH/Swiss-Webster mice were immunized in the presence of adjuvant with the following: PBS, viable, normal thymocytes (Viable), and dexamethasone-treated apoptotic thymocytes (Apoptotic). (C) Binding of IgM from pooled immune sera of C57BL/6 mice immunized in the absence of adjuvant with the following: PBS, viable, normal thymocytes (Viable), dexamethasone treated apoptotic thymocytes (Apoptotic), or suspension of primary necrotic thymocytes generated by repeated freeze-thaw cycles (Necrotic). Sera from NIH/Swiss-Webster and C57BL/6 mice were diluted at 1:400 and 1:250 in PBS with 2% BSA, respectively, and IgM and IgG bindings were measured as described in Materials and Methods. Data are expressed as relative light units (RLU) per 100 ms. Each point is the mean of triplicate determinations.
Using competition immunoassay studies, we demonstrated that the IgM binding of antisera from immunized mice to OxLDL was nearly completely inhibited by both OxLDL and MDA-LDL, but not native LDL, indicating high specificity of the induced antibodies for oxidation-specific epitopes (Fig. 3 A). PC-KLH also very efficiently inhibited the IgM binding of the antisera to OxLDL, whereas KLH alone did not, clearly demonstrating the specificity of some of these antibodies for PC (Fig. 3 A). Therefore, immunization with apoptotic cells induced antibodies that are highly specific for epitopes derived from phospholipid oxidation.
Figure 3.

Competition immunoassay for immune sera binding to OxLDL. (A) Competition immunoassay for binding of IgM from a representative serum of NIH/Swiss-Webster mice immunized with apoptotic thymocytes as described in Materials and Methods. The serum was diluted at 1:5,000 in PBS with 1% BSA and was incubated in the absence or presence of indicated concentrations of native LDL, OxLDL, MDA-LDL, or PC-KLH as competitors. Immune complexes were pelleted by centrifugation, and supernatants were tested for binding of IgM to OxLDL using chemiluminescent immunoassay. Competition immunoassay for IgM (B) and IgG (C) from another serum of NIH/Swiss-Webster mice immunized with apoptotic thymocytes was performed to determine the binding affinity of the antisera for OxLDL as described in Materials and Methods. IgM binding was performed with 1:2,000 dilution and IgG binding was performed with 1:300 dilution. Data are expressed as a ratio of the binding of serum in the presence of competitor (B) over the binding in the absence of competitor (Bo). Each point is the mean of triplicate determinations.
To compare the apparent binding affinities of the IgM and IgG of antisera to OxLDL, we performed competition immunoassays with these antisera using soluble OxLDL as a competitor, and determined the dissociation constants (Kds) of antibodies according to the Klotz method (23). The Kds values for IgM and IgG binding were 1.5 × 10−9 mol/l and 6.13 × 10−8 mol/l, respectively (Fig. 3, B and C).
To further evaluate the immunogenicity of apoptotic cells, we also immunized C57BL/6 mice with syngeneic apoptotic thymocytes in PBS alone (i.e., without adjuvant), as well as with viable thymocytes, or primary necrotic thymocytes generated by repeated freeze-thaw cycles (all without adjuvant). Fig. 2 C demonstrates that even immunization with apoptotic cells without adjuvant induced specific IgM antibodies against oxidation-specific epitopes. However, in contrast with mice immunized in the presence of adjuvant, in these mice, we did not observe equivalent IgG responses (unpublished data). Here as well, the specificity of the IgM binding to OxLDL was confirmed by competition immunoassays and found to be similar to the specificity of the antisera obtained from aforementioned immunized NIH/Swiss-Webster mice (unpublished data). This response was specific for apoptotic cells as immunization with neither viable thymocytes nor with primary necrotic thymocytes induced such immune responses. Thus, the process of programmed cell death seems to be a requisite for the generation of immunogenic oxidation-specific epitopes, as primary necrotic cells or cellular components leaking from damaged cells are not sufficient to induce humoral responses against oxidation-specific epitopes.
Immune Sera Bind to Apoptotic Thymocytes via Oxidation-specific Epitopes.
Next, we characterized the binding and specificity of IgM and IgG from antisera to apoptotic cells. Using deconvolution microscopy, we demonstrated specific binding of the immune sera to apoptotic cells (Fig. 4 A). Using immune sera from NIH/Swiss-Webster mice, we noted both IgM and IgG binding to syngeneic apoptotic cells seen as surface staining in a specific discrete pattern (Fig. 4 A, green), consistent with membrane blebbing that occurs with apoptosis (Fig. 4 A). The condensed nuclei characteristic of apoptotic cells were detected by Hoechst DNA staining (Fig. 4 A, blue). Preimmune sera did not stain the apoptotic cells (Fig. 4 B), and immune sera did not show reactivity with viable thymocytes (Fig. 4 C). Moreover, flow cytometry analyses demonstrated that the binding of these immune sera (IgM) was especially pronounced in apoptotic thymocytes that were highly positive for PI staining, indicating a later stage of apoptosis (Fig. 5 A, R3). Because apoptotic cells generate OxPLs in their membranes (Fig. 1), we tested whether the binding of immune sera to apoptotic cells was in part due to binding to oxidation-specific epitopes. We performed competition studies of the IgM binding to apoptotic cells by flow cytometry and determined the intensity of binding of the antisera after preabsorption with indicated antigens. IgM binding of immune sera to the gated cells (Fig. 5 A, R3) was strongly decreased by absorption with either OxLDL or MDA-LDL, but not by native LDL, suggesting the importance of these epitopes for the immunoreactivity of apoptotic cells (Fig. 5 C). Moreover, preabsorption of immune sera with both OxLDL and MDA-LDL together led to an even stronger and dose-dependent (∼70%) inhibition of the binding to apoptotic cells (Fig. 5 C). Thus, a large part of the apoptotic cell–reactive IgM antibodies induced by immunization with apoptotic cells bind via oxidation-specific epitopes. In similar studies, IgG binding to apoptotic cells was also decreased by preabsorption of immune sera with either OxLDL or MDA-LDL by 50%, and both OxLDL and MDA-LDL together showed similar inhibition (∼60%) as was seen for the IgM binding to apoptotic cells (unpublished data).
Figure 4.

Immunofluorescence deconvolution microscopy of sera binding to apoptotic cells: Pooled (IgM) or a representative (IgG) preimmune and postimmune sera from NIH/Swiss-Webster mice immunized with apoptotic thymocytes were diluted 1:200 in PBS with 1% BSA and incubated with apoptotic or normal thymocytes. IgM or IgG binding to the cells was detected by fluorescein-conjugated F(ab)2 fragments against mouse IgM or IgG, respectively. (A) Note the marked binding of postimmune sera (green) to apoptotic thymocytes with their characteristic condensed, fragmented nuclei detected by Hoechst staining (blue). (B) Note the negative staining of preimmune sera to apoptotic cells and (C) postimmune sera to normal thymocytes. Bar, 5 μm.
Figure 5.

Flow cytometry analysis of sera binding to apoptotic thymocytes and competition immunoassay for immune sera binding to apoptotic cells. (A) Apoptosis-induced thymocytes were gated into three populations according to forward scatter and the intensity of PI staining. (R1) Region 1: viable cells or cells at the very early stage of apoptosis; (R2) region 2: cells at intermediate stage of apoptosis with dim PI staining, and (R3) region 3: cells at later stage of apoptosis with shrunken cell size and bright PI staining. (B) Pooled preimmune or postimmune sera from NIH/Swiss-Webster mice immunized with apoptotic thymocytes were diluted at 1:500 in PBS with 1% BSA and tested for IgM binding to apoptotic thymocytes in region 3. (C) Aliquots of diluted, pooled sera were incubated in the absence or presence of OxLDL or MDA-LDL, or both as competitors at indicated concentrations (μg/ml). After the incubations, immune complexes were pelleted by centrifugation and supernatants were tested for IgM binding to apoptotic thymocytes using flow cytometry. Mean fluorescence intensity of binding of immune sera incubated in the absence (No) or presence of competitors to the apoptotic thymocytes in region 3 was measured. Data display the inhibition of binding by competitors as percent of control in the absence of competitors and representative of three independent experiments.
Splenocyte Cultures from Immunized Mice Secrete Both Th1 and Th2 Cytokines.
Because NIH/Swiss-Webster mice immunized with apoptotic cells in the presence of adjuvant developed high titers of IgG antibodies to oxidation-specific epitopes, we tested whether cellular immune responses were induced in these mice as well. Therefore, we determined the cytokine secretion of splenocyte cultures obtained from all immunized NIH/Swiss-Webster mice.
In the initial studies, splenocytes were cultured in the absence or presence of syngeneic apoptotic or viable thymocytes to assess antigen-specific cytokine secretion. After 72 h of incubation, cytokine levels were measured in culture supernatants. Surprisingly, we found high cytokine secretion in the culture supernatants of immunized mice, but not naive mice, even in the absence of any added antigenic stimuli. We speculated that splenocytes themselves could undergo apoptosis during the in vitro culture for 72 h, which in turn could provide autoantigenic stimuli. Indeed, splenocytes cultured for 72 h had definite evidence of apoptosis, which was detected by DNA electrophoresis documenting the typical ladder pattern of DNA fragmentation (unpublished data). Indeed, a similar degree of apoptosis was found in the splenocyte cultures obtained from all three experimental groups.
As shown in Fig. 6, splenocyte cultures from mice immunized with apoptotic cells in the presence of adjuvant showed both Th1 (IFNγ) and Th2 (IL-5, IL-10, and IL-13) cytokine release in the absence of added antigen. In sharp contrast, splenocyte cultures from mice immunized with PBS or viable thymocytes (which also contained adjuvant) showed only very low levels of cytokine responses, except for INFγ in the case of viable cells (even though these cultures contained apoptotic cells as well). Additional control cultures were stimulated with plate-bound anti-CD3 to assess maximal nonspecific secretion of cytokines, which yielded similar results among splenocyte cultures of all three groups (unpublished data). These findings clearly demonstrate that the resulting cytokine secretion was specific (with the possible exception of INFγ, which may be related in part to the adjuvant) for the immunization with apoptotic cells, which induced strong cellular immune responses after immunization in the presence of adjuvant.
Figure 6.

Cytokine assay of splenocyte cultures. Splenocytes from each mouse in each group of NIH/Swiss-Webster mice immunized with PBS, viable cells, or apoptotic cells were harvested. Splenocytes were seeded at 5 × 106 cells per well into a 96-well tissue culture plate and were cultured in triplicates in the absence of any added antigens for 72 h. After the incubation, the amount of cytokine released into the supernatants was determined using chemiluminescent immunoassays as described in Materials and Methods. Data reflect mean ± SEM values of each group. *, P < 0.04 compared with groups immunized with either PBS or normal cells.
Apoptotic Cells with Oxidation-specific Epitopes Induce Monocyte Adhesion to Endothelial Cells.
Monocytes are the primary cells that are localized to chronic inflammatory lesions, including atherosclerosis. Initially, monocytes roll along the vascular endothelium where they become activated by soluble or surface-bound chemokines. The activated monocytes adhere firmly to the endothelium and transmigrate through the endothelial cell monolayer. This occurs by the expression of adhesion molecules on overlying endothelial cells induced by inflammatory stimuli. OxLDL and OxPLs, such as POVPC and PEIPC, have been shown to be capable of activating endothelial cells, leading to monocyte-endothelial cell interaction (21, 27, 28).
To determine whether OxPLs that are present in the context of apoptotic cells (14) may also exert such proinflammatory properties, we tested the ability of the apoptotic cells to activate PAECs, as assessed by the ability of PAECs to bind monocytes. Preincubation of PAECs with apoptotic thymocytes led to the subsequent enhanced adhesion of THP-1 cells, a human monocyte-like cell line frequently used in these studies (24, 29), to the confluent layer of PAECs (Fig. 7 A). In contrast, preincubation of viable thymocytes did not lead to activation of the PAECs (unpublished data). Furthermore, the monocyte adhesion induced by apoptotic thymocytes was almost abolished by the inclusion of EO6 during the preincubation, which binds specifically to PC-containing OxPLs; however, note that EO6 does not bind to lyso-PC (20). EO6 did not inhibit LPS-induced monocyte adhesion, implying that OxPLs that were bound by EO6, such as POVPC or PEIPC, were responsible for this effect. We also used HCAECs in a similar study. As shown in Fig. 7 B, we observed a significant ability of apoptotic cells to activate HCAECs to bind THP-1 monocytes, and again viable thymocytes did not.
Figure 7.

Monocyte adhesion to endothelial cells induced by apoptotic cells. (A) The ability of apoptotic cells to stimulate endothelial cells for monocyte binding was tested. Porcine aortic endothelial cells (PAECs) were incubated for 4 h at 37°C with culture media alone or EO6 antibody in the absence or presence of apoptotic thymocytes (5 × 106 per well) generated by incubation with serum-starved media for 18 h as stimulant. 100 ng/ml LPS was used as positive control. For these experiments, EO6 antibody was obtained from the culture supernatant of EO6 hybridoma that was maintained under LPS-free culture condition. After the incubation, stimulants were washed off with PBS, and PAECs were incubated with THP-1 cells for 30 min at 37°C. Nonadherent THP-1 cells were removed by washing, and the number of adherent THP-1 cells was determined in 10× high-power field per well. The results of four to six separate wells were averaged for each experiment. Data reflect mean ± SEM values of data from three different experiments. (B) Human coronary artery endothelial cells (HCAECs) were tested. In this experiment, viable, normal thymocytes were used as another control.
To directly address the mechanism by which apoptotic cells induce monocyte–endothelial cell interaction, we tested the expression of adhesion molecules on HCAECs using flow cytometry. We did not observe enhanced expression of VCAM-1 and CS-1 fibronectin on the stimulated endothelial cells (10). However, we did observe HCAECs to secrete a significantly higher level of IL-8 when they were incubated with apoptotic cells compared with that in the absence of stimulant or in the presence of normal, viable cells (Fig. 8). Recently, IL-8 has been reported to mediate firm monocyte adhesion to endothelial cells even under flow conditions (30, 31). Furthermore, the ability of PC-containing OxPLs to induce expression of IL-8 in endothelial cells has been documented (21).
Figure 8.

Apoptotic cells stimulate endothelial cells to secrete IL-8. HCAECs were incubated for 4 h at 37°C with culture media alone; viable, normal thymocytes; or apoptotic thymocytes induced by UV irradiation as stimulant. Apoptotic thymocytes themselves did not release IL-8. 200 ng/ml LPS was used as a positive control. After the incubation, IL-8 in the supernatants was determined as described in Materials and Methods. Data represent the mean ± SEM of quadruplicates.
Discussion
Because the oxidative modification of LDL renders it immunogenic and induces robust autoantibody responses to oxidized lipids and/or lipid-protein adducts (11), and because apoptotic cells display such oxidized moieties on their plasma membranes (14), we investigated whether oxidation-specific epitopes of apoptotic cells could also give rise to similar autoimmune responses. Cell membranes contain phospholipids with polyunsaturated fatty acids that would be susceptible to oxidation when cells undergo apoptosis. Indeed, a substantial body of evidence suggests that cells undergoing apoptosis are under increased oxidative stress, and that reactive oxygen species and/or OxPLs mediate an apoptotic program and recognition of dying cells by phagocytes (32–34).
In the present paper, we demonstrate directly that apoptotic cells have an increased content of biologically active OxPLs and that apoptotic cells bearing such oxidatively modified moieties are immunogenic. Indeed, immunization of mice with syngeneic apoptotic cells induced high titers of autoantibodies to various oxidation-specific epitopes of OxLDL, whereas immunization with neither viable, normal cells nor primary necrotic cells did (Fig. 2). Thus, the process of apoptosis is requisite for the generation of immunostimulatory, oxidation-specific neoepitopes that induce such autoimmune responses. Reciprocally, we also demonstrate that immune sera bind to the cells at later stages of apoptosis, in large part through the recognition of oxidation-specific epitopes, implying that these epitopes can also serve as antigenic determinants for the recognition of apoptotic cells by autoantibodies. Although it has been demonstrated previously that syngeneic apoptotic cells can be immunogenic under appropriate conditions, our data demonstrate that, remarkably, up to 70% of the IgM from such murine sera (and ∼60% of IgG) are directed at oxidation-specific epitopes of apoptotic cells. These data demonstrate that such oxidation-specific epitopes are immunodominant neoepitopes, just as they are when LDL undergoes oxidative modification.
Apoptosis is a physiological form of cell death responsible for removal of unwanted cells in diverse physiological and pathological processes (7). Normally, apoptotic cells are cleared swiftly by phagocytes without inciting inflammatory or immune responses. However, under certain conditions, the pathological accumulation of increased numbers of apoptotic cells may incite inflammatory and immune responses (6, 7, 35). Indeed, an increased rate of formation, and/or a defective clearance of apoptotic cells has been postulated as a potential mechanism that may be responsible for autoimmune responses (7, 36). The systemic exposure of apoptotic cells in mice by immunization with syngeneic apoptotic cells, but not viable cells, has been shown to initiate autoantibodies to apoptotic cells or autoantigens, such as ssDNA and cardiolipin (4, 5). Moreover, studies have demonstrated that humans and mice with C1q deficiency, or mice lacking function of the c-mer membrane tyrosine kinase show defective clearance of apoptotic cells, and develop a lupuslike syndrome as well as anti-DNA antibodies (37–39). Similarly, mice deficient in MFG-E8, which led to impaired uptake of apoptotic cells, were also noted to have autoantibody generation and glomerulonephritis (40).
Therefore, we speculate that in conditions that lead to defective clearance of apoptotic cells and advancement to later stages of apoptosis, novel antigenic determinants are generated, which in turn stimulate such autoimmune responses. Consistent with this, autoantibodies found in patients with SLE and aPL syndrome have been shown to recognize apoptotic cells as well as OxLDL (1–3, 41). In collaboration with others, we demonstrated enhanced titers of autoantibodies to oxidation-specific epitopes of OxLDL in patients with SLE (42), even in young children who have no evidence for clinical cardiovascular disease (43). However, the precise nature of the antigenic determinants of apoptotic cells that lead to such autoimmunity has not yet been clearly elucidated. Several studies have suggested that the autoantigens inducing autoimmune response during apoptosis might be structurally modified proteins, generated as a result of proteolytic cleavage and/or phosphorylation (36). Furthermore, earlier studies in our laboratory have shown that many so-called aPLs are in fact directed against OxPL and/or oxidized lipid-protein adducts, but not against non-OxPLs (44–46). These observations support our demonstration that oxidative modification serves to generate antigenic determinants. Another group also has shown that monoclonal anticardiolipin antibodies established from the (NZWxBXSB)F1 mouse model of aPL syndrome cross-reacted with OxLDL (47), and we too have made similar observations (unpublished data). Thus, our present studies provide direct evidence that as a consequence of apoptosis, oxidative modification of lipid components generates neoself-oxidation–specific epitopes, which in turn lead to an autoimmune response and serve as antigenic determinants targeted by the autoantibodies.
As noted in the Introduction, EO6 is a classic natural antibody specific to PC, and we have demonstrated previously that they bind specifically to apoptotic cells, but not to viable cells (14, 18). In mice, such natural anti-PC antibodies provide a first line of innatelike immune defense against a variety of microbial infections, such as S. pneumoniae (19), and mice deficient in T15 antibodies are uniquely susceptible to S. pneumoniae infection (48). Similar antibodies have been demonstrated in the plasma of healthy humans as well (49, 50). Because these germline encoded antibodies are naturally selected and are present within the first week of life even in mice grown in germ-free conditions (51), we have postulated that they are positively selected by PC exposed by oxidized phosphatidylcholine present on apoptotic cells and/or oxidized lipoproteins (18). In the present paper, we directly demonstrate that apoptotic cells can stimulate such anti-PC antibodies (Figs. 2 and 3). Therefore, we propose that oxidative modification of phosphatidylcholine in the membranes of cells undergoing apoptosis reveals PC as a natural antigenic determinant, which in turn stimulates B cell immune responses to PC. Furthermore, these data suggest that apoptotic cells bearing PC might play a role in the ontogeny of anti-PC antibodies.
The functional capacity of the immune system represents a sophisticated balance between the defense from invading infectious pathogens, and the avoidance of autoreactivity that can damage the host. Certainly, some B-1 cell–derived IgM antibodies may exert adverse effects (52). However, it is now also generally accepted that certain aspects of autoimmunity may occur in, and perhaps even contribute to, health (53). Several studies suggest that the recognition of IgM anti-PC antibodies to apoptotic cells is important in clearance of apoptotic cells as well as immune modulation. One consequence that occurs as a result of induction of apoptosis is the generation of enhanced oxidative mechanisms, resulting in enhanced content of oxidized phosphatidylserine (34). These likely form the important recognition signals for binding to the phosphatidylserine receptor on macrophages (34). We now add the generation of oxidized phosphatidylcholines in apoptotic cells as well. The presence of such PC-containing OxPLs are known ligands mediating CD36 binding (16, 17) and explains why antibody EO6 inhibits binding and uptake of apoptotic cells by macrophages (14).
In addition, our studies demonstrated the enhanced generation of lyso-PC (Fig. 1). The latter may occur as oxidized phosphatidylcholines contain oxidized sn-2 fatty acids that become preferred substrates for several PLA2-type enzymes, which lead to lyso-PC generation. A recent paper by Kim et al. has shown that human natural anti-PC IgM antibodies from sera of normal individuals bind to late apoptotic cells through recognition of lyso-PC. Subsequently, their binding recruited C1q on the cell surface leading to C3 activation (54), which in turn would enhance clearance of opsonized apoptotic cells (55). Moreover, because macrophages or dendritic cells phagocytosing apoptotic cells opsonized with C3 produce immunosuppressive cytokines, complement activation mediated by these IgM antibodies may play a crucial role in modulating the immune response (56, 57). In our paper, we directly demonstrate another potential protective capacity of such natural IgM anti-PC antibodies, such as EO6, which have the capacity to block proinflammatory properties (Fig. 7) and, thus, may represent another conserved arc of innate immunity that is recruited to dampen inflammatory responses. In a similar manner, EO6 binds to apoptotic cell blebs and abrogates some of their proinflammatory properties (58). Therefore, the possible scenario would be that these anti-PC antibodies stimulated by PC exposed in the context of apoptotic cells may confer protection to the host from more devastating inflammatory and autoimmune responses by promoting the clearance of dying cells and also by modulating these responses.
Alternately, it should be pointed out that autoimmune responses to PC and other oxidation-specific epitopes could bring harmful outcomes, in particular in the presence of other proinflammatory stimuli. As shown in Fig. 2 B, immunization of apoptotic cells in the presence of Freund's adjuvant, a potent proinflammatory stimulant, induced high titers of IgG to oxidation-specific epitopes, but such IgG immune responses were not observed in the absence of the adjuvant. Were such IgG immune responses to occur in vivo as a consequence of inflammatory stimuli, this could lead to the generation of IgG responses, engagement of IgG immune complexes to macrophage Fcγ receptors, and the production of inflammatory cytokines. This phenomenon may play a role in the pathogenesis of autoimmune diseases when the defective clearance, and hence the pathological accumulation of apoptotic cells, occur coupled with various proinflammatory stimuli, such as infectious agents or toxic oxidized lipids (i.e., as found in the atherosclerotic plaque. Indeed, it was recently shown that mice with impaired clearance of apoptotic cells due to Fas ligand deficiency, when crossed into apoE −/− mice, developed accelerated atherosclerosis and attendant autoimmune phenomenon, including enhanced autoantibody titers to epitopes found in OxLDL (59).
Atherosclerosis is now considered a chronic inflammatory disease of the vascular wall (8, 60) and there is a substantial body of evidence that OxPLs in OxLDL induce proinflammatory responses promoting the atherogenic process (10). PC-containing OxPLs activate endothelial cells to recruit monocytes into the vascular wall and stimulate them by regulating inflammatory gene expression (10, 61) OxLDL with such OxPLs accumulate in atherosclerotic lesions (13, 27). Furthermore, there is also a marked accumulation of apoptotic cells in atherosclerotic lesions (62). In the present paper, we demonstrate directly that the ability of apoptotic cells to activate endothelial cells to tightly bind monocytes is completely abrogated by EO6. This implies that the enhanced content of biologically active oxidized phosphatidylcholines in apoptotic cells activates endothelial cells to induce adhesion and recruitment of monocytes. Adhesion of monocytes to endothelial cells is a crucial, rate-limiting step in the pathogenesis of atherosclerosis in particular, and an initial process for the recruitment of macrophages into damaged tissues and inflammatory lesions in general. In this paper, we demonstrated further that such monocyte–endothelial interaction can be mediated in part through IL-8. IL-8 is a CXC chemokine produced by endothelial cells that has recently been reported to mediate firm adhesion of monocytes to endothelial cells, even under flow conditions (30, 63). Oxidized PAPC (1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine), and specifically isomers of PEIPC as well as POVPC (products readily bound by EO6), are known to be present in the atherosclerotic lesion and can induce such IL-8 expression (21). In support of an important role of IL-8 is the observation that deletion of the IL-8 receptor in LDLR −/− mice markedly decreases atherosclerosis (64).
Consistent with our finding, recent studies have demonstrated that apoptotic cells have the ability to provoke an inflammatory response by unidentified proinflammatory mediators (65, 66), suggesting that under certain conditions, apoptotic cells themselves may provide endogenous “danger signals” triggering an inflammatory response. Lauber et al. have shown recently that apoptotic cells release lyso-PC to serve as a chemotactic signal that induces attraction of monocytic cells to apoptotic cells, presumably to ensure their efficient removal (67). Lyso-PC released from OxLDL has been demonstrated already to be a potent chemotactic factor for monocytes and T cells (68, 69). Because EO6 does not bind to lyso-PC (20), yet abrogated the ability of apoptotic cells to induce firm monocyte adhesion to endothelial cells (Fig. 7), our findings suggest that oxidized phosphatidylcholines present on apoptotic cells are also prominent mediators that lead to recruitment of inflammatory cells. Thus, they too serve as endogenous inflammatory danger signals, ensuring an ensuing host response that could play an important role in limiting the danger posed by the accumulated dying cells.
It is tempting to postulate that oxidative modification of lipids in cells that occur during apoptosis is a novel mechanism by which apoptotic cells could promote autoimmune and proinflammatory responses. Alternately, such responses may be protective by enhancing the clearance of apoptotic cells and/or repression of such proinflammatory response. However, they could also participate in the pathology of a variety of diseases in which the abnormal accumulation of such cells occurs, for instance, in the atherosclerotic lesion (62) and other chronic inflammatory diseases, including autoimmune disorders such as SLE (7).
Acknowledgments
We thank F. Almazan for excellent technical assistance.
This work was supported by National Institutes of Health grant nos. HL56989 (Specialized Centers of Research in Molecular Medicine and Atherosclerosis), HL69464, and HL57505. M.-K. Chang was supported by a fellowship from Tobacco-Related Disease Research Program (TRDRP) and a Scientist Development grant from the American Heart Association (AHA). C.J. Binder was supported by a Ph.D. scholarship from the Boehringer Ingelheim Fonds, a scholarship from the Austrian Academy of Science, and a fellowship from the AHA. Y.I. Miller and J.A. Berliner were supported by TRDRP grant no. 12KT-0104 and United States Public Health Service grant no. HL30568, respectively.
The authors have no conflicting financial interests.
M.-K. Chang and C.J. Binder contributed equally to this work.
Abbreviations used in this paper: aPL, antiphospholipid antibody; LC/MRM, liquid chromatography multiple reaction monitoring; LDL, low density lipoprotein; lyso-PC, 1-palmitoyl-2-lyso-sn-glycero-3-PC; MDA, malondialdehyde; OxLDL, oxidized LDL; OxPL, oxidized phospholipid; PAEC, porcine aortic endothelial cell; PC, phosphorylcholine; PEIPC, 1-palmitoyl-2-epoxyisoprostane-sn-glycero-3-PC; POVPC, 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-PC; SLE, systemic lupus erythematosus.
References
- 1.Casciola-Rosen, L. 1994. Autoantigen targeted in systemic lupus erythematosus are clustered in two population of surface structure on apoptotic keratinocyte. J. Exp. Med. 179:1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cocca, B.A., A.M. Cline, and M.Z. Radic. 2002. Blebs and apoptotic bodies are B cell autoantigens. J. Immunol. 169:159–166. [DOI] [PubMed] [Google Scholar]
- 3.Price, B.E., J. Rauch, M.A. Shia, M.T. Walsh, W. Lieberthal, H.M. Gilligan, T. O'Laughlin, J.S. Koh, and J.S. Levine. 1996. Anti-phospholipid autoantibodies bind to apoptotic, but not viable, thymocytes in a beta 2-glycoprotein I-dependent manner. J. Immunol. 157:2201–2208. [PubMed] [Google Scholar]
- 4.Levine, J.S., J.S. Koh, R. Subang, and J. Rauch. 1999. Apoptotic cells as immunogen and antigen in the antiphospholipid syndrome. Exp. Mol. Pathol. 66:82–98. [DOI] [PubMed] [Google Scholar]
- 5.Mevorach, D., J.L. Zhou, X. Song, and K.B. Elkon. 1998. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J. Exp. Med. 188:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fadok, V.A., D.L. Bratton, and P.M. Henson. 2001. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J. Clin. Invest. 108:957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savill, J., I. Dransfield, C. Gregory, and C. Haslett. 2002. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2:965–975. [DOI] [PubMed] [Google Scholar]
- 8.Glass, C.K., and J.L. Witztum. 2001. Atherosclerosis. The road ahead. Cell. 104:503–516. [DOI] [PubMed] [Google Scholar]
- 9.Binder, C.J., M.K. Chang, P.X. Shaw, Y.I. Miller, K. Hartvigsen, A. Dewan, and J.L. Witztum. 2002. Innate and acquired immunity in atherogenesis. Nat. Med. 8:1218–1226. [DOI] [PubMed] [Google Scholar]
- 10.Navab, M., G.M. Ananthramaiah, S.T. Reddy, B.J. Van Lenten, B.J. Ansell, G.C. Fonarow, K. Vahabzadeh, S. Hama, G. Hough, N. Kamranpour, et al. 2004. Thematic review series: the pathogenesis of atherosclerosis: the oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J. Lipid Res. 45:993–1007. [DOI] [PubMed] [Google Scholar]
- 11.Hörkkö, S., C. Binder, P.X. Shaw, M.K. Chang, G. Silverman, W. Palinski, and J.L. Witztum. 2000. Immunological responses to oxidized LDL. Free Radic. Biol. Med. 28:1771–1779. [DOI] [PubMed] [Google Scholar]
- 12.Palinski, W., V.A. Ord, A.S. Plump, J.L. Breslow, D. Steinberg, and J.L. Witztum. 1994. ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler. Thromb. 14:605–616. [DOI] [PubMed] [Google Scholar]
- 13.Palinski, W., S. Hörkkö, E. Miller, U.P. Steinbrecher, H.C. Powell, L.K. Curtiss, and J.L. Witztum. 1996. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apo E-deficient mice. Demonstration of epitopes of oxidized LDL in human plasma. J. Clin. Invest. 98:800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang, M.-K., C. Bergmark, A. Laurila, S. Hörkkö, K.H. Han, P. Friedman, E.A. Dennis, and J.L. Witztum. 1999. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc. Natl. Acad. Sci. USA. 96:6353–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hörkkö, S., D.A. Bird, E. Miller, H. Itabe, N. Leitinger, G. Subbanagounder, J.A. Berliner, P. Friedman, E.A. Dennis, L.K. Curtiss, et al. 1999. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J. Clin. Invest. 103:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boullier, A., K.L. Gillotte, S. Hörkkö, D.R. Green, P. Friedman, E.A. Dennis, J.L. Witztum, D. Steinberg, and O. Quehenberger. 2000. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of lipoprotein. J. Biol. Chem. 275:9163–9169. [DOI] [PubMed] [Google Scholar]
- 17.Podrez, E.A., E. Poliakov, Z. Shen, R. Zhang, Y. Deng, M. Sun, P.J. Finton, L. Shan, B. Gugiu, P.L. Fox, et al. 2002. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J. Biol. Chem. 277:38503–38516. [DOI] [PubMed] [Google Scholar]
- 18.Shaw, P.X., S. Hörkkö, M.-K. Chang, L.K. Curtiss, W. Palinski, G.J. Silverman, and J.L. Witztum. 2000. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Invest. 105:1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Briles, D.E., C. Forman, S. Hudak, and J.L. Claflin. 1982. Antiphosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae. J. Exp. Med. 156:1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman, P., S. Hörkkö, D. Steinberg, J.L. Witztum, and E.A. Dennis. 2002. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. Importance of schiff base formation and aldol condensation. J. Biol. Chem. 277:7010–7020. [DOI] [PubMed] [Google Scholar]
- 21.Subbanagounder, G., J.W. Wong, H. Lee, K.F. Faull, E. Miller, J.L. Witztum, and J.A. Berliner. 2002. Epoxyisoprostane and epoxycyclopentenone phospholipids regulate monocyte chemotactic protein-1 and interleukin-8 synthesis. Formation of these oxidized phospholipids in response to interleukin-1beta. J. Biol. Chem. 277:7271–7281. [DOI] [PubMed] [Google Scholar]
- 22.Abbas, A.K., A.H. Litchman, and J.S. Pober. 2000. Cellular and Molecular Immunology. 4th edition. W.B. Saunders Co., Philadelphia, PA.
- 23.Friguet, B., A.F. Chaffotte, L. Djavadi-Ohaniance, and M.E. Goldberg. 1985. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J. Immunol. Methods. 77:305–319. [DOI] [PubMed] [Google Scholar]
- 24.Lee, C., F. Sagari, T. Segrado, S. Hörkkö, S.H.P.V. Subbaiah, M. Mowa, M. Navab, J.L. Witztum, and P. Reaven. 1999. All ApoB-containing lipoproteins induce monocyte chemotaxis and adhesion when minimally modified: modulation of lipoprotein bioactivity by platelet-activating factor acetylhydrolase. Arterioscler. Thromb. Vasc. Biol. 19:1437–1446. [DOI] [PubMed] [Google Scholar]
- 25.Shaw, P.X., C.S. Goodyear, M.K. Chang, J.L. Witztum, and G.J. Silverman. 2003. The autoreactivity of anti-phosphorylcholine antibodies for atherosclerosis-associated neo-antigens and apoptotic cells. J. Immunol. 170:6151–6157. [DOI] [PubMed] [Google Scholar]
- 26.Binder, C.J., S. Hörkkö, A. Dewan, M.K. Chang, E.P. Kieu, C.S. Goodyear, P.X. Shaw, W. Palinski, J.L. Witztum, and G.J. Silverman. 2003. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 9:736–743. [DOI] [PubMed] [Google Scholar]
- 27.Watson, A.D., N. Leitinger, M. Navab, K.F. Faull, S. Hörkkö, J.L. Witztum, W. Palinski, D.C. Schwenke, R.G. Salomon, W. Sha, et al. 1997. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J. Biol. Chem. 272:13597–13607. [DOI] [PubMed] [Google Scholar]
- 28.Leitinger, N., T.R. Tyner, L. Oslund, C. Rizza, G. Subbanagounder, H. Lee, P.T. Shih, N. Mackman, G. Tigyi, M.C. Territo, et al. 1999. Structurally similar oxidized phospholipids differentially regulate endothelial binding of monocytes and neutrophils. Proc. Natl. Acad. Sci. USA. 96:12010–12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cole, A.L., G. Subbanagounder, S. Mukhopadhyay, J.A. Berliner, and D.K. Vora. 2003. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMP-dependent R-Ras/PI3-kinase pathway. Arterioscler. Thromb. Vasc. Biol. 23:1384–1390. [DOI] [PubMed] [Google Scholar]
- 30.Gerszten, R.E., E.A. Garcia-Zepeda, Y.C. Lim, M. Yoshida, H.A. Ding, M.A. Gimbrone Jr., A.D. Luster, F.W. Luscinskas, and A. Rosenzweig. 1999. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 398:718–723. [DOI] [PubMed] [Google Scholar]
- 31.Huo, Y., C. Weber, S.B. Forlow, M. Sperandio, J. Thatte, M. Mack, S. Jung, D.R. Littman, and K. Ley. 2001. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J. Clin. Invest. 108:1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green, D.R., and J.C. Reed. 1998. Mitochondria and apoptosis. Science. 281:1309–1312. [DOI] [PubMed] [Google Scholar]
- 33.Fleury, C., B. Mignotte, and J.L. Vayssiere. 2002. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 84:131–141. [DOI] [PubMed] [Google Scholar]
- 34.Kagan, V.E., G.G. Borisenko, B.F. Serinkan, Y.Y. Tyurina, V.A. Tyurin, J. Jiang, S.X. Liu, A.A. Shvedova, J.P. Fabisiak, W. Uthaisang, and B. Fadeel. 2003. Appetizing rancidity of apoptotic cells for macrophages: oxidation, externalization, and recognition of phosphatidylserine. Am. J. Physiol. Lung Cell. Mol. Physiol. 285:L1–L17. [DOI] [PubMed] [Google Scholar]
- 35.Green, D.R., and H.M. Beere. 2000. Gone but not forgotten. Nature. 405:28–29. [DOI] [PubMed] [Google Scholar]
- 36.Rosen, A., and L. Casciola-Rosen. 1999. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ. 6:6–12. [DOI] [PubMed] [Google Scholar]
- 37.Botto, M., C. Dell'Agnola, A.E. Bygrave, E.M. Thompson, H.T. Cook, F. Petry, M. Loos, P.P. Pandolfi, and M.J. Walport. 1998. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 19:56–59. [DOI] [PubMed] [Google Scholar]
- 38.Cohen, P.L., R. Caricchio, V. Abraham, T.D. Camenisch, J.C. Jennette, R.A. Roubey, H.S. Earp, G. Matsushima, and E.A. Reap. 2002. Delayed apoptotic cell clearance and lupuslike autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 196:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scott, R.S., E.J. McMahon, S.M. Pop, E.A. Reap, R. Caricchio, P.L. Cohen, H.S. Earp, and G.K. Matsushima. 2001. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 411:207–211. [DOI] [PubMed] [Google Scholar]
- 40.Hanayama, R., M. Tanaka, K. Miyasaka, K. Aozasa, M. Koike, Y. Uchiyama, and S. Nagata. 2004. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 304:1147–1150. [DOI] [PubMed] [Google Scholar]
- 41.Vaarala, O. 2000. Antibodies to oxidised LDL. Lupus. 9:202–205. [DOI] [PubMed] [Google Scholar]
- 42.Svenungsson, E., K. Jensen-Urstad, M. Heimburger, A. Silveira, A. Hamsten, U. de Faire, J.L. Witztum, and J. Frostegard. 2001. Risk factors for cardiovascular disease in systemic lupus erythematosus. Circulation. 104:1887–1893. [DOI] [PubMed] [Google Scholar]
- 43.Soep, J.B., M. Mietus-Snyder, M.J. Malloy, J.L. Witztum, and E. von Scheven. 2004. Assessment of atherosclerotic risk factors and endothelial function in children and young adults with pediatric-onset systemic lupus erythematosus. Arthritis Rheum. 51:451–457. [DOI] [PubMed] [Google Scholar]
- 44.Hörkkö, S., E. Miller, E. Dudl, P. Reaven, L.K. Curtiss, N.J. Zvaifler, R. Terkeltaub, S.S. Pierangeli, D.W. Branch, W. Palinski, and J.L. Witztum. 1996. Antiphospholipid antibodies are directed against epitopes of oxidized phospholipids. Recognition of cardiolipin by monoclonal antibodies to epitopes of oxidized low density lipoprotein. J. Clin. Invest. 98:815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hörkkö, S., E. Miller, D.W. Branch, W. Palinski, and J.L. Witztum. 1997. The epitopes for some antiphospholipid antibodies are adducts of oxidized phospholipid and β2 glycoprotein 1 (and other proteins). Proc. Natl. Acad. Sci. USA. 94:10356–10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hörkkö, S., T. Olee, L. Mo, D.W. Branch, V.L. Woods Jr., W. Palinski, P.P. Chen, and J.L. Witztum. 2001. Anticardiolipin antibodies from patients with the antiphospholipid antibody syndrome recognize epitopes in both beta(2)-glycoprotein 1 and oxidized low-density lipoprotein. Circulation. 103:941–946. [DOI] [PubMed] [Google Scholar]
- 47.Mizutani, H., Y. Kurata, S. Kosugi, M. Shiraga, H. Kashiwagi, Y. Tomiyama, Y. Kanakura, R.A. Good, and Y. Matsuzawa. 1995. Monoclonal anticardiolipin autoantibodies established from the (New Zealand white x BXSB)F1 mouse model of antiphospholipid syndrome cross-react with oxidized low-density lipoprotein. Arthritis Rheum. 38:1382–1388. [DOI] [PubMed] [Google Scholar]
- 48.Guo, W.X., A.M. Burger, R.T. Fischer, D.G. Sieckmann, D.L. Longo, and J.J. Kenny. 1997. Sequence changes at the V-D junction of the VH1 heavy chain of anti-phosphocholine antibodies alter binding to and protection against Streptococcus pneumoniae. Int. Immunol. 9:665–677. [DOI] [PubMed] [Google Scholar]
- 49.Brown, M., G. Schiffman, and M.B. Rittenberg. 1984. Subpopulations of antibodies to phosphocholine in human serum. J. Immunol. 132:1323–1328. [PubMed] [Google Scholar]
- 50.Halpern, R., S.V. Kaveri, and H. Kohler. 1991. Human anti-phosphorylcholine antibodies share idiotopes and are self-binding. J. Clin. Invest. 88:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sigal, N.H., P.J. Gearhart, and N.R. Klinman. 1975. The frequency of phosphorylcholine-specific B cells in conventional and germfree BALB/C mice. J. Immunol. 114:1354–1358. [PubMed] [Google Scholar]
- 52.Zhang, M., W.G. Austen Jr., I. Chiu, E.M. Alicot, R. Hung, M. Ma, N. Verna, M. Xu, H.B. Hechtman, F.D. Moore Jr., and M.C. Carroll. 2004. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA. 101:3886–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silverstein, A.M. 2002. The clonal selection theory: what it really is and why modern challenges are misplaced. Nat. Immunol. 3:793–796. [DOI] [PubMed] [Google Scholar]
- 54.Kim, S.J., D. Gershov, X. Ma, N. Brot, and K.B. Elkon. 2002. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J. Exp. Med. 196:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takizawa, F., S. Tsuji, and S. Nagasawa. 1996. Enhancement of macrophage phagocytosis upon iC3b deposition on apoptotic cells. FEBS Lett. 397:269–272. [DOI] [PubMed] [Google Scholar]
- 56.Verbovetski, I., H. Bychkov, U. Trahtemberg, I. Shapira, M. Hareuveni, O. Ben Tal, I. Kutikov, O. Gill, and D. Mevorach. 2002. Opsonization of apoptotic cells by autologous iC3b facilitates clearance by immature dendritic cells, down-regulates DR and CD86, and up-regulates CC chemokine receptor 7. J. Exp. Med. 196:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morelli, A.E., A.T. Larregina, W.J. Shufesky, A.F. Zahorchak, A.J. Logar, G.D. Papworth, Z. Wang, S.C. Watkins, L.D. Falo Jr., and A.W. Thomson. 2003. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood. 101:611–620. [DOI] [PubMed] [Google Scholar]
- 58.Huber, J., A. Vales, G. Mitulovic, M. Blumer, R. Schmid, J.L. Witztum, B.R. Binder, and N. Leitinger. 2002. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 22:101–107. [DOI] [PubMed] [Google Scholar]
- 59.Aprahamian, T., I. Rifkin, R. Bonegio, B. Hugel, J.M. Freyssinet, K. Sato, J.J. Castellot Jr., and K. Walsh. 2004. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J. Exp. Med. 199:1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross, R. 1999. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 340:115–126. [DOI] [PubMed] [Google Scholar]
- 61.Marathe, G.K., K.A. Harrison, R.C. Murphy, S.M. Prescott, G.A. Zimmerman, and T.M. McIntyre. 2000. Bioactive phospholipid oxidation products. Free Radic. Biol. Med. 28:1762–1770. [DOI] [PubMed] [Google Scholar]
- 62.Martinet, W., and M.M. Kockx. 2001. Apoptosis in atherosclerosis: focus on oxidized lipids and inflammation. Curr. Opin. Lipidol. 12:535–541. [DOI] [PubMed] [Google Scholar]
- 63.Luscinskas, F.W., R.E. Gerszten, E.A. Garcia-Zepeda, Y.C. Lim, M. Yoshida, H.A. Ding, M.A. Gimbrone Jr., A.D. Luster, and A. Rosenzweig. 2000. C-C and C-X-C chemokines trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Ann. NY Acad. Sci. 902:288–293. [DOI] [PubMed] [Google Scholar]
- 64.Boisvert, W.A., R. Santiago, L.K. Curtiss, and R.A. Terkeltaub. 1998. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J. Clin. Invest. 101:353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lorimore, S.A., P.J. Coates, G.E. Scobie, G. Milne, and E.G. Wright. 2001. Inflammatory-type responses after exposure to ionizing radiation in vivo: a mechanism for radiation-induced bystander effects? Oncogene. 20:7085–7095. [DOI] [PubMed] [Google Scholar]
- 66.Lucas, M., L.M. Stuart, J. Savill, and A. Lacy-Hulbert. 2003. Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion. J. Immunol. 171:2610–2615. [DOI] [PubMed] [Google Scholar]
- 67.Lauber, K., E. Bohn, S.M. Krober, Y.J. Xiao, S.G. Blumenthal, R.K. Lindemann, P. Marini, C. Wiedig, A. Zobywalski, S. Baksh, et al. 2003. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 113:717–730. [DOI] [PubMed] [Google Scholar]
- 68.Quinn, M.T., S. Parthasarathy, and D. Steinberg. 1988. Lysophosphatidylcholine: a chemotactic factor for human monocytes and its potential role in atherogenesis. Proc. Natl. Acad. Sci. USA. 85:2805–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ryborg, A.K., B. Deleuran, K. Thestrup-Pedersen, and K. Kragballe. 1994. Lysophosphatidylcholine: a chemoattractant to human T lymphocytes. Arch. Dermatol. Res. 286:462–465. [DOI] [PubMed] [Google Scholar]